

Abstract
Purpose of review
The pathogenesis of acute pancreatitis (AP) is still not well understood. This articles reviews recent advances in our understanding of AP with emphasis on literature published during the last year.
Recent Findings
Zymogen activation was shown to be sufficient to induce AP. Another key early event, NFkB activation has previously been shown to induce AP. The relationship between these two key early steps is beginning to be clarified. The mechanisms responsible for zymogen activation- pathologic calcium signaling, pH changes, colocalization and autophagy; mechanisms of NFkB activation and potential therapeutic targets both upstream and downstream of these key events have been explored. Additional key findings have been elucidation of the dual role of oxidative stress in AP and role of bioenergetics in determining mode of cell death, recognition of endoplasmic reticulum stress as an early step and duct cells as important players in pancreatic injury.
Summary
Current findings have provided further insight into the roles and mechanisms of zymogen activation and inflammatory pathways in pancreatic injury. Future studies are being undertaken to establish the relative contributions of these pathways during acute pancreatitis which will be critical to identifying successful therapeutic targets.
Keywords: Acute pancreatitis, Trypsin, NFkB, ER stress
Introduction
The pathogenesis of Acute Pancreatitis (AP) remains to be elusive despite significant advances in the last 25 years. Beginning with proposition of Chiari (1) more than a century back that autodigestion by prematurely activated digestive enzymes is responsible for this debilitating disease, significant research has and continues to be focused on mechanisms responsible for this premature zymogen activation (2–5). Additionally, for over a decade now, NFkB activation has been observed early paralleling trypsinogen activation (6, 7). The relationship between these key events has been a matter of hot debate (7–10) and their relative contribution to pancreatic injury is currently not established. In this brief review, we first summarize current research on the role of zymogen activation and NFkB activation in pancreatic injury and then discuss recent literature exploring the mechanisms responsible for these key events and how these have guided our search for potential therapeutic targets.
Zymogen Activation and NFkB activation- parallel players capable of pancreatic injury
Intracellular trypsinogen activation has consistently been observed early during the course of pancreatitis in experimental models (2, 3, 5, 11–16). The association of hereditary pancreatitis with mutations leading to high intracellular trypsin activity supports that trypsinogen activation is crucial in pancreatic injury (17, 18). In-vitro experiments using adenoviral gene transfer techniques to express active trypsin or mutated trypsinogens within pancreatic acini have provided further confirmation as well as mechanistic insights into acinar cell death induction by persistent intra-acinar trypsinogen activation (19–21).
In a landmark study published this year, Gaiser et al demonstrated that intra-acinar expression of active trypsin in-vivo was sufficient to induce cell death and inflammation resulting in AP (22). This study used a conditionally inducible pancreas acini-specific trypsinogen construct which was activated during post-translational modification. On the other hand, activation of NFkB, early event paralleling trypsinogen activation in time course, has also been shown to result in AP (23–25).
Both trypsinogen activation and NFkB activation are therefore sufficient to induce AP though their relationship has been debated for a long time (7–10). In-vitro expression of active trypsin failed to activate NF-kB (19) suggesting that these two events are independent which is further supported by our data (26). Thus it remains to be established whether these two independent and parallel players, each sufficient to result in pancreatic injury, are prerequisite for development of AP (figure 1). The elucidation of the relative contribution of these events is crucial in advancing our understanding of pancreatitis. Our group has recently developed novel knockout mice which lack trypsinogen7 gene, the mouse correlate of human cationic trypsinogen. These mice lack pathologic trypsinogen activation. Using these mice, studies are currently underway to study these crucial issues in the pathogenesis of AP.
Figure 1. Two key parallel and independent events occurring early during pancreatitis.
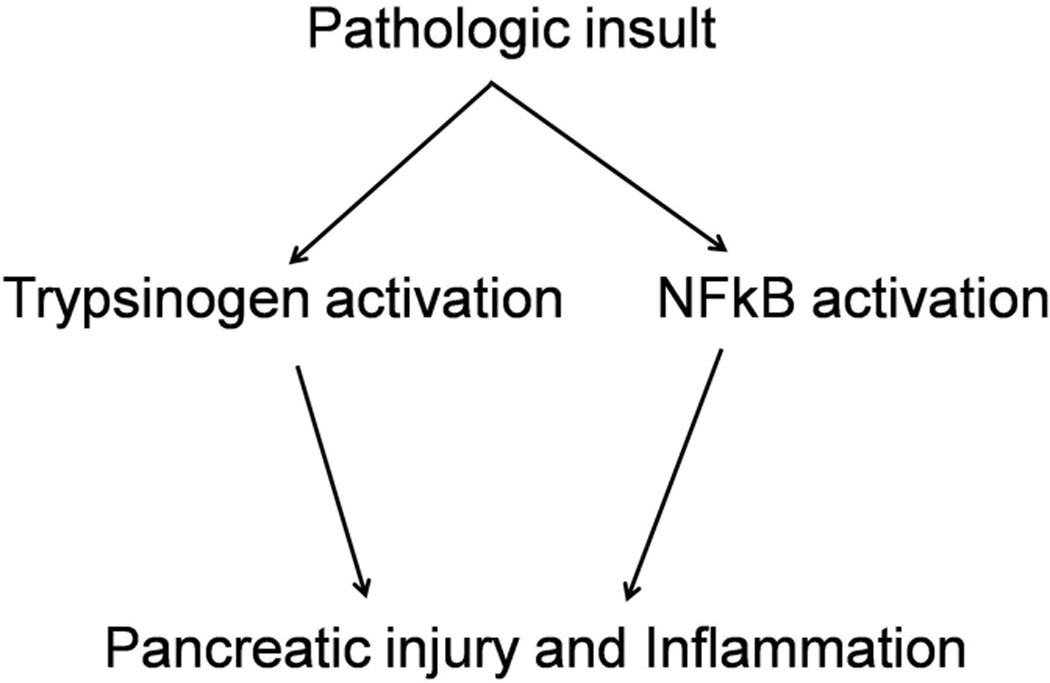
Both these events are capable of causing pancreatic damage leading to acute pancreatitis. The relative contribution of these events in acute pancreatitis is one of the central questions in the pathogenesis of pancreatic injury at present.
Mechanisms of Zymogen activation
a) Pathologic calcium signaling
Cytosolic calcium influx [Ca2+i] is crucial in both physiological and pathological responses in the acinar cell, and understanding this differential response has been a great accomplishment in recent times. Unlike physiologic response which involves localized transient spikes in Ca2+i, pathologic response involves a sustained global rise in Ca2+i (27–29). This response is seen in experimental models of pancreatitis and its blockade leads to inhibition of trypsinogen activation (3, 11).
Recent research has focused on the sources of this pathologic calcium response. Our current understanding of the sources as well as elimination routes of Ca2+i and their relevance in pancreatic injury has been depicted in figure 2 (30–33). Endoplasmic reticulum (ER) membrane Ryanodine Receptors (RyR) (34) and plasma membrane store operated calcium channels (SOCs) (35, 36) have been implicated as important sources. In this context, pharmacologic antagonism of RyR using Dantrolene (37), and genetic and pharmacologic inhibition of TRPC3, a recently recognized SOC have been shown to reduce zymogen activation as well as pancreatic damage. Prolongation of the pathologic Ca2+i by inhibition of SERCA (38) (figure 2) or by ATP depletion (39) (figure 2 and 3) have been recognized as important mechanisms of pancreatic injury by bile acids and ethanol metabolites.
Figure 2. Sources and clearance routes of pathologic cytoplasmic calcium response [Ca2+i].
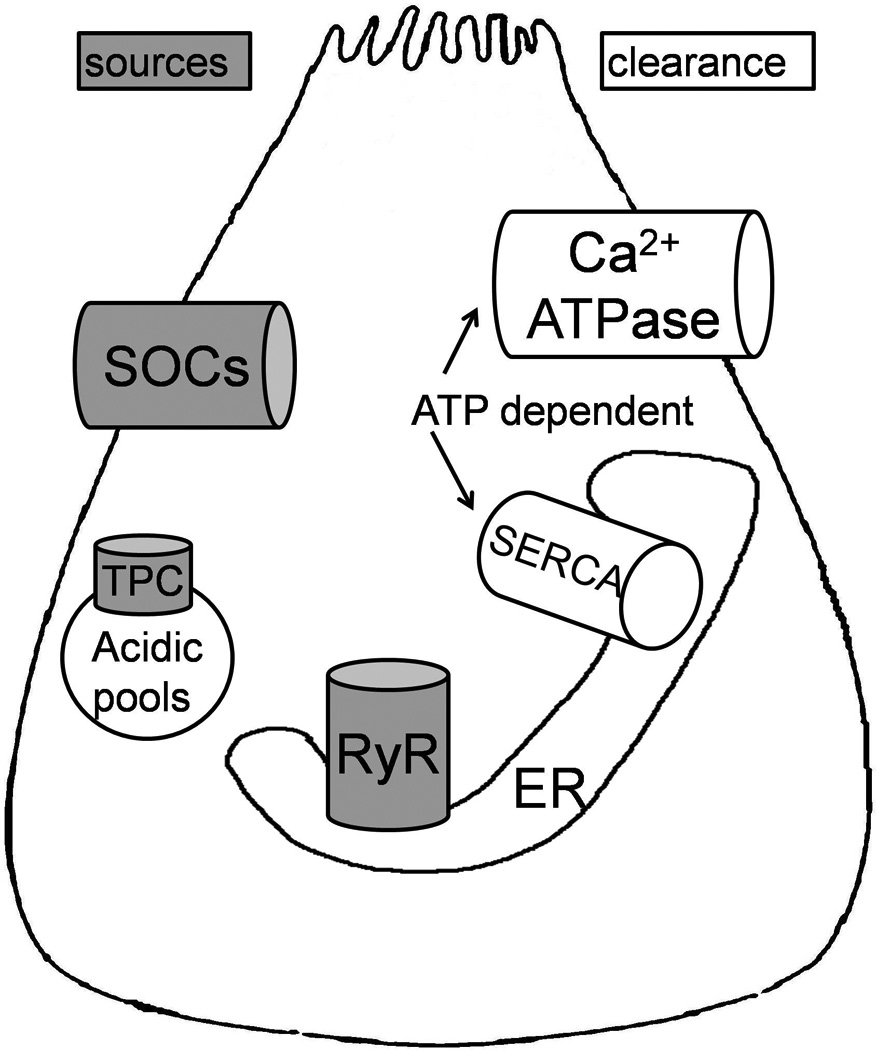
Ryanodine Receptors (RyR) (34) and store operated calcium channels (SOCs) (35, 36) are major sources of Ca2+i. RyRs are calcium sensitive channels, and open in response to mild rise in Ca2+i although cADPR and NADDP are also possible RyR ligands (27–29, 33). The identity of SOCs and the mechanism of their regulation by ER calcium signals has been a field of active research. Recently TRPC3 and ORAI channels have been identified as important SOCs (35, 36) and it is postulated that STIMs that sit on ER membrane sense calcium depletion within the ER (as would occur after opening of RyRs) and migrate to plasma membrane where they open the SOCs (35, 36). Acidic pools are thought to be important in alcohol induced injury (30) and include organelles with low pH such as lysosomes, endososomes and zymogens. Recently recognized Two Pore Channels (TPCs) release calcium from acid pools (31–33). Mitochondria have been recognized as another source of calcium (not shown in the figure). Note that clearance of Ca2+i is an ATP requiring process, and ATP depletion or direct inhibition of SERCA prolongs Ca2+i, a mechanism thought to be important in pancreatic injury due to bile acids and ethanol metabolites.
Figure 3. Signal transduction events resulting in pathologic trypsinogen activation and NFkB activation.
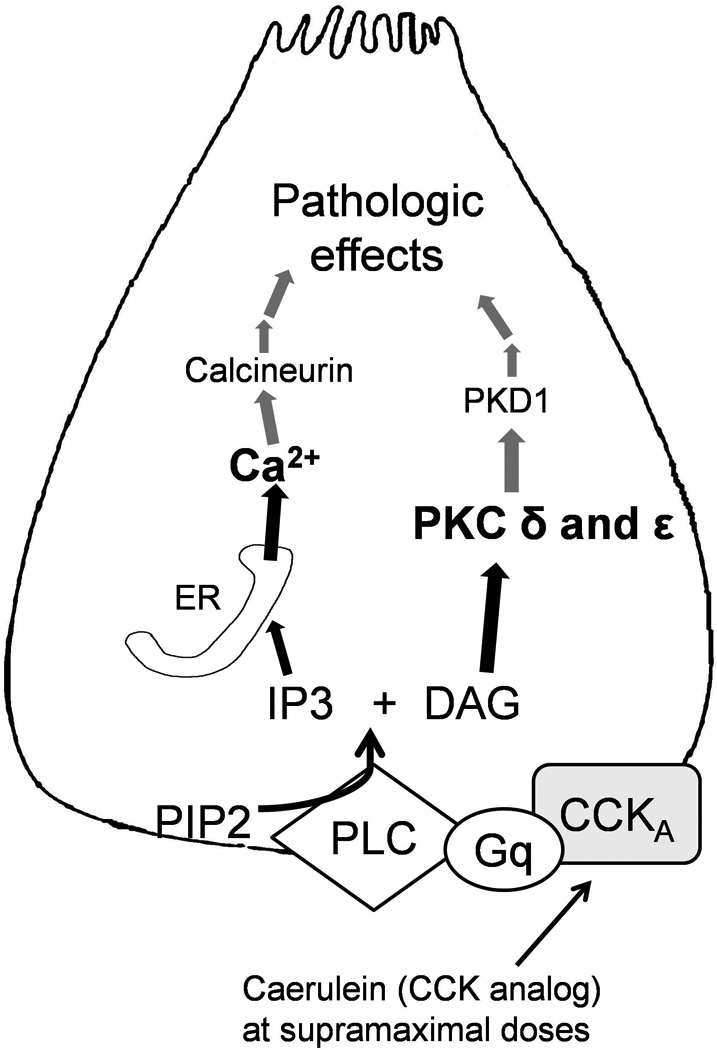
Cholecystokinin analog Caerulein induced pancreatitis has been used as a model in this schematic. CCKA: Cholecystokinin receptor subtype A, Gq: G-protein q subtype; PLC: phospholipase C, PIP2: Phosphoinositol 4-phosphate, IP3: Inositol-3 Phosphate, DAG: Diacylglycerol; PKC: protein kinase C, PKD: protein kinase D. Caerulein (CCK analog) binds to its receptor as shown and leads to generation of IP3 and DAG. IP3 opens ER membrane IP3 receptors which are implicated in physiologic calcium signaling. Calcium released through IP3R leads to opening of RyRs as described in figure 2. The grey lines in the figure depict either unknown steps or proposed mechanisms awaiting verification in future studies.
The downstream targets of Ca2+i are currently unknown though recently Calcineurin has been proposed as one such target (figure 3) (40). Calcineurin is a well recognized downstream effector of Ca2+i in several inflammatory processes, especially in T-cell activation and its inhibitors have been clinically successful. Of note, Calcineurin inhibitor tacrolimus resulted in decreased zymogen activation and reduced parameters of pancreatic damage (41).
b) Colocalization of lysosomes and zymogens
We and others have shown that premature trypsinogen activation takes place in membrane-bound compartments of autophagic nature where zymogen and lysosomal contents colocalize (13, 42–44). Lysosomal cathepsin B is believed to activate trypsinogen in these colocalization vacuoles (figure 3) (13, 45). However, it appears that this process requires additional conditions, most likely low pH. Support for this hypothesis comes from the finding that missorting of cathepsin B into the secretory compartment alone failed to activate trypsinogen but only enhanced trypsin activity during AP (46). In fact, in-vitro experiments indicate that a low pH significantly enhances catalytic activity of cathepsin B to activate trypsinogen (43). The mechanism that could account for the lowering of pH in these vacuoles was recently identified as vacuolar ATPase (vATPase) which pumps protons into the vacuoles (figure 3) (4, 47).
c) Sensitizing effects of low extracellular pH (pHe)
Acidemia has been known to a precipitant of acute pancreatitis. But until now it was not clear how extracellular pH affects the acinar cell. RyR mediated increased the amplitude of pathologic Ca2+i (48), enhanced proton transfer into colocalization vacuoles by vATPase (49) and disruption of intercellular junctional coupling possibly facilitating spread of activated zymogens into intra-cellular spaces (50) are the so far described pathologic effects of low pHe (figure 4). Interestingly, Behrendorff et al recently showed that physiological secretion causes acidification of pancreatic lumen and the released protons appeared to function as a negative-feedback regulating acinar secretion (50). However, supramaximal stimulation with caerulein led to extreme prolonged acidification of lumen, thus causing low pHe (50). Further inhibited bicarbonate secretion by duct cells appears to be another mechanism leading to low pHe (described later).
Figure 4. Effect of low extracellular pH, role of bioenergetics in determining cell fate and dual role of oxidative stress.
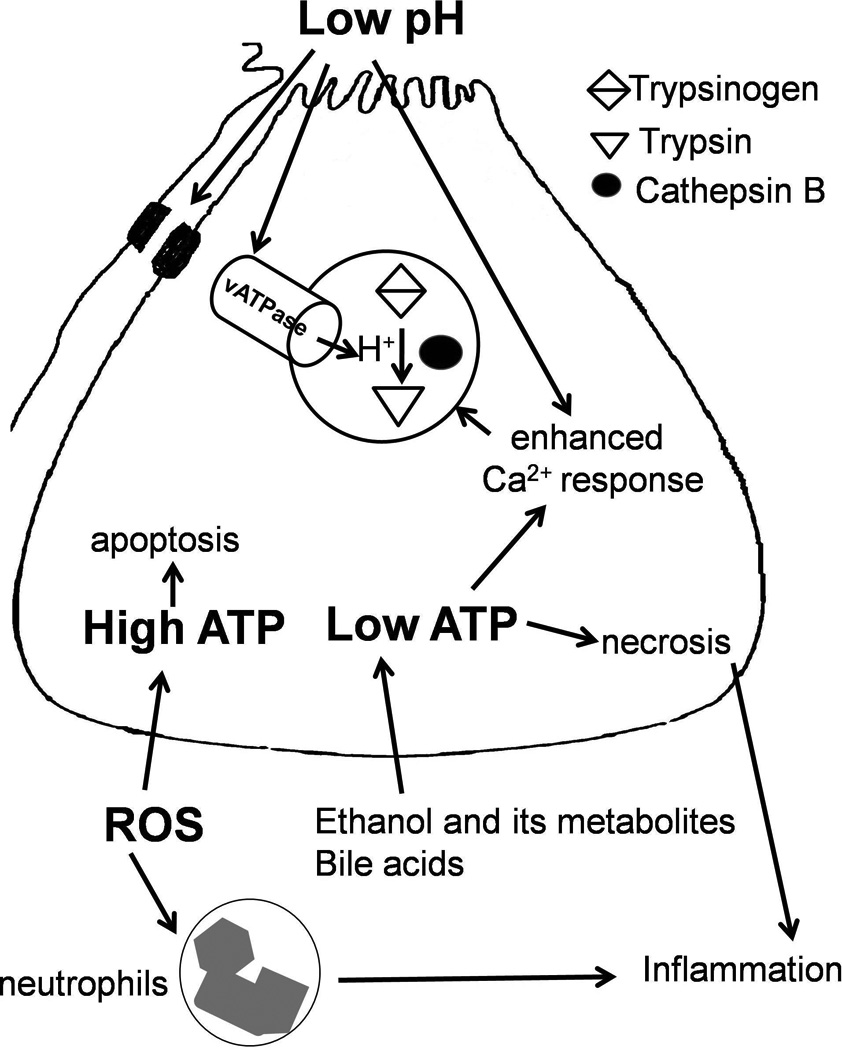
Low pH in the lumen leads to enhanced activity of vATPase, enhanced pathologic calcium response and disruption of intercellular junctions leading to zymogen activation and spread of activated zymogens causing further damage (48–50). High ATP states favor apoptosis and this mode of death avoids inflammatory response leading to relatively less severe injury compared to necrosis which elicits intense inflammation (39). Necrosis is the mode of death during low ATP states and causes severe pancreatic injury. ROS depletion in the acinar cells leads to low ATP state and favors necrosis while ROS induction favors apoptosis avoiding severe pancreatic damage, and therefore seems to be protective to the acinar cell (72). At the same time, ROS in the neutrophils leading to inflammation may contribute to pancreatic injury. Thus oxidative stress spears to have dual role in pancreatitis.
d) Impaired autophagy, Zymophagy and lysosomal cathepsins
It is currently believed that the colocalization organelles are of autophagic nature as demonstrated by presence of LC3-II on the membrane of these organelles (51, 52). However the role of autophagy during pancreatitis has been controversial. Genetic inhibition of autophagy reduced trypsinogen activation and pancreatic damage indicating that autophagy is a harmful response (51). In contrast, Grasso et al described a selective autophagy which is a protective response to sequester and degrade potentially deleterious activated zymogens during early pancreatitis response (53). This protective autophagy was termed zymophagy. Inhibiting zymophagy reduced acinar cell survival while upregulating zymophagy led to reduced trypsinogen activation and improvement in all parameters of pancreatic injury (53). Another group suggested that during pancreatitis autophagy is retarded because of imbalance in cathepsin B (activates trypsinogen) and cathepsin L (degrades trypsin) due to misprocessing (52). Under physiological conditions, prematurely activated trypsinogen would undergo degradation by cathepsin L but during pancreatitis this autophagic degradation is retarded due to imbalance between cathepsins B and L (52).
Mechanisms of inflammatory pathways activation
NFkB activation has been observed in experimental models paralleling trypsinogen activation in time course (6–10, 54). Current data suggests that pathologic Ca2+i as well as activation of novel isoforms of protein kinase C are responsible for NFkB activation. The evidence for the role of pathologic Ca2+i comes from experiment using pharmacologic agents to block Ca2+i which led to inhibition of NFkB (7). Recent studies have established that activation of novel protein kinase C (PKC) isoforms ε and ζ are involved in NFkB activation using caerulein and ethanol based models of pancreatitis (55–59). These novel PKC isoforms also appear to be involved in zymogen activation (56, 60). It is currently unknown if the effect of pathologic Ca2+i and PKC in activation of these early pancreatitis responses are independent. The signal transduction steps leading to PKC activation and pathologic Ca2+i have been depicted in figure 3 based on the caerulein model of pancreatitis. The downstream targets of PKC as well as Ca2+i have so far been unknown. Protein kinase D1 has been proposed as the downstream target of PKC isoforms (61) recently, though PKD1 activation might also occur by other PKC independent mechanisms (62). Interestingly, pharmacologic inhibition of PKD1 resulted in reduced trypsinogen activation and NFkB activation but cell injury was not affected (63). Similarly, as described earlier, calcinerin has been proposed as downstream target of Ca2+i though its effects on NFkB activation during pancreatic is not known.
Mechanisms other than Calcium or PKC dependent pathways have also been put forth, notable among these being angiotensin 2 acting through AT2 receptors (64) and p38/MAPK pathways (65, 66). In a recent study, the role of acinar cell injury by itself to activate inflammatory cascades was explored. It was found that cell injury may lead to activation of damage-associated molecular pattern (DAMP) receptors (TLR-9 and P2X-7 recognized as important ones). Activation of DAMP receptors induces formation of a cytosolic complex termed the inflammasome that is implicated in initiating inflammatory cascade during pancreatic injury (67).
Bioenergetics, oxidative stress, determination of cell fate and its effects on pancreatic injury
Apoptosis is known to eliminate damaged cells without eliciting much inflammation while overwhelming cell injury shifts the cell death to necrosis which leads to widespread inflammation. However, apoptosis is an active process requiring energy. Therefore bioenergetics of the acinar cell is crucial in determining whether apoptosis or necrosis occurred during pancreatic injury (figure 4). Though it is misleading to say that apoptosis is a protective response, severity of pancreatitis is considerably less when apoptosis is the predominant mechanism compared to when necrosis is predominant (68). In an interesting study, Voronina et al found that physiological secretagouges (CCK, Acetyl choline, Bombesin) led to increase in ATP production while bile acid and non-oxidative metabolites of alcohol led to ATP depletion (figure 4) (39). This novel finding explains why bile acid and ethanol metabolites cause severe pancreatic injury (low ATP state leading to necrosis). In fact, intracellular administration of ATP abolished acinar injury in response to ethanol metabolites (69). On the other hand, positive bioenergetics resulting from physiological secretagouges would support normal physiological function of enzyme secretion (39). Thus bioenergetics alterations by toxic agents seem to be crucial in determining extent of pancreatic injury.
Oxidative stress is generally believed to be detrimental and going by this logic, several antioxidants have been tried in pancreatitis although with limited success (70, 71). A recent study investigating the role of Reactive Oxygen Species (ROS) in pancreatitis produced surprising findings (72). In this study, ROS induction in the acinar cells promoted apoptosis while inhibition of ROS generation led to increase in necrosis accompanied by reduced ATP (figure 4) (72). These findings suggest that ROS generation within acinar cells may be a protective response during pancreatitis. At the same time, oxidative stress in the neutrophils activated during inflammatory response to acinar injury may be responsible for further propagation of local and systemic inflammation (figure 4) (72). Therefore oxidative stress appears to have a dual role in pancreatic injury.
Endoplasmic Reticulum (ER) Stress- an early event
ER stress is observed early paralleling trypsinogen activation in caerluein model as well as L-arginine model of AP (73, 74). It is currently unknown whether ER stress is dependent on trypsinogen activation or is an independent event. However ER stress is known to be able to induce cell death pathways as well as activate inflammatory pathways in other cell systems (75). Szmola and Sahin-Toth recently showed that expression of mutant chymotrypsin (CTRC) associated with hereditary pancreatitis in acinar cells led to accumulation of misfolded CTRC in the ER leading to ER stress activation and eventually cell death by apoptosis (76). This study proposes ER stress as a novel mechanism of pancreatic injury. Reducing ER stress using tauroursodeoxycholic acid, a chemical chaperone, or by using genetic manipulation of GRP78 showed protective effect in AP (77) (78).
The role of ER stress responses, also collectively also known as Unfolded Protein Response (UPR), has been explored in alcohol related pancreatitis in considerable detail (79). It appears that a functional UPR effectively clears ethanol induced oxidative ER stress and provides resistance to ethanol induced pancreatic damage (79). However, XBP1+/− mice fed with ethanol had significantly increased pancreatic damage (79). XBP1 is an essential component in the induction of UPR, and its partial deletion in XBP1+/− led to a defective UPR. Thus a defective UPR unmasked ethanol induced pancreatic damage (79). The role of the functional ER responses in other forms of pancreatic injury is currently unclear.
Duct cells – not silent bystanders
The elucidation of pathophysiology of ductal cells and its relevance during pancreatitis has been a major advance in recent years (80). Current research suggests that injurious stimulus alter normal physiological functions of duct cells as much as it does for the acinar cells. For example, bile acids at lower doses leads to enhanced ductal secretion while at higher doses ductal secretion is inhibited (81). Similar response occurs in response to ethanol related models (82). Based on these findings, Hegyi et al have put forth an interesting hypothesis that enhanced ductal fluid secretion during early stages of pancreatitis may defend against damage by washing out toxic agents and digestive enzymes while overwhelming of this ductal defense mechanism leads to pancreatic injury (80). Inhibited bicarbonate secretion may also lead to reduction in luminal pH which has recently been shown to contribute to intra-acinar zymogen activation, as described in figure 4 (48, 50, 80). In addition to inhibited bicarbonate secretion, it was recently shown that non-conjugated chenodeoxycholate at high doses led to mitochondrial injury and ductal ATP depletion (83). While membrane GPBAR1 receptors may mediate noxious effects of bile acids on acinar cells (38), duct cells lack these membrane receptors for bile acids (84). Instead duct cells have strong expression of large-conductance Ca2+ activated potassium channels (BK channels) which regulate bicarbonate secretion and mediate the bile acid induced responses in duct cells (84).
The search for therapy
Inflammation has been a major target of developing therapy for pancreatitis. Numerous anti-inflammatory agents – thalidomide (believed to target TNFα) (85, 86), panhaematin (which decreases leukocyte infiltration and chemokine release) (87), leukotriene receptor 1 antagonist montelukast (88), leukocyte function antigen (which was shown to inhibit neutrophil chemotaxis) (89), MCP1 inhibitors (90), COX-2 inhibiotor flavocoxid (91), calcineurin inhibitor tacrolimus (41, 92), calcium channel RyR antagonist Dantrolene (37), Rho-kinase inhibitor (93), activator of stress kinases and apoptotis Betacellulin (94), vitamin K3 (shown to inhibit autophagy) (95) and broad anti-inflammatory agents like quercetin (96), resveratrol (97), pentoxyphylline (98), circumin (99, 100), and even human bone marrow derived clonal mesenchymal stem cells (101, 102) have all been tried within the last two years in experimental pancreatitis, and remarkably each one of them has shown beneficial response.
The biggest challenge in the field of pancreatitis research lies in finding clinical utility of these potential targets. A recent trial of Activated protein C (103, 104) failed to show any benefit reminding of the failure of the much-hyped Lexipafant in clinical trial more than a decade ago (105). The failure of valdecoxib in combination with glyceryl trinitrate in preventing post ERCP pancreatitis is another such example (106). These are constant reminders that there is a lot still to be learnt about the pathogenesis of pancreatic injury.
Conclusion
The understanding of mechanisms of pancreatic injury has advanced considerably though there is a lot more that remains to be explored. Lack of any therapy for this debilitating disease so far has been a serious setback in the field. Targeting multiple targets may be more effective than the current approach of single target directed approach. Further insight into the mechanisms and contributions of the key events - activation of zymogen and inflammatory pathways activation will be crucial in our understanding of pancreatitis.
Footnotes
Financial Disclosures: None.
Conflict of Interest: None to disclose.
References
- 1.Chiari H. ÜberdieSelbstverdauung des menschlichen Pankreas. Zeitschriftfür Heilkunde. 1896;17:69–96. [Google Scholar]
- 2.Halangk W, Lerch MM. Early events in acute pancreatitis. Clin Lab Med. 2005;25(1):1–15. doi: 10.1016/j.cll.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Saluja AK, Lerch MM, Phillips PA, Dudeja V. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol. 2007;69:249–269. doi: 10.1146/annurev.physiol.69.031905.161253. [DOI] [PubMed] [Google Scholar]
- 4.Gorelick FS, Thrower E. The acinar cell and early pancreatitis responses. Clin Gastroenterol Hepatol. 2009;7(11 Suppl):S10–S14. doi: 10.1016/j.cgh.2009.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofbauer B, Saluja AK, Lerch MM, Bhagat L, Bhatia M, Lee HS, Frossard JL, Adler G, Steer ML. Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am J Physiol. 1998;275(2 Pt 1):G352–G362. doi: 10.1152/ajpgi.1998.275.2.G352. [DOI] [PubMed] [Google Scholar]
- 6.Gukovsky I, Gukovskaya AS, Blinman TA, Zaninovic V, Pandol SJ. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol. 1998;275(6 Pt 1):G1402–G1414. doi: 10.1152/ajpgi.1998.275.6.G1402. [DOI] [PubMed] [Google Scholar]
- 7.Hietaranta AJ, Saluja AK, Bhagat L, Singh VP, Song AM, Steer ML. Relationship between NF-kappaB and trypsinogen activation in rat pancreas after supramaximal caerulein stimulation. Biochem Biophys Res Commun. 2001;280(1):388–395. doi: 10.1006/bbrc.2000.4120. [DOI] [PubMed] [Google Scholar]
- 8.Rakonczay Z, Jr, Hegyi P, Takacs T, McCarroll J, Saluja AK. The role of NF-kappaB activation in the pathogenesis of acute pancreatitis. Gut. 2008;57(2):259–267. doi: 10.1136/gut.2007.124115. [DOI] [PubMed] [Google Scholar]
- 9.Han B, Ji B, Logsdon CD. CCK independently activates intracellular trypsinogen and NF-kappaB in rat pancreatic acinar cells. Am J Physiol Cell Physiol. 2001;280(3):C465–C472. doi: 10.1152/ajpcell.2001.280.3.C465. [DOI] [PubMed] [Google Scholar]
- 10.Tando Y, Algul H, Schneider G, Weber CK, Weidenbach H, Adler G, Schmid RM. Induction of IkappaB-kinase by cholecystokinin is mediated by trypsinogen activation in rat pancreatic lobules. Digestion. 2002;66(4):237–245. doi: 10.1159/000068364. [DOI] [PubMed] [Google Scholar]
- 11.Saluja AK, Bhagat L, Lee HS, Bhatia M, Frossard JL, Steer ML. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol. 1999;276(4 Pt 1):G835–G842. doi: 10.1152/ajpgi.1999.276.4.G835. [DOI] [PubMed] [Google Scholar]
- 12.Saluja A, Saluja M, Villa A, Leli U, Rutledge P, Meldolesi J, Steer M. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J Clin Invest. 1989;84(4):1260–1266. doi: 10.1172/JCI114293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saluja AK, Donovan EA, Yamanaka K, Yamaguchi Y, Hofbauer B, Steer ML. Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology. 1997;113(1):304–310. doi: 10.1016/s0016-5085(97)70108-2. [DOI] [PubMed] [Google Scholar]
- 14.Lerch MM, Gorelick FS. Early trypsinogen activation in acute pancreatitis. Med Clin North Am. 2000;84(3):549–563. viii. doi: 10.1016/s0025-7125(05)70239-x. [DOI] [PubMed] [Google Scholar]
- 15.Kloppel G, Dreyer T, Willemer S, Kern HF, Adler G. Human acute pancreatitis: its pathogenesis in the light of immunocytochemical and ultrastructural findings in acinar cells. Virchows Arch A Pathol Anat Histopathol. 1986;409(6):791–803. doi: 10.1007/BF00710764. [DOI] [PubMed] [Google Scholar]
- 16.Willemer S, Kloppel G, Kern HF, Adler G. Immunocytochemical and morphometric analysis of acinar zymogen granules in human acute pancreatitis. Virchows Arch A Pathol Anat Histopathol. 1989;415(2):115–123. doi: 10.1007/BF00784348. [DOI] [PubMed] [Google Scholar]
- 17.Whitcomb DC. Genetic aspects of pancreatitis. Annu Rev Med. 2010;61:413–424. doi: 10.1146/annurev.med.041608.121416. [DOI] [PubMed] [Google Scholar]
- 18.Chen JM, Ferec C. Chronic pancreatitis: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2009;10:63–87. doi: 10.1146/annurev-genom-082908-150009. [DOI] [PubMed] [Google Scholar]
- 19.Ji B, Gaiser S, Chen X, Ernst SA, Logsdon CD. Intracellular trypsin induces pancreatic acinar cell death but not NF-kappaB activation. J Biol Chem. 2009;284(26):17488–17498. doi: 10.1074/jbc.M109.005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kereszturi E, Sahin-Toth M. Intracellular autoactivation of human cationic trypsinogen mutants causes reduced trypsinogen secretion and acinar cell death. J Biol Chem. 2009;284(48):33392–33399. doi: 10.1074/jbc.M109.056812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaiser S, Ahler A, Gundling F, Kruse ML, Savkovic V, Selig L, Teich N, Tomasini R, Dagorn JC, Mossner J, Keim V, Bodeker H. Expression of mutated cationic trypsinogen reduces cellular viability in AR4-2J cells. Biochem Biophys Res Commun. 2005;334(2):721–728. doi: 10.1016/j.bbrc.2005.06.148. [DOI] [PubMed] [Google Scholar]
- 22.Gaiser S, Daniluk J, Liu Y, Tsou L, Chu J, Lee W, Longnecker DS, Logsdon CD, Ji B. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut. 2011 doi: 10.1136/gut.2010.226175. gut.2010.226175 [pii]10.1136/gut.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Ji B, Han B, Ernst SA, Simeone D, Logsdon CD. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology. 2002;122(2):448–457. doi: 10.1053/gast.2002.31060. [DOI] [PubMed] [Google Scholar]
- 24.Baumann B, Wagner M, Aleksic T, von Wichert G, Weber CK, Adler G, Wirth T. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J Clin Invest. 2007;117(6):1502–1513. doi: 10.1172/JCI30876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aleksic T, Baumann B, Wagner M, Adler G, Wirth T, Weber CK. Cellular immune reaction in the pancreas is induced by constitutively active IkappaB kinase-2. Gut. 2007;56(2):227–236. doi: 10.1136/gut.2005.084665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dawra RK, Delori LR, Bekolay A, Sah RP, Saluja AK. Deletion of Cationic Trypsinogen or Cathepsin B Gene in Mice Does not Influence Caerulein Induced Activation of Nfkb in Pancreas. Pancreas. 2010;39(8):1317–1318. [Google Scholar]
- 27.Criddle DN, Gerasimenko JV, Baumgartner HK, Jaffar M, Voronina S, Sutton R, Petersen OH, Gerasimenko OV. Calcium signalling and pancreatic cell death: apoptosis or necrosis? Cell Death Differ. 2007;14(7):1285–1294. doi: 10.1038/sj.cdd.4402150. [DOI] [PubMed] [Google Scholar]
- 28.Petersen OH. Ca2+-induced pancreatic cell death: roles of the endoplasmic reticulum, zymogen granules, lysosomes and endosomes. J Gastroenterol Hepatol. 2008;23(Suppl 1):S31–S36. doi: 10.1111/j.1440-1746.2007.05281.x. [DOI] [PubMed] [Google Scholar]
- 29.Straub SV, Giovannucci DR, Yule DI. Calcium wave propagation in pancreatic acinar cells: functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J Gen Physiol. 2000;116(4):547–560. doi: 10.1085/jgp.116.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerasimenko JV, Lur G, Sherwood MW, Ebisui E, Tepikin AV, Mikoshiba K, Gerasimenko OV, Petersen OH. Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc Natl Acad Sci U S A. 2009;106(26):10758–10763. doi: 10.1073/pnas.0904818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, Tang J, Rietdorf K, Teboul L, Chuang KT, Lin P, Xiao R, Wang C, Zhu Y, Lin Y, Wyatt CN, Parrington J, Ma J, Evans AM, Galione A, Zhu MX. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459(7246):596–600. doi: 10.1038/nature08030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu MX, Ma J, Parrington J, Galione A, Evans AM. TPCs: Endolysosomal channels for Ca2+ mobilization from acidic organelles triggered by NAADP. FEBS Lett. 584(10):1966–1974. doi: 10.1016/j.febslet.2010.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogunbayo OA, Zhu Y, Rossi D, Sorrentino V, Ma J, Zhu MX, Evans AM. Cyclic adenosine diphosphate ribose activates ryanodine receptors, whereas NAADP activates two-pore domain channels. J Biol Chem. 286(11):9136–9140. doi: 10.1074/jbc.M110.202002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Husain SZ, Prasad P, Grant WM, Kolodecik TR, Nathanson MH, Gorelick FS. The ryanodine receptor mediates early zymogen activation in pancreatitis. Proc Natl Acad Sci U S A. 2005;102(40):14386–14391. doi: 10.1073/pnas.0503215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cahalan MD. STIMulating store-operated Ca(2+) entry. Nat Cell Biol. 2009;11(6):669–677. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee KP, Yuan JP, Hong JH, So I, Worley PF, Muallem S. An endoplasmic reticulum/plasma membrane junction: STIM1/Orai1/TRPCs. FEBS Lett. 2010;584(10):2022–2027. doi: 10.1016/j.febslet.2009.11.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orabi AI, Shah AU, Ahmad MU, Choo-Wing R, Parness J, Jain D, Bhandari V, Husain SZ. Dantrolene mitigates caerulein-induced pancreatitis in vivo in mice. Am J Physiol Gastrointest Liver Physiol. 2010;299(1):G196–G204. doi: 10.1152/ajpgi.00498.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perides G, Laukkarinen JM, Vassileva G, Steer ML. Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology. 2010;138(2):715–725. doi: 10.1053/j.gastro.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voronina SG, Barrow SL, Simpson AW, Gerasimenko OV, da Silva Xavier G, Rutter GA, Petersen OH, Tepikin AV. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology. 2010;138(5):1976–1987. doi: 10.1053/j.gastro.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Husain SZ, Grant WM, Gorelick FS, Nathanson MH, Shah AU. Caerulein-induced intracellular pancreatic zymogen activation is dependent on calcineurin. Am J Physiol Gastrointest Liver Physiol. 2007;292(6):G1594–G1599. doi: 10.1152/ajpgi.00500.2006. [DOI] [PubMed] [Google Scholar]
- 41.Shah AU, Sarwar A, Orabi AI, Gautam S, Grant WM, Park AJ, Liu J, Mistry PK, Jain D, Husain SZ. Protease activation during in vivo pancreatitis is dependent on calcineurin activation. Am J Physiol Gastrointest Liver Physiol. 2009;297(5):G967–G973. doi: 10.1152/ajpgi.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe O, Baccino FM, Steer ML, Meldolesi J. Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: early morphological changes during development of experimental pancreatitis. Am J Physiol. 1984;246(4 Pt 1):G457–G467. doi: 10.1152/ajpgi.1984.246.4.G457. [DOI] [PubMed] [Google Scholar]
- 43.Lerch MM, Saluja AK, Dawra R, Saluja M, Steer ML. The effect of chloroquine administration on two experimental models of acute pancreatitis. Gastroenterology. 1993;104(6):1768–1779. doi: 10.1016/0016-5085(93)90658-y. [DOI] [PubMed] [Google Scholar]
- 44.Grady T, Saluja A, Kaiser A, Steer M. Edema and intrapancreatic trypsinogen activation precede glutathione depletion during caerulein pancreatitis. Am J Physiol. 1996;271(1 Pt 1):G20–G26. doi: 10.1152/ajpgi.1996.271.1.G20. [DOI] [PubMed] [Google Scholar]
- 45.Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, Domschke W, Lippert H, Peters C, Deussing J. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106(6):773–781. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meister T, Niehues R, Hahn D, Domschke W, Sendler M, Lerch MM, Schnekenburger J. Missorting of cathepsin B into the secretory compartment of CI-MPR/IGFII-deficient mice does not induce spontaneous trypsinogen activation but leads to enhanced trypsin activity during experimental pancreatitis--without affecting disease severity. J Physiol Pharmacol. 2010;61(5):565–575. [PubMed] [Google Scholar]
- 47.Waterford SD, Kolodecik TR, Thrower EC, Gorelick FS. Vacuolar ATPase regulates zymogen activation in pancreatic acini. J Biol Chem. 2005;280(7):5430–5434. doi: 10.1074/jbc.M413513200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reed AM, Husain SZ, Thrower E, Alexandre M, Shah A, Gorelick FS, Nathanson MH. Low extracellular pH induces damage in the pancreatic acinar cell by enhancing calcium signaling. J Biol Chem. 2010;286(3):1919–1926. doi: 10.1074/jbc.M110.158329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhoomagoud M, Jung T, Atladottir J, Kolodecik TR, Shugrue C, Chaudhuri A, Thrower EC, Gorelick FS. Reducing extracellular pH sensitizes the acinar cell to secretagogue-induced pancreatitis responses in rats. Gastroenterology. 2009;137(3):1083–1092. doi: 10.1053/j.gastro.2009.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Behrendorff N, Floetenmeyer M, Schwiening C, Thorn P. Protons released during pancreatic acinar cell secretion acidify the lumen and contribute to pancreatitis in mice. Gastroenterology. 2010;139(5):1711–1720. 20, e1–e5. doi: 10.1053/j.gastro.2010.07.051. [DOI] [PubMed] [Google Scholar]
- 51.Hashimoto D, Ohmuraya M, Hirota M, Yamamoto A, Suyama K, Ida S, Okumura Y, Takahashi E, Kido H, Araki K, Baba H, Mizushima N, Yamamura K. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J Cell Biol. 2008;181(7):1065–1072. doi: 10.1083/jcb.200712156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mareninova OA, Hermann K, French SW, O'Konski MS, Pandol SJ, Webster P, Erickson AH, Katunuma N, Gorelick FS, Gukovsky I, Gukovskaya AS. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J Clin Invest. 2009;119(11):3340–3355. doi: 10.1172/JCI38674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grasso D, Ropolo A, Lo Re A, Boggio V, Molejon MI, Iovanna JL, Gonzalez CD, Urrutia R, Vaccaro MI. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J Biol Chem. 2011;286(10):8308–8324. doi: 10.1074/jbc.M110.197301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tando Y, Algul H, Wagner M, Weidenbach H, Adler G, Schmid RM. Caerulein-induced NF-kappaB/Rel activation requires both Ca2+ and protein kinase C as messengers. Am J Physiol. 1999;277(3 Pt 1):G678–G686. doi: 10.1152/ajpgi.1999.277.3.G678. [DOI] [PubMed] [Google Scholar]
- 55.Satoh A, Gukovskaya AS, Nieto JM, Cheng JH, Gukovsky I, Reeve JR, Jr, Shimosegawa T, Pandol SJ. PKC-delta and -epsilon regulate NF-kappaB activation induced by cholecystokinin and TNF-alpha in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2004;287(3):G582–G591. doi: 10.1152/ajpgi.00087.2004. [DOI] [PubMed] [Google Scholar]
- 56.Thrower EC, Wang J, Cheriyan S, Lugea A, Kolodecik TR, Yuan J, Reeve JR, Jr, Gorelick FS, Pandol SJ. Protein kinase C delta-mediated processes in cholecystokinin-8-stimulated pancreatic acini. Pancreas. 2009;38(8):930–935. doi: 10.1097/MPA.0b013e3181b8476a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gukovskaya AS, Hosseini S, Satoh A, Cheng JH, Nam KJ, Gukovsky I, Pandol SJ. Ethanol differentially regulates NF-kappaB activation in pancreatic acinar cells through calcium and protein kinase C pathways. Am J Physiol Gastrointest Liver Physiol. 2004;286(2):G204–G213. doi: 10.1152/ajpgi.00088.2003. [DOI] [PubMed] [Google Scholar]
- 58.Satoh A, Gukovskaya AS, Reeve JR, Jr, Shimosegawa T, Pandol SJ. Ethanol sensitizes NF-kappaB activation in pancreatic acinar cells through effects on protein kinase C-epsilon. Am J Physiol Gastrointest Liver Physiol. 2006;291(3):G432–G438. doi: 10.1152/ajpgi.00579.2005. [DOI] [PubMed] [Google Scholar]
- 59.Gorelick F, Pandol S, Thrower E. Protein kinase C in the pancreatic acinar cell. J Gastroenterol Hepatol. 2008;23(Suppl 1):S37–S41. doi: 10.1111/j.1440-1746.2007.05282.x. [DOI] [PubMed] [Google Scholar]
- 60.Thrower EC, Osgood S, Shugrue CA, Kolodecik TR, Chaudhuri AM, Reeve JR, Jr, Pandol SJ, Gorelick FS. The novel protein kinase C isoforms -delta and -epsilon modulate caerulein-induced zymogen activation in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2008;294(6):G1344–G1353. doi: 10.1152/ajpgi.00020.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yuan J, Lugea A, Zheng L, Gukovsky I, Edderkaoui M, Rozengurt E, Pandol SJ. Protein kinase D1 mediates NF-kappaB activation induced by cholecystokinin and cholinergic signaling in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2008;295(6):G1190–G1201. doi: 10.1152/ajpgi.90452.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berna MJ, Hoffmann KM, Tapia JA, Thill M, Pace A, Mantey SA, Jensen RT. CCK causes PKD1 activation in pancreatic acini by signaling through PKC-delta and PKC-independent pathways. Biochim Biophys Acta. 2007;1773(4):483–501. doi: 10.1016/j.bbamcr.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thrower EC, Yuan J, Usmani A, Liu Y, Jones C, Minervini SN, Alexandre M, Pandol SJ, Guha S. A novel protein kinase D inhibitor attenuates early events of experimental pancreatitis in isolated rat acini. Am J Physiol Gastrointest Liver Physiol. 2011;300(1):G120–G129. doi: 10.1152/ajpgi.00300.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan YC, Leung PS. Co-operative effects of angiotensin II and caerulein in NFkappaB activation in pancreatic acinar cells in vitro. Regul Pept. 2011;166(1–3):128–134. doi: 10.1016/j.regpep.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 65.Williard DE, Twait E, Yuan Z, Carter AB, Samuel I. Nuclear factor kappa B-dependent gene transcription in cholecystokinin- and tumor necrosis factor-alpha-stimulated isolated acinar cells is regulated by p38 mitogen-activated protein kinase. Am J Surg. 2010;200(2):283–290. doi: 10.1016/j.amjsurg.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Twait E, Williard DE, Samuel I. Dominant negative p38 mitogen-activated protein kinase expression inhibits NF-kappaB activation in AR42J cells. Pancreatology. 2010;10(2–3):119–128. doi: 10.1159/000290656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S, Mehal W. TLR9 and the NLRP3 Inflammasome Link Acinar Cell Death With Inflammation in Acute Pancreatitis. Gastroenterology. 2011 doi: 10.1053/j.gastro.2011.03.041. S0016-5085(11)00384-2 [pii]10.1053/j.gastro.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bhatia M, Wallig MA, Hofbauer B, Lee HS, Frossard JL, Steer ML, Saluja AK. Induction of apoptosis in pancreatic acinar cells reduces the severity of acute pancreatitis. Biochem Biophys Res Commun. 1998;246(2):476–483. doi: 10.1006/bbrc.1998.8519. [DOI] [PubMed] [Google Scholar]
- 69.Criddle DN, Murphy J, Fistetto G, Barrow S, Tepikin AV, Neoptolemos JP, Sutton R, Petersen OH. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology. 2006;130(3):781–793. doi: 10.1053/j.gastro.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 70.Mohseni Salehi Monfared SS, Vahidi H, Abdolghaffari AH, Nikfar S, Abdollahi M. Antioxidant therapy in the management of acute, chronic and post-ERCP pancreatitis: a systematic review. World J Gastroenterol. 2009;15(36):4481–4490. doi: 10.3748/wjg.15.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sateesh J, Bhardwaj P, Singh N, Saraya A. Effect of antioxidant therapy on hospital stay and complications in patients with early acute pancreatitis: a randomised controlled trial. Trop Gastroenterol. 2009;30(4):201–206. [PubMed] [Google Scholar]
- 72.Booth DM, Murphy JA, Mukherjee R, Awais M, Neoptolemos JP, Gerasimenko OV, Tepikin AV, Petersen OH, Sutton R, Criddle DN. Reactive Oxygen Species Induced by Bile Acid Induce Apoptosis and Protect Against Necrosis in Pancreatic Acinar Cells. Gastroenterology. 2011 doi: 10.1053/j.gastro.2011.02.054. S0016-5085(11)00265-4 [pii]10.1053/j.gastro.2011.02.054. [DOI] [PubMed] [Google Scholar]
- 73.Kubisch CH, Sans MD, Arumugam T, Ernst SA, Williams JA, Logsdon CD. Early activation of endoplasmic reticulum stress is associated with arginine-induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2006;291(2):G238–G245. doi: 10.1152/ajpgi.00471.2005. [DOI] [PubMed] [Google Scholar]
- 74.Kubisch CH, Logsdon CD. Secretagogues differentially activate endoplasmic reticulum stress responses in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2007;292(6):G1804–G1812. doi: 10.1152/ajpgi.00078.2007. [DOI] [PubMed] [Google Scholar]
- 75.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115(10):2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Szmola R, Sahin-Toth M. Pancreatitis-associated chymotrypsinogen C (CTRC) mutant elicits endoplasmic reticulum stress in pancreatic acinar cells. Gut. 2009;59(3):365–372. doi: 10.1136/gut.2009.198903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Malo A, Kruger B, Seyhun E, Schafer C, Hoffmann RT, Goke B, Kubisch CH. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, trypsin activation, and acinar cell apoptosis while increasing secretion in rat pancreatic acini. Am J Physiol Gastrointest Liver Physiol. 299(4):G877–G886. doi: 10.1152/ajpgi.00423.2009. [DOI] [PubMed] [Google Scholar]
- 78.Ye R, Mareninova OA, Barron E, Wang M, Hinton DR, Pandol SJ, Lee AS. Grp78 heterozygosity regulates chaperone balance in exocrine pancreas with differential response to cerulein-induced acute pancreatitis. Am J Pathol. 2010;177(6):2827–2836. doi: 10.2353/ajpath.2010.100368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lugea A, Tischler D, Nguyen J, Gong J, Gukovsky I, French SW, Gorelick FS, Pandol SJ. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology. 2011;140(3):987–997. doi: 10.1053/j.gastro.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hegyi P, Pandol S, Venglovecz V, Rakonczay Z., Jr The acinar-ductal tango in the pathogenesis of acute pancreatitis. Gut. 2011;60(4):544–552. doi: 10.1136/gut.2010.218461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Venglovecz V, Rakonczay Z, Jr, Ozsvari B, Takacs T, Lonovics J, Varro A, Gray MA, Argent BE, Hegyi P. Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut. 2008;57(8):1102–1112. doi: 10.1136/gut.2007.134361. [DOI] [PubMed] [Google Scholar]
- 82.Yamamoto A, Ishiguro H, Ko SB, Suzuki A, Wang Y, Hamada H, Mizuno N, Kitagawa M, Hayakawa T, Naruse S. Ethanol induces fluid hypersecretion from guinea-pig pancreatic duct cells. J Physiol. 2003;551(Pt 3):917–926. doi: 10.1113/jphysiol.2003.048827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maleth J, Venglovecz V, Razga Z, Tiszlavicz L, Rakonczay Z, Jr, Hegyi P. Non-conjugated chenodeoxycholate induces severe mitochondrial damage and inhibits bicarbonate transport in pancreatic duct cells. Gut. 2011;60(1):136–138. doi: 10.1136/gut.2009.192153. [DOI] [PubMed] [Google Scholar]
- 84.Venglovecz V, Hegyi P, Rakonczay Z, Jr, Tiszlavicz L, Nardi A, Grunnet M, Gray MA. Pathophysiological relevance of apical large-conductance Ca(2)+-activated potassium channels in pancreatic duct epithelial cells. Gut. 2011;60(3):361–369. doi: 10.1136/gut.2010.214213. [DOI] [PubMed] [Google Scholar]
- 85.Malleo G, Mazzon E, Genovese T, Di Paola R, Muia C, Crisafulli C, Siriwardena AK, Cuzzocrea S. Effects of thalidomide in a mouse model of cerulein-induced acute pancreatitis. Shock. 2008;29(1):89–97. doi: 10.1097/shk.0b013e318067df68. [DOI] [PubMed] [Google Scholar]
- 86.Tsai MJ, Chen C, Chen SH, Huang YT, Chiu TH. Pomalidomide suppresses cerulein-induced acute pancreatitis in mice. J Gastroenterol. 2011 doi: 10.1007/s00535-011-0394-x. 10.1007/s00535-011-0394-x. [DOI] [PubMed] [Google Scholar]
- 87.Habtezion A, Kwan R, Akhtar E, Wanaski SP, Collins SD, Wong RJ, Stevenson DK, Butcher EC, Omary MB. Panhematin provides a therapeutic benefit in experimental pancreatitis. Gut. 2011;60(5):671–679. doi: 10.1136/gut.2010.217208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ozkan E, Akyuz C, Sehirli AO, Topaloglu U, Ercan F, Sener G. Montelukast, a selective cysteinyl leukotriene receptor 1 antagonist, reduces cerulein-induced pancreatic injury in rats. Pancreas. 2010;39(7):1041–1046. doi: 10.1097/MPA.0b013e3181db2dfd. [DOI] [PubMed] [Google Scholar]
- 89.Awla D, Abdulla A, Zhang S, Roller J, Menger MD, Regner S, Thorlacius H. Lymphocyte function antigen-1 regulates neutrophil recruitment and tissue damage in acute pancreatitis. Br J Pharmacol. 2011;163(2):413–423. doi: 10.1111/j.1476-5381.2011.01225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou GX, Zhu XJ, Ding XL, Zhang H, Chen JP, Qiang H, Zhang HF, Wei Q. Protective effects of MCP-1 inhibitor on a rat model of severe acute pancreatitis. Hepatobiliary Pancreat Dis Int. 2010;9(2):201–207. [PubMed] [Google Scholar]
- 91.Polito F, Bitto A, Irrera N, Squadrito F, Fazzari C, Minutoli L, Altavilla D. Flavocoxid, a dual inhibitor of cyclooxygenase-2 and 5-lipoxygenase, reduces pancreatic damage in an experimental model of acute pancreatitis. Br J Pharmacol. 2010;161(5):1002–1011. doi: 10.1111/j.1476-5381.2010.00933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mayer JM, Laine VJ, Gezgin A, Kolodziej S, Nevalainen TJ, Storck M, Beger HG. Single doses of FK506 and OKT3 reduce severity in early experimental acute pancreatitis. Eur J Surg. 2000;166(9):734–741. doi: 10.1080/110241500750008501. [DOI] [PubMed] [Google Scholar]
- 93.Awla D, Hartman H, Abdulla A, Zhang S, Rahman M, Regner S, Thorlacius H. Rho-kinase signalling regulates trypsinogen activation and tissue damage in severe acute pancreatitis. Br J Pharmacol. 2011;162(3):648–658. doi: 10.1111/j.1476-5381.2010.01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dahlhoff M, Algul H, Siveke JT, Lesina M, Wanke R, Wartmann T, Halangk W, Schmid RM, Wolf E, Schneider MR. Betacellulin protects from pancreatitis by activating stress-activated protein kinase. Gastroenterology. 2010;138(4):1585–1594. 94, e1–e3. doi: 10.1053/j.gastro.2009.12.045. [DOI] [PubMed] [Google Scholar]
- 95.Chinzei R, Masuda A, Nishiumi S, Nishida M, Onoyama M, Sanuki T, Fujita T, Moritoh S, Itoh T, Kutsumi H, Mizuno S, Azuma T, Yoshida M. Vitamin K3 attenuates cerulein-induced acute pancreatitis through inhibition of the autophagic pathway. Pancreas. 2011;40(1):84–94. doi: 10.1097/MPA.0b013e3181f69fc9. [DOI] [PubMed] [Google Scholar]
- 96.Carvalho KM, Morais TC, de Melo TS, de Castro Brito GA, de Andrade GM, Rao VS, Santos FA. The natural flavonoid quercetin ameliorates cerulein-induced acute pancreatitis in mice. Biol Pharm Bull. 2010;33(9):1534–1539. doi: 10.1248/bpb.33.1534. [DOI] [PubMed] [Google Scholar]
- 97.Ma Q, Zhang M, Wang Z, Ma Z, Sha H. The beneficial effect of resveratrol on severe acute pancreatitis. Ann N Y Acad Sci. 2011;1215:96–102. doi: 10.1111/j.1749-6632.2010.05847.x. [DOI] [PubMed] [Google Scholar]
- 98.Sandoval J, Escobar J, Pereda J, Sacilotto N, Rodriguez JL, Sabater L, Aparisi L, Franco L, Lopez-Rodas G, Sastre J. Pentoxifylline prevents loss of PP2A phosphatase activity and recruitment of histone acetyltransferases to proinflammatory genes in acute pancreatitis. J Pharmacol Exp Ther. 2009;331(2):609–617. doi: 10.1124/jpet.109.157537. [DOI] [PubMed] [Google Scholar]
- 99.Chen KH, Chao D, Liu CF, Chen CF, Wang D. Curcumin attenuates airway hyperreactivity induced by ischemia-reperfusion of the pancreas in rats. Transplant Proc. 2010;42(3):744–747. doi: 10.1016/j.transproceed.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 100.Seo SW, Bae GS, Kim SG, Yun SW, Kim MS, Yun KJ, Park RK, Song HJ, Park SJ. Protective effects of Curcuma longa against cerulein-induced acute pancreatitis and pancreatitis-associated lung injury. Int J Mol Med. 2011;27(1):53–61. doi: 10.3892/ijmm.2010.548. [DOI] [PubMed] [Google Scholar]
- 101.Jung KH, Song SU, Yi T, Jeon MS, Hong SW, Zheng HM, Lee HS, Choi MJ, Lee DH, Hong SS. Human bone marrow-derived clonal mesenchymal stem cells inhibit inflammation and reduce acute pancreatitis in rats. Gastroenterology. 2011;140(3):998–1008. doi: 10.1053/j.gastro.2010.11.047. [DOI] [PubMed] [Google Scholar]
- 102.Schneider G, Saur D. Mesenchymal stem cells: therapeutic potential for acute pancreatitis. Gastroenterology. 2011;140(3):779–782. doi: 10.1053/j.gastro.2011.01.026. [DOI] [PubMed] [Google Scholar]
- 103.Pettila V, Kyhala L, Kylanpaa ML, Leppaniemi A, Tallgren M, Markkola A, Puolakkainen P, Repo H, Kemppainen E. APCAP--activated protein C in acute pancreatitis: a double-blind randomized human pilot trial. Crit Care. 2010;14(4):R139. doi: 10.1186/cc9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shankar-Hari M, Wyncoll D. Activated protein C in severe acute pancreatitis without sepsis? Not just yet. Crit Care. 2010;14(4):188. doi: 10.1186/cc9190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Johnson CD, Kingsnorth AN, Imrie CW, McMahon MJ, Neoptolemos JP, McKay C, Toh SK, Skaife P, Leeder PC, Wilson P, Larvin M, Curtis LD. Double blind, randomised, placebo controlled study of a platelet activating factor antagonist, lexipafant, in the treatment and prevention of organ failure in predicted severe acute pancreatitis. Gut. 2001;48(1):62–69. doi: 10.1136/gut.48.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bhatia V, Ahuja V, Acharya SK, Garg PK. A randomized controlled trial of valdecoxib and glyceryl trinitrate for the prevention of post-ERCP pancreatitis. J Clin Gastroenterol. 2011;45(2):170–176. doi: 10.1097/MCG.0b013e3181eb600e. [DOI] [PubMed] [Google Scholar]