

SUMMARY
Tuberculosis remains a major threat as drug resistance continues to increase. Pulmonary tuberculosis in adults is responsible for 80% of clinical cases and nearly 100% of transmission of infection. Unfortunately, since we have no animal models of adult type pulmonary tuberculosis, the most important type of disease remains largely out of reach of modern science and many fundamental questions remain unanswered. This paper reviews research dating back to the 1950’s providing compelling evidence that cord factor (trehalose 6,6′ dimycolate [TDM]) is essential for understanding tuberculosis. However, the original papers by Bloch and Noll were too far ahead of their time to have immediate impact. We can now recognize that the physical and biologic properties of cord factor are unprecedented in science, especially its ability to switch between two sets of biologic activities with changes in conformation. While TDM remains on organisms, it protects them from killing within macrophages, reduces antibiotic effectiveness and inhibits the stimulation of protective immune responses. If it comes off organisms and associates with lipid, TDM becomes a driver of tissue damage and necrosis. Studies emanating from cord factor research have produced (1) a rationale for improving vaccines, (2) an approach to new drugs that overcome natural resistance to antibiotics, (3) models of caseating granulomas that reproduce multiple manifestations of human tuberculosis. (4) evidence that TDM is a key T cell antigen in destructive lesions of tuberculosis, and (5) a new understanding of the pathology and pathogenesis of postprimary tuberculosis that can guide more informative studies of long standing mysteries of tuberculosis.
Keywords: Tuberculosis, Trehalose dimycolate, Cord factor, Caseating granuloma, Cavity, Drug
1. Introduction
In spite of the best efforts of modern science, M. tuberculosis (MTB) persists throughout the world and kills more people than any other bacterial infection. Drug resistance continues to rise. Genetic analysis, molecular biology, combinatorial chemistry and high-throughput screening have produced no new drugs. Nor have they been able to improve on a vaccine, BCG, developed in the 1920’s. Similarly, key questions of the disease remain unanswered. Why can’t immunity control established infection when most infected people never get sick? How can MTB be an obligate human parasite when people are more resistant to disease than most animals? How does MTB damage tissue? How can it grow in vast numbers in one part of a lung, while the rest of the lung and all other parts of the body remain highly resistant? These questions remain unanswered because they are seldom addressed since we lack experimental models and methods to study them.
Scientists since Koch have looked for virulence factors of MTB. A virulence factor is a component or attribute of an organism that facilitates disease. The oldest, most durable and best documented virulence factor of MTB is cord formation. Koch noted that virulent MTB grow in twisted rope-like aggregates known as serpentine cords. Middlebrook wrote that cord formation was an “essential accompaniment of virulence”.1 Bloch and Noll identified a lipid, cord factor (trehalose 6,6′ dimycolate [TDM]), on the surface of virulent MTB and published the following evidence that it is a virulence factor:2–4
TDM is the most abundant and most toxic lipid produced by MTB. Since MTB is an obligate human parasite, it is safe to assume that every component has been selected to promote its survival in humans.
Much more TDM can be extracted from the surface of virulent than avirulent MTB.
Removal of TDM from MTB abolishes its ability to produce progressive infection in mice.
Injections of TDM too small to cause toxicity by themselves enhance both acute and chronic tuberculosis causing mice to die sooner of massive infections. The effect is specific for tuberculosis.
One might expect the scientific community to vigorously pursue such compelling evidence. In actuality, the opposite happened. The concept that TDM might be a virulence factor for MTB was vigorously attacked by the leading scientists of the day, none of whom ever attempted to repeat the studies.5–7 The concept was denounced as “unphysiologic” because scientists could neither understand nor study it further with technologies of the day. Bloch and Noll were too far ahead of their time with a revolutionary new concept of science. As Kuhn explained, when faced with the choice of adapting to a new idea or proving it wrong, most people work on the proof.8
2. The toxic surface of TDM – new concept in biology
Our studies began with investigations of surface-active vaccine adjuvants before we learned the troubled history of TDM.9,10 We became fascinated by the observation that the toxicity of TDM was due to an extremely rigid insoluble crystalline monolayer that formed spontaneously at water–hydrophobe interfaces.11 The LD50 of the toxic TDM monolayer approximates 30 μg/mouse while doses of 50,000 μg of the nontoxic TDM micelles produce no overt toxicity. We realized that such activity had never been described for any biologic substance and that TDM did not fit into any know category of toxin. The structure of both the toxic monolayer and nontoxic micelles of TDM were deduced from physical chemical studies and confirmed by scanning tunneling microscopy,12,13 Fig. 1. The toxic monolayer of TDM is the most stable and ridged biologic monolayer ever described. It consists of linear arrays of trehalose head groups alternating with exposed hydrophobic mycolic acid domains. This structure forms spontaneously at water–hydrophobe interfaces including oil, polystyrene and air. The intensity of inflammation induced by the TDM monolayer is a function of its surface area, but is not otherwise related to dose.14 The surface of the non-toxic cylindrical micelles of TDM is composed entirely of trehalose head groups. The extreme insolubility of TDM contributes to the long-term stability of these structures.
Fig. 1.

Toxic monolayer and non-toxic micelle structures of TDM.13 TDM molecules consist of one trehalose attached to two mycolic acid moieties each of which has a short and long hydrocarbon chain. In aqueous suspension, TDM forms non-toxic cylindrical micelles as shown in a diagram and by scanning tunneling microscopy at 500,000× magnification (left-hand panel). The surface consists entirely of trehalose moieties with the fatty acid chains on the inside. These micelles are non toxic, but have important functions on MTB. TDM forms a toxic crystalline monolayer at hydrophobe–aqueous interfaces as shown in a diagram and by scanning tunneling microscopy at 1,840,000× magnification (right-hand panel). The major dark lines, spaced approximately 90 Å, are elevated arrays of trehalose head groups. A secondary pattern intersecting the main one at an angle of 45° has a periodicity approximating 65 Å and represents the mycolic acid residues. Susceptible mice are killed by as little as 10 μg of TDM in this configuration.
The concept of toxic surfaces is well established for mineral crystals, but had never before been described for a microbiologic product. SiO2 exists in four crystalline forms. Its toxicity varies from extreme (tridimyte) to none (stitsovite) depending on the crystal structure.15,16 Quartz has intermediate structure and toxicity. In another example, pure carbon can exist as graphite that is non toxic or as carbon nanotubes that are very toxic.17 Other examples are asbestos and monosodium urate. The biologic activity of each of these materials is dependent on the configuration of atoms on the surface of insoluble crystals.
Over the years, we and others have gradually repeated all of Bloch & Noll’s studies and learned much more about TDM. Several investigators have studied it as a conventional lipid. While interesting things have been learned, such studies miss the significance of its surface-dependent activities. We proposed that TDM is ‘the key’ to understanding the pathophysiology of tuberculosis for several reasons:18 First, it the most abundant component of the organism. Second, it is the largest lipid known. Non-pathogenic mycobacteria have smaller and less toxic species of TDM. Third, its two sets of surface-dependent activities are unique in biology. Finally, since MTB is an obligate human parasite, it is safe to assume that every component of MTB has been selected to facilitate its survival in man. Virulent MTB expend great energy to produce far more TDM during infection than is required for survival in artificial culture. Much of the TDM is released outside the organism where it directly interacts with the host. The benefit to the organism must be worth the expenditure of resources.
These studies have progressively come together to suggest answers to long standing mysteries of tuberculosis. The ability of TDM to switch between two vastly different sets of biologic activities is essential. While TDM remains on organisms, it protects them from killing by macrophages. Once it comes off the organisms and associates with lipid, TDM becomes the driver of caseation necrosis.19 This led to correction of long standing misconceptions of the pathology of adult pulmonary tuberculosis, to development of prototype drugs that overcome natural resistance of MTB to antibiotics, and to vaccines that counter MTB’s ability to suppress immunization. Each of these findings will be described.20–22
3. Effects of TDM on colony morphology
We published evidence confirming the hypothesis that TDM causes cording of mycobacteria.23 TDM is released free by organisms but is so insoluble that much remains stuck to them. MTB grown in liquid medium without surfactants preferentially grows floating on the surface as a pellicle. The pellicle of virulent organisms spreads rapidly across the surface as a thin film and friable climbs the sides of the container, Fig. 2. Avirulent organisms tend to grow as discrete clumps. We demonstrated that this spreading climbing pellicle growth of virulent MTB is due to TDM that forms a monolayer and then multilayers on the surface of the media.23 The rigidity of TDM layers causes the pellicle of virulent MTB to spread and climb the walls of a container.
Fig. 2.
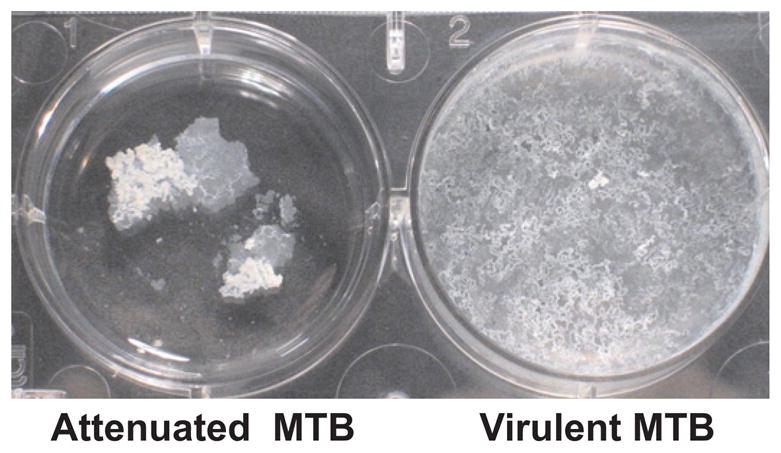
Pellicle cultures of MTB.23 Attenuated MTB form easily crumbled aggregates with no tendency to spread. Fragments could be easily broken off without disturbing the rest of the surface. Virulent MTB typically form pellicles that spread and climb the walls of flasks. The pellicle surface has multiple holes that give it an appearance resembling lace. Touching any part of this pellicle causes the entire surface to move. The spreading, climbing behavior of the pellicle is due to excess TDM.
We observed that organisms isolated from pulmonary cavities typically grow more rapidly and produce more TDM than organisms of the same clade isolated from non-pulmonary sites.24 These growth patterns could be demonstrated only with fresh clinical isolates. Once the organisms were passaged in culture, the growth patterns changed. This implies that MTB frequently grow rapidly and produce large amounts of TDM in a cavity while they grow much more slowly producing less TDM in other organs. These observations are consistent with the observations that very little TDM is required to protect MTB in macrophages and that MTB appear to grow as pellicles on the walls of cavities. We proposed that large amounts of the TDM in the toxic monolayer configuration are required for maintenance of cavities.18
4. New drugs directed at natural resistance of MTB to antibiotics
MTB can display at least three different types of resistance to antibiotics. The first and most familiar is genetic resistance in which mutations produce resistance to particular drugs. The second, phenotypic resistance, occurs when MTB become dormant making them refractory to agents that inhibit active metabolic processes. Finally, natural resistance consists of barriers and efflux pumps that retard penetration of antibiotics into organisms.25 Many investigations are scrutinizing the metabolic pathways of MTB in an effort to identify new targets for antibiotics to combat genetic and phenotypic resistance. However, very little attention has ever been paid to the possibility of overcoming natural resistance.
Since natural resistance of MTB has been related to lipid barriers, we proposed that surface-active agents that disrupt surface lipids of MTB might facilitate penetration of antibiotics. Following the lead of D’Arcy Hart that certain surfactants could potentiate the activity of antibiotics against MTB, we conducted structure–activity studies that identified a poloxamer surfactant, CRL-1072, as an optimal agent that caused drug-resistant MTB to become susceptible to therapeutic serum levels of several antibiotics.21,26 In the presence of CRL-1072, drug-resistant MTB were rendered highly sensitive to both first and second line antibiotics. The greatest effect was observed with two drug-resistant strains in which CRL-1072 completely restored susceptibility to first line drugs, INH and rifampin. CRL-1072 reduced the MIC of one strain 66-fold from 10 μg/mL to 0.15 μg/mL and the other nearly as much from 10 μg/mL to 0.2 μg/mL.
CRL-1072 significantly enhanced the activity of INH, rifampin, streptomycin, pyrazinamide and ethambutol on acute tuberculosis in mice, Fig. 3. These studies progressed to the point of filing an IND that was approved for phase I studies in 1996. However, at that time MDR MTB in New York was being controlled by DOTS and no one was interested in pursuing new drugs for a small and shrinking market. Subsequently, WHO evaluated worldwide R&D for drugs against MTB to identify a shortlist for development.27 CRL-1072 was on the shortlist with the recommendation to “pursue further with a view to encouraging further efficacy and toxicity studies”. With the continuing resurgence of multidrug-resistant tuberculosis and paucity of new drug candidates, we believe that agents, like CRL-1072, that overcome natural resistance to tuberculosis deserve reconsideration.
Fig. 3.
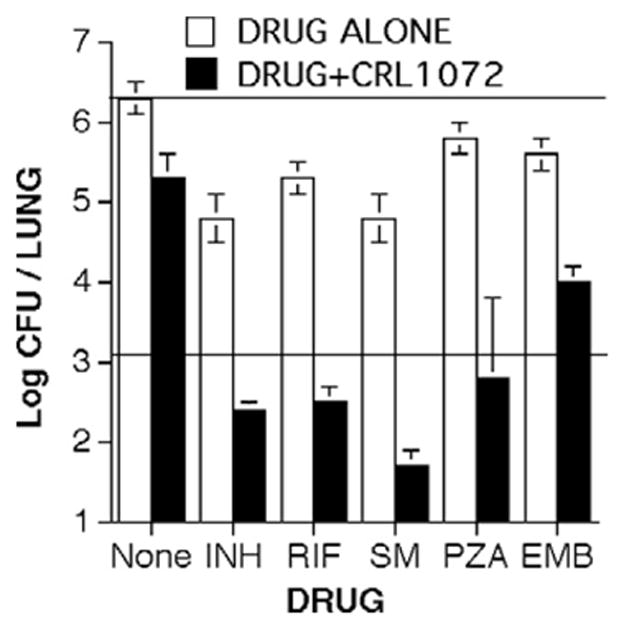
Overcoming natural resistance of MTB to antibiotics.21 The combination of CRL-1072 with each drug reduced CFUs in lungs below the levels achieved by the drug alone. The combinations of CRL-1072 with isoniazid (INH), rifampin (RIF), streptomycin (SM) and pyrazinamide (PZA) produced significant bactericidal effects while the antibiotic alone was only mildly bacteriostatic. Ethambutol (ETH) had a significant bacteriostatic effect. Top horizontal line = 28 day growth in untreated controls. The lower horizontal line = CFU counts at day 0 baseline.
4.1. Drugs effective against MTB in cavities
MTB in cavities are particularly difficult to eradicate with antibiotics and are the primary site of generation of new antibiotic-resistant strains. MTB in cavities characteristically grow on the air–tissue surface as pellicles.28,29 The pellicle of MTB is a type of biofilm, and bacteria, including mycobacteria, in biofilms have increased resistance to antibiotics.23,30 We have shown that non-toxic surfactants can disrupt pellicles of MTB and either kill the organisms or make them more susceptible to antibiotics.
5. Biologic activities of TDM on MTB organisms
The most unusual property of TDM is that its biologic activity changes with its conformation from non-toxic micelles to a highly toxic monolayer.11–14 While the structure of TDM on MTB is not completely characterized, it does consist, at least in part, of micelles. While non toxic, micelles are not inactive. It has been known since the 1950’s that removal of TDM from the surface of MTB reduces their ability to survive in mice.31,32 The experimental model of extracting surface lipids from viable organisms with wet petroleum ether and reconstituting them with purified TDM has proved highly informative for studies of the mechanisms of these activities. Jessica Indrigo demonstrated that removal of lipids from the surface of MTB caused over 99% of organisms to be killed in macrophages in tissue culture within three days, Fig. 4.33,34 Reconstituting the organisms with purified TDM restored their ability to survive in macrophages almost completely. Surprisingly, MTB produced far more TDM than was required to restore their protection within macrophages. Reconstitution of MTB with less than 10% of the TDM removed was sufficient to completely restore protection of the organisms within macrophages. Since MTB is an obligate human pathogen and TDM synthesis is energy intensive, this implies that TDM has additional functions that are necessary for survival of MTB in humans.
Fig. 4.
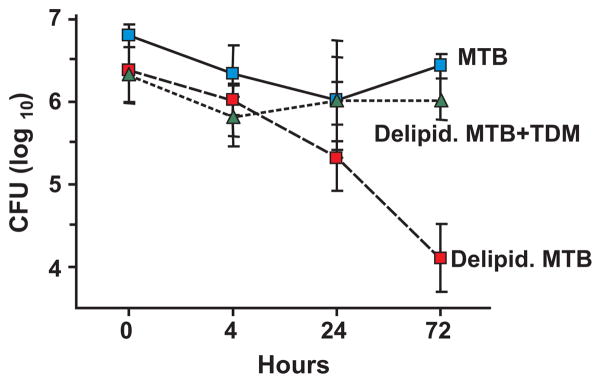
TDM protects MTB from killing by macrophages.33 J774A.1 macrophages were infected with native MTB, delipidated MTB (delipid. MTB) or delipidated MTB reconstituted with TDM (delipid. MTB + TDM), m.o.i. 5:1. Mycobacterial survival was measured by colony-forming units (CFU) at 4, 24, and 72 hours after infection. Data are expressed as average log10 CFU ± one SD, 3 replicates per time point. 99% reduction in CFU of delipid. MTB occurred by 72 hours (P < 0.025). TDM reconstitution protected the MTB almost completely.
Further studies revealed that TDM on mycobacteria inhibited phagosome–lysosome fusion and acidification of phagosomes.33,34 Recent studies have demonstrated that TDM on MTB also inhibits the ability of MTB to present antigens to T cells, thereby preventing the induction of strong immune responses.35 TDM even inhibits the ability of interferon gamma to enhance antigen presentation. Celestine Kan demonstrated that interaction of TDM with toll-like receptors induces Suppressors Of Cytokine Signaling (SOCS-1 and SOCS-3) by TDM on MTB. SOCS-1 and SOCS-3 then inhibit antigen presentation and induction of T cell immunity.
Dr Jagannath recently used these concepts to produce an improved vaccine for tuberculosis.22 Many attempts have been made to improve BCG by incorporating additional antigens to induce stronger immune responses. However, the organisms’ evasive mechanisms prevented the development of optimal immune responses. Our work suggested two means to neutralize BCG’s evasive mechanisms. The first was to reduce MTB’s ability to synthesize TDM. The final step in synthesis of TDM is mediated by a mycolyl transferase enzyme, Antigen 85a. Deletion of the gene, fpbA, that encodes for Antigen 85a produces an organism deficient in TDM production.36 Dr. Jagannath demonstrated that this organism is more immunogenic than the wild-type BCG.37 He then hypothesized that a drug, rapamycin, that modulates the movement of particles in cells, would cause mycobacterial antigens to more effectively enter pathways leading to immunization. Immunization with this dual-approach vaccine produced a tenfold increase in the number of organisms killed and a threefold increase in the duration of protection. These findings “break new ground in vaccine research in general and make improvements for antiTB vaccines in particular, because they provide a simple and powerful strategy to enhance vaccine efficiency”.22
In other studies, Hwang and Actor used a novel lactoferrin adjuvant with BCG to produce a vaccine that reduced the chronic pathologic response to MTB.38 This vaccine caused reduction of the lipid pneumonia of progressive tuberculosis that we believe is a valuable model of postprimary tuberculosis in humans. This pathology in mice, like human postprimary tuberculosis, develops preferentially in individuals with sufficient immunity to control primary tuberculosis. The facts that no vaccine has ever shown efficacy against postprimary tuberculosis and that young immunocompetent people with strong tuberculin skin tests are the most likely to die rapidly from postprimary tuberculosis have led some to question the ability of the immune system to control this form of tuberculosis. To our knowledge, this is the first vaccination study in animals to suggest that that it may be possible to produce a vaccine against postprimary tuberculosis.
5.1. TDM causes caseation necrosis
If tightly clumped cords of virulent MTB come in contact with droplets of oil, the organisms enter the oil drop and become dispersed.2 When this happens, TDM, being a surface-active agent, comes off the organisms and forms a monolayer on the surface of the oil. In this configuration, it exhibits an entirely different set of biologic activities. For many years, the toxicity of TDM on oil was considered unphysiologic because mineral oil is not a normal constituent of the body.7 However, increasing evidence demonstrates that this is exactly how it works to produce caseation necrosis in tuberculosis.18 Oil is provided by lipids in the body and the combination of TDM with lipid appears to be central to the pathogenesis of tuberculosis.
5.2. Macrophage toxicity
The toxic monolayer of TDM forms spontaneously at water–hydrophobe interfaces including oil, polystyrene and air. Human or mouse macrophages adhere to polystyrene tissue culture dishes and spread as pseudopods within a period of hours. If a dish is coated with a single molecular monolayer of TDM, the macrophages spread in minutes in 360 degrees of arc without forming pseudopods.18 Many are killed within minutes by a process of membrane blebbing that resembles oncosis.39 If TDM is spread as a molecular monolayer on hydrophobic beads, the beads are rapidly taken up by macrophages. The subsequent effects depends upon the size of the beads. Small beads are less toxic.40 Larger beads are taken into phagosomes. A short time later tight adherence is observed between the phagosomal membrane and the bead, resulting in disruption of the membrane and death of the macrophages.41 It may be that macrophage membranes can withstand rapid adhesion and spreading on a small surface, but are torn apart by such spreading on large surfaces.
5.3. Nonallergic, and allergic granulomas
If TDM is injected intravenously into mice as oil emulsion or coated on beads, it induces activated foreign body granulomas in the lung.42,43 These granulomatous responses are associated with production of TNF and other cytokines,44–46 as well as complement factors.47–49 TDM also produces local inhibition of corticosteroid activity that could be a factor in developing tuberculosis.50 The granulomas heal over a period of weeks. Injection of TDM into immune animals produces very different effects. Animals can be immunized with BCG, chronic MTB infection or with TDM complexed with methylated BSA, but not by TDM alone. In appropriately immunized animals, TDM produces hypersensitivity granulomas.51–53 These are larger granulomas cuffed by a rim of lymphocytes with more activated appearing macrophages and necrosis at their centers. Immunization to produce such granulomas requires CD1-mediated antigen presentation for full development of an immune response.53 The effector cells are CD3+ CD4+ positive T cells since ability to produce hypersensitivity granulomas can only be transferred from animal to animal by these cells, but not by macrophages, CD8 or CD19 positive cells.52
5.4. Caseating granulomas in mice
The literature for many decades has reported that mice are a flawed model of tuberculosis because they fail to produce caseating granulomas following infection with a low dose of MTB. However, a TDM–mineral oil emulsion, injected intraperitoneally into mice with chronic tuberculosis, induced classic caseating granulomas.19 A series of experiments were conducted to characterize these lesions. Variations in protocols produced a series of caseating granulomas each of which resembled a particular phase of human disease. They could be induced either by a TDM emulsion or by a dose of virulent MTB containing sufficient TDM. Typical caseating granulomas were produced in the lung, pancreas, mediastinum and mesentery. Caseating granulomas in the lung formed in adipose tissue unless the MTB were injected with oil in which case they formed in association with the oil in lung parenchyma. Those in the mesentery closely resembled human mesenteric tuberculosis in that they were located in adipose tissue densely infiltrated with lymphocytes with central caseation necrosis and Langhans-type giant cells.18 Early in the formation of such lesions, acid fast bacilli could be found within fat cells demonstrating that association of TDM with lipid droplets could happen during infections. Other protocols produced caseating granulomas which persisted for months containing large numbers of acid fast organisms after the bacteria had been largely cleared from other parts of the body, Fig. 5. These lesions were encapsulated by dense fibrosis tissue in a pattern characteristic of chronic lesions in humans.
Fig. 5.
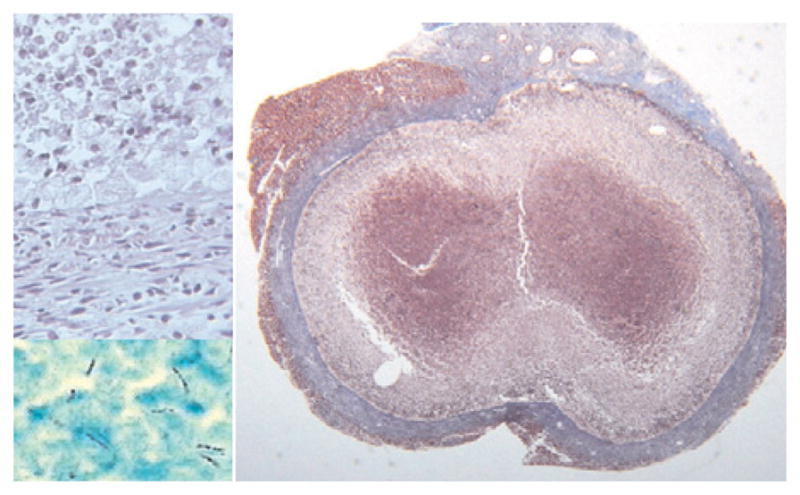
Persistent caseating granuloma in a mouse.19 This mouse was injected i.p. with 100 μg TDM in an oil-in-water emulsion and with 106 MTB i.v. one day later. The lesions were examined after 80 days. The large section (right-hand panel) shows an encapsulated lesion consisting of dense connective tissue, fibroblasts and macrophages surrounding caseation necrosis with focal calcification (Trichrome connective tissue stain, 20× magnification). Upper left section shows a thin layer of foamy macrophages between the capsule and interior caseous necrotic material (H&E stain, 400× magnification). Lower left section shows that many more MTB were present in this lesion than in any other part of the body (AFB stain, 1000×).
5.5. Caseating granulomas in humans
The observation that mice can produce a spectrum of typical caseating granulomas stimulated investigations of how they form in mice and the relevance of these findings for tuberculosis in humans. It has long been known that caseation necrosis in humans is largely lipid of host origin.28 Active caseating granulomas in humans characteristically contain prominent lipid droplets in association with epithelioid cells. This was true of all specimens we examined, including brain, bone, liver, lung, lymph node and kidney. In addition, caseating granulomas stained strongly for fat.54 In mice, caseating granulomas formed by association of MTB or purified TDM with lipid. We were able to distinguish two patterns of necrosis: caseation and ischemic.19 TDM alone produced only the caseation while MTB produced both. Evaluation of microscopic sections of human tuberculosis demonstrated that caseating granulomas in humans also display evidence of both caseation and ischemic necrosis. It was even possible to demonstrate acid fast organisms within lipid droplets and fat cells in newly forming human caseating granulomas, Fig. 6. Collectively, these data demonstrated that mice can produce a spectrum of caseating granulomas each of which reproduces a particular manifestation of human tuberculosis.19
Fig. 6.
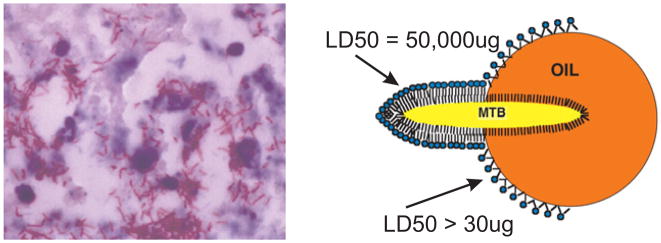
Interaction of MTB with lipid in developing human caseation necrosis.18 AFB in lipid droplets of foci of developing necrosis in the lung of a human with postprimary tuberculosis (AFB stain 1000×) (left). These sections demonstrate that MTB associates with lipid in foci of developing necrosis of tuberculosis. TDM remains with MTB until it encounters a lipid interface. It then comes off the organisms and assumes a stable monolayer configuration on the interface thereby activating its toxicity and antigenicity. The diagram (right) shows MTB transferring TDM to the surface of an oil drop. Observation of this phenomenon led Bloch to discovery of cord factor.2 Gentle extraction with oily solvents is still used to purify TDM and to produce delipidated organisms that have reduced virulence.
6. Pathology of postprimary tuberculosis
Modern science has produced powerful tools for dissecting molecular and cellular mechanisms of disease. However, even the best tools require appropriate targets. The choice of molecular targets for study of a disease depends on understanding cell biology and tissue pathology. It seems incomprehensible, but it is true, that the contemporary scientific literature could completely fail to understand the basic pathology of postprimary tuberculosis and, as a result, be unable to approach long standing questions. This situation developed as follows:
Postprimary tuberculosis occurs preferentially in people with sufficient immunity to resolve primary tuberculosis. It is a different disease from primary tuberculosis on clinical, radiologic, immunologic, epidemiologic, pathologic and genetic grounds.20 Postprimary tuberculosis is responsible for 80% of clinical disease and nearly 100% of transmission from person to person. Typically an upper lobe of lung becomes infected, undergoes necrosis and develops a cavity while the entire rest of the body remains highly immune. Immunization with BCG effectively prevents dissemination of primary tuberculosis, but has never been shown to have any effect on postprimary tuberculosis. Unfortunately, postprimary tuberculosis is strictly a human disease. Several animals develop progressive primary tuberculosis, but none develop postprimary disease. We know a great deal about primary tuberculosis, but without animal models, postprimary tuberculosis has remained largely unapproachable by modern science. Most of the major unresolved questions of tuberculosis involve postprimary disease. Why is it restricted to the lung? How does it produce tissue damage? Why don’t cavities heal? Why doesn’t immunity heal cavities or prevent their development? Why is strong immunity detrimental in that young immunocompetent adults are the most likely to die?55 The inability of immunity to control postprimary tuberculosis has never been explained.
The pathologies of both primary and postprimary tuberculosis were clearly described by the end of the 19th century.56 The advent of antibiotics in the 1950’s produced a sharp drop in lung tissues for study of postprimary tuberculosis and diminution of interest in the disease. The few researchers remaining were forced to study animal models, particularly the rabbit.57 Unfortunately, rabbits are resistant to MTB, but they are susceptible to M. bovis, the organism that causes tuberculosis in cattle. Many new facts about macrophages, granulomas and immunity were learned from studies of M. bovis. However, the older descriptions of pathology were forgotten. Consequently, virtually all modern descriptions of the pathogenesis of postprimary tuberculosis are descriptions of M. bovis infection of rabbits. Postprimary tuberculosis is almost universally described today as an accelerated primary tuberculosis in which caseating granulomas progressively enlarge, erode into bronchi, undergo softening and discharge their contents to leave a cavity. This pathologic process has been reported in humans infected with M. bovis, but has never been reported and probably never happens in people infected with MTB.58
Our finding of caseating granulomas in mice stimulated search for literature and pathologic specimens of postprimary tuberculosis. We collected histopathologic slides from over 100 autopsy cases of primary and postprimary tuberculosis and reviewed the extensive literature from the preantibiotic era. This was made possible because Google Books can search old libraries almost as well as current literature. Modern textbooks provide an accurate description of the development of cavities in progressive primary tuberculosis in rabbits. Human postprimary tuberculosis, however, develops through a different pathologic process that is nowhere found in contemporary literature.20
Postprimary tuberculosis occurs as an endogenous lipid pneumonia in people with sufficient immunity to control primary tuberculosis and heal caseating granulomas, Fig. 7. Infection is restricted to alveolar macrophages and may cause little inflammation in the early stages. It is occasionally diagnosed by finding X-ray evidence of upper lobe pneumonia in people who are not sick. Postprimary tuberculosis begins in the upper lobes of the lung and progresses slowly for about a year before rapidly undergoing necrosis to produce a cavity.20,28 The lungs characteristically develop caseous pneumonia that develops from lipid pneumonia, not from granulomas. Cavities typically develop in lungs with no granulomas. Granulomas and caseating granulomas are characteristic lesions of primary tuberculosis. Lipid pneumonia is the characteristic lesion of postprimary tuberculosis. A consequence of the failure to understand the pathology of postprimary tuberculosis is that the most important and troublesome lesions of tuberculosis lie largely beyond the reach of modern science.
Fig. 7.
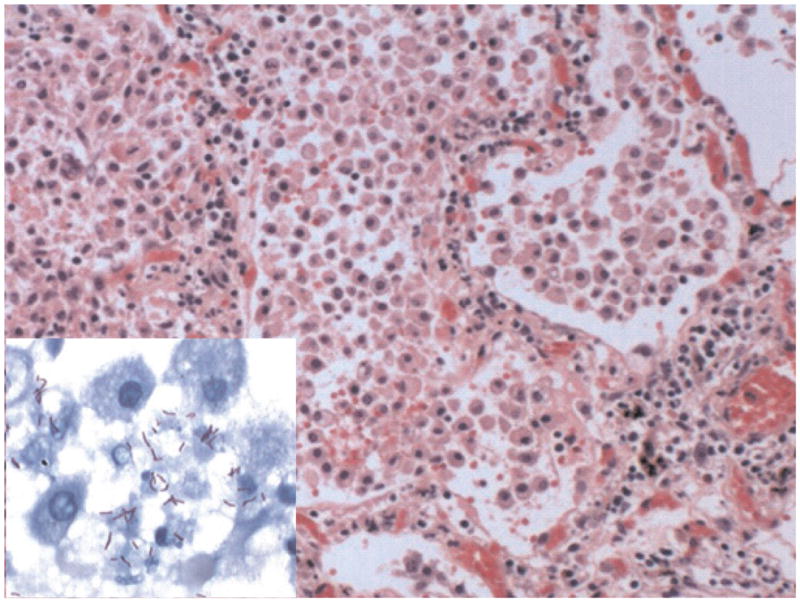
Histopathology of postprimary tuberculosis.20 This section is from a person who died suddenly of hemoptysis. Autopsy revealed tuberculosis in the upper lobes of both lungs with early cavitation that caused massive bleeding. The section illustrates the typical pathology of developing postprimary tuberculosis (H&E, 200×). It is an endogenous lipid pneumonia. The alveoli are filled with foamy macrophages, fibrin, and cell debris. Alveolar walls are infiltrated by lymphocytes. No granulomas were present anywhere in these lungs. MTB were present only in alveoli associated with alveolar macrophages and debris (insert; 1000× acid fast stain).
Postprimary tuberculosis resolves spontaneously in approximately 90% of people prior to development of cavities. Once cavities develop, however, they almost never heal spontaneously. Almost nothing is known of the factors that determine whether postprimary tuberculosis resolves or progresses to cavitation. Chronic cavitary tuberculosis is a complex process that can assume multiple histopathologic forms.28
The recognition that postprimary tuberculosis begins as a lipid pneumonia immediately suggests models and hypotheses to study it further. Available data identifies at least two factors that contribute to development of tuberculous lipid pneumonia. The first is bronchial obstruction that occurs in 100% of cases of early postprimary tuberculosis.59 In its early stages, tuberculous pneumonia closely resembles post obstructive or golden pneumonia that occurs distal to bronchial obstructions and probably nerve damage by cancer.56,60 We have found such cases with large cavities resulting from coagulation necrosis of lipid pneumonia remarkably similar to caseation and cavities of tuberculosis. This suggests that bronchial obstruction and nerve damage may contribute to cavity formation in tuberculosis. Second, mycolic acids cause macrophages, especially alveolar macrophages, to accumulate lipid and become tolerant to ingested mycobacteria.61 Much more study is needed.
Chronic slowly progressive tuberculosis in the mouse has been criticized as a model of human tuberculosis because it does not produce caseating granulomas.62 However, it is a progressive lipid pneumonia with many similarities to the human disease. We proposed, therefore, that it is an excellent model of early postprimary tuberculosis.20
7. The role of TDM in formation and maintenance of cavities
While much remains unknown, available data support the hypothesis that the monolayer form of TDM is a major factor in both the pathogenesis and maintenance of cavities. In most mouse and human studies, lipid progressively accumulates in endogenous lipid pneumonia over a period of months before the lesions rapidly undergo necrosis.19,62 In murine studies, mycobacterial components have been shown to accumulate with host lipids.63 Our hypothesis is that conditions eventually develop for activating the toxicity of the TDM by interaction with lipid droplets. Once activated, the toxicity of TDM increases several orders of magnitude and it becomes highly immunogenic leading to the rapid necrosis and cavity formation. We have found that injections of TDM in the toxic form accelerate development of necrosis of murine tuberculous lipid pneumonia.
As described above, MTB isolated from cavities typically grow much faster and produce much more TDM than those isolated from other organs. Since MTB can only be efficiently transmitted to new hosts from cavities in the lung, rapid growth of organisms in any other part of the body is likely to kill the host and the organisms as well. Consequently, cavities are the only place where MTB benefits from rapid multiplication. The production of large amounts of TDM by organisms in cavities suggest that TDM is essential for the maintenance of cavities. Laennec observed long ago that a smooth velvety layer lines mature cavities.64 Histologically, this layer is a mass of organisms resembling a pellicle.18 Consequently, it seems that spreading, climbing behavior of pellicles in culture reproduces their behavior in a cavity. This is also consistent with Indrigo and Actor’s observation that the amount of TDM on the surface of organisms is far more than is required to prevent their killing by macrophages.33,34
8. Conclusion
In summary, many fundamental questions of the pathogenesis of tuberculosis have remained out of reach of modern science because of lack of informative models. By focusing on TDM, our studies have developed several new models, new understanding of the disease and new approaches to combat it. We began with decades-old evidence that TDM is necessary for virulence of MTB. Continuing studies demonstrate that TDM is truly unique. Neither micellar nor monolayer TDM conforms to paradigms of modern biology. There is no other known biologic substance that has similar properties or activities. By changing conformation, TDM can switch between two very different sets of biologic activities. While surface-dependent toxicity is well established for certain mineral crystals, it is unique among biologic substances. On organisms, TDM is non toxic and protects the organisms from host defenses by several mechanisms. If it comes off organisms and associates with lipid, TDM becomes the most toxic, immunogenic and destructive component of the organism. MTB is an obligate human parasite. It can infect other species, but is transmitted only by people. Since virulent MTB expend large amounts of energy synthesizing far more TDM in our lungs than they need to survive in culture, it is safe to assume that it TDM is necessary for its survival as an infectious agent. We propose that understanding the activities of the two conformations of TDM is the key to unraveling several of the most perplexing mysteries of tuberculosis.
Acknowledgments
These studies were supported by Grants from the National Institutes of Health AI49534, AI068520, AI058247, HL068537, GM079810 and CytRx Corporation. This work was presented in part at the Texas Tuberculosis Research Symposium (TTRS) 2009, Houston, TX, co-sponsored by the University of Texas Health Sciences Center-Houston and the Methodist Hospital Research Institute.
Footnotes
Competing interests: No conflict of interest declared.
References
- 1.Middlebrook G, Dubos RG, Pierce C. Virulence and morphological characteristics of mammalian tubercle bacilli. J Exp Med. 1947;86:175–84. doi: 10.1084/jem.86.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bloch H. Studies on the virulence of tubercle bacilli: Isolation and biological properties of a constituent of virulent organisms. J Exp Med. 1950;91:197–219. doi: 10.1084/jem.91.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bloch H. Virulence of mycobacteria. Adv Tuberc Res. 1955;6:49–61. [PubMed] [Google Scholar]
- 4.Bloch H, Noll H. Studies on the virulence of tubercle bacilli. The effect of cord factor on murine tuberculosis. British J Exp Path. 1955;36:8–17. [PMC free article] [PubMed] [Google Scholar]
- 5.Dubos R, Dubos G. The White Plague. Tuberculosis, man, and society. Brunswick, NJ: Rutgers University Press; 1987. [Google Scholar]
- 6.Spitznagel JK, Dubos RJ. A fraction of tubercle bacilli possessing primary toxicity. J Exp Med. 1955;101:291–311. doi: 10.1084/jem.101.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Youmans GP. Chapter 3. Biologic activities of mycobacterial cells and cell components. In: Youmans GP, editor. Tuberculosis. Philadelphia, PA: W.B. Saunders; 1979. pp. 46–62. [Google Scholar]
- 8.Kuhn TS. The structure of scientific revolutions. Chicago, IL: University of Chicago Press; 1966. [Google Scholar]
- 9.Hunter R, Strickland F, Kezdy F. The adjuvant activity of nonionic block polymer surfactants I. The role of hydrophile–lipophile balance. J Immunol. 1981;127:1244–50. [PubMed] [Google Scholar]
- 10.Newman MJ, Actor JK, Balusubramanian M, Jagannath C. Use of nonionic block copolymers in vaccines and therapeutics. Crit Rev Ther Drug Carrier Syst. 1998;15:89–142. doi: 10.1615/critrevtherdrugcarriersyst.v15.i2.10. [DOI] [PubMed] [Google Scholar]
- 11.Retzinger GS, Meredith SC, Takayama K, Hunter RL, Kezdy FJ. The role of surface in the biological activities of trehalose 6,6′-dimycolate. Surface properties and development of a model system. J Biol Chem. 1981;256:8208–16. [PubMed] [Google Scholar]
- 12.Behling CA, Bennett B, Takayama K, Hunter RL. Development of a trehalose 6,6′-dimycolate model which explains cord formation by Mycobacterium tuberculosis. Infect Immun. 1993;61:2296–303. doi: 10.1128/iai.61.6.2296-2303.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schabbing RW, Garcia A, Hunter RL. Characterization of the trehalose 6,6′-di-mycolate surface monolayer by scanning tunneling microscopy. Infect Immun. 1994;62:754–6. doi: 10.1128/iai.62.2.754-756.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Retzinger GS, Meredith SC, Hunter RL, Takayama K, Kezdy FJ. Identification of the physiologically active state of the mycobacterial glycolipid trehalose 6,6′-di-mycolate and the role of fibrinogen in the biologic activities of trehalose 6,6′-di-mycolate monolayers. J Immunol. 1982;129:735–44. [PubMed] [Google Scholar]
- 15.Wiessner JH, Henderson JJ, Sohnle PG, Mandel NS, Mandel GS. The effect of crystal structure on mouse lung inflammation and fibrosis. Am Rev Respir Dis. 1988;138:445–50. doi: 10.1164/ajrccm/138.2.445. [DOI] [PubMed] [Google Scholar]
- 16.Syed SS, Hunter RL. The toxicity of TDM for macrophages depends on its surface structure. FASEB J. 1995;9:A509. [Google Scholar]
- 17.Lam CW, James JT, McCluskey R, Hunter RL. Pulmonary toxicity of single-wall carbon nanotubes in mice 7 and 90 days after intratracheal instillation. Toxicol Sci. 2004;77:126–34. doi: 10.1093/toxsci/kfg243. [DOI] [PubMed] [Google Scholar]
- 18.Hunter RL, Olsen MR, Jagannath C, Actor JK. Multiple roles of cord factor in the pathogenesis of primary, secondary, and cavitary tuberculosis, including a revised description of the pathology of secondary disease. Ann Clin Lab Sci. 2006;36:371–86. [PubMed] [Google Scholar]
- 19.Hunter RL, Olsen M, Jagannath C, Actor JK. Trehalose 6,6′-dimycolate and lipid in the pathogenesis of caseating granulomas of tuberculosis in mice. Am J Pathol. 2006;168:1249–61. doi: 10.2353/ajpath.2006.050848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunter RL, Jagannath C, Actor JK. Pathology of postprimary tuberculosis in humans and mice: contradiction of long-held beliefs. Tuberculosis (Edinb) 2007;87:267–78. doi: 10.1016/j.tube.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Jagannath C, Emanuele MR, Hunter RL. Activity of poloxamer CRL-1072 against drug-sensitive and resistant strains of Mycobacterium tuberculosis in macrophages and in mice. Int J Antimicrob Agents. 2000;15:55–63. doi: 10.1016/s0924-8579(00)00118-7. [DOI] [PubMed] [Google Scholar]
- 22.Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15:267–76. doi: 10.1038/nm.1928. [DOI] [PubMed] [Google Scholar]
- 23.Hunter RL, Venkataprasad N, Olsen MR. The role of trehalose dimycolate (cord factor) on morphology of virulent M. tuberculosis in vitro. Tuberculosis. 2006 Sep;86(5):349–56. doi: 10.1016/j.tube.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 24.Arora A, Armitige LY, Wanger A, Hunter R. Production of trehalose dimycolate (TDM) by pulmonary and extrapulmonary isolates of. Mycobacterium. ASM Abstracts 108th General Meetimg; 2008. p. U-053/0933. [Google Scholar]
- 25.Jarlier V, Nikaido H. Mycobacterial cell wall: structure and role in natural resistance to antibiotics [Review] FEMS Microbiol Lett. 1994;123:11–8. doi: 10.1111/j.1574-6968.1994.tb07194.x. [DOI] [PubMed] [Google Scholar]
- 26.Hart DP. Mycobacterium tuberculosis in macrophages: Effect of certain surfactants and other membrane active compounds. Science. 1968;162:686–9. doi: 10.1126/science.162.3854.686. [DOI] [PubMed] [Google Scholar]
- 27.Anonymous. WHO Report 2006. Global tuberculosis control – surveillance, planning, financing. WHO/HTM/TB/2006362. Geneva, Switzerland: World Health Organization; 2006. [Google Scholar]
- 28.Pagel W, Simmonds F, MacDonald N, Nassau E. Pulmonary tuberculosis, bacteriology, pathology, management, epidemiology and prevention. London: Oxford University Press; 1964. Chapter 3. The morbid anatomy and histology of tuberculosis, an introduction in simple terms; pp. 36–63. [Google Scholar]
- 29.Rich A. The Pathogenesis of tuberculosis, second edition. Springfield, Illinois: Charles C Thomas; 1951. [Google Scholar]
- 30.Ojha AK, Baughn AD, Sambandan D, Hsu T, Trivelli X, Guerardel Y, et al. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol Microbiol. 2008;69:164–74. doi: 10.1111/j.1365-2958.2008.06274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bloch H, Noll H. Studies on the virulence of tubercle bacilli. Variations in the virulence effect elicited by Tween 80 and thiosemicarbazone. Br J Exp Pathol. 1953;97:1–16. doi: 10.1084/jem.97.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silva CL. Participation of tumor necrosis factor in the antitumor activity of mycobacterial trehalose dimycolate (cord factor) Braz J Med Biol Res. 1989;22:341–4. [PubMed] [Google Scholar]
- 33.Indrigo J, Hunter RL, Jr, Actor JK. Cord factor trehalose 6,6′-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology. 2003;149:2049–59. doi: 10.1099/mic.0.26226-0. [DOI] [PubMed] [Google Scholar]
- 34.Indrigo J, Hunter RL, Jr, Actor JK. Influence of trehalose 6,6′-dimycolate (TDM) during mycobacterial infection of bone marrow macrophages. Microbiology. 2002;148:1991–8. doi: 10.1099/00221287-148-7-1991. [DOI] [PubMed] [Google Scholar]
- 35.Kan-Sutton C, Jagannath C, Hunter RL., Jr Trehalose 6,6′-dimycolate on the surface of Mycobacterium tuberculosis modulates surface marker expression for antigen presentation and costimulation in murine macrophages. Microbes Infect. 2009;11:40–8. doi: 10.1016/j.micinf.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Armitige LY, Jagannath C, Wanger AR, Norris SJ. Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: effect on growth in culture and in macrophages. Infect Immun. 2000;68:767–78. doi: 10.1128/iai.68.2.767-778.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Copenhaver RH, Sepulveda E, Armitige LY, Actor JK, Wanger A, Norris SJ, et al. A mutant of Mycobacterium tuberculosis H37Rv that lacks expression of antigen 85A is attenuated in mice but retains vaccinogenic potential. Infect Immun. 2004;72:7084–95. doi: 10.1128/IAI.72.12.7084-7095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwang SA, Wilk K, Kruzel ML, Actor JK. A novel recombinant human lactoferrin augments the BCG vaccine and protects alveolar integrity upon infection with Mycobacterium tuberculosis in mice. Vaccine. 2009;27:3026–34. doi: 10.1016/j.vaccine.2009.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trump BF, Berezesky IK, Chang SH, Phelps PC. The pathways of cell death: oncosis, apoptosis, and necrosis. Toxicol Pathol. 1997;25:82–8. doi: 10.1177/019262339702500116. [DOI] [PubMed] [Google Scholar]
- 40.Geisel RE, Sakamoto K, Russell DG, Rhoades ER. In vivo activity of released cell wall lipids of Mycobacterium bovis Bacillus Calmette-Guerin is due principally to trehalose mycolates. J Immunol. 2005;174:5007–15. doi: 10.4049/jimmunol.174.8.5007. [DOI] [PubMed] [Google Scholar]
- 41.Hunter RL, Bennett B. Am Soc Microbiol Abstracts. 1987. Mycobacterial cord factor kills macrophages by a membrane adhesive mechanism; p. 129. [Google Scholar]
- 42.Bekierkunst A, Levij IS, Yarkoni E, Vilkas E, Adam A, Lederer E. Granuloma formation induced in mice by chemically defined mycobacterial fractions. J Bacteriol. 1969;100:95–102. doi: 10.1128/jb.100.1.95-102.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bekierkunst A, Yarkoni E. Granulomatous hypersensitivity to trehalose 6,6′-di-mycolate (cord factor) in mice infected with BCG. Infect Immun. 1973;7:631–8. doi: 10.1128/iai.7.4.631-638.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Actor JK, Olsen M, Hunter RL, Jr, Geng YJ. Dysregulated response to mycobacterial cord factor trehalose-6,6′-dimycolate in CD1D−/− mice. J Interferon Cytokine Res. 2001;21:1089–96. doi: 10.1089/107999001317205222. [DOI] [PubMed] [Google Scholar]
- 45.Perez RL, Roman J, Roser S, Little C, Olsen M, Indrigo J, et al. Cytokine message and protein expression during lung granuloma formation and resolution induced by the mycobacterial cord factor trehalose-6,6′-dimycolate. J Interferon Cytokine Res. 2000;20:795–804. doi: 10.1089/10799900050151067. [DOI] [PubMed] [Google Scholar]
- 46.Welsh KJ, Abbott AN, Hwang SA, Indrigo J, Armitige LY, Blackburn MR, et al. A role for tumour necrosis factor-alpha, complement C5 and interleukin-6 in the initiation and development of the mycobacterial cord factor trehalose 6,6′-dimycolate induced granulomatous response. Microbiology. 2008;154:1813–24. doi: 10.1099/mic.0.2008/016923-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jagannath C, Hoffmann H, Sepulveda E, Actor JK, Wetsel RA, Hunter RL. Hypersusceptibility of A/J mice to tuberculosis is in part due to a deficiency of the fifth complement component (C5) Scand J Immunol. 2000;52:369–79. doi: 10.1046/j.1365-3083.2000.00770.x. [DOI] [PubMed] [Google Scholar]
- 48.Actor JK, Breij E, Wetsel RA, Hoffmann H, Hunter RL, Jr, Jagannath C. A role for complement C5 in organism containment and granulomatous response during murine tuberculosis. Scand J Immunol. 2001;53:464–74. doi: 10.1046/j.1365-3083.2001.00902.x. [DOI] [PubMed] [Google Scholar]
- 49.Borders CW, Courtney A, Ronen K, Pilar Laborde-Lahoz M, Guidry TV, Hwang SA, et al. Requisite role for complement C5 and the C5a receptor in granulomatous response to mycobacterial glycolipid trehalose 6,6′-dimycolate. Scand J Immunol. 2005;62:123–30. doi: 10.1111/j.1365-3083.2005.01643.x. [DOI] [PubMed] [Google Scholar]
- 50.Abbott AN, Guidry TV, Welsh KJ, Thomas AM, Kling MA, Hunter RL, et al. 11beta-hydroxysteroid dehydrogenases are regulated during the pulmonary granulomatous response to the mycobacterial glycolipid trehalose-6,6′-dimycolate. Neuroimmunomodulation. 2009;16:147–54. doi: 10.1159/000204227. [DOI] [PubMed] [Google Scholar]
- 51.Guidry TV, Hunter RL, Jr, Actor JK. CD3+ cells transfer the hypersensitive granulomatous response to mycobacterial glycolipid trehalose 6,6′-dimycolate in mice. Microbiology. 2006;152:3765–75. doi: 10.1099/mic.0.29290-0. [DOI] [PubMed] [Google Scholar]
- 52.Guidry TV, Hunter RL, Jr, Actor JK. Mycobacterial glycolipid trehalose 6,6′-dimycolate-induced hypersensitive granulomas: contribution of CD4+ lymphocytes. Microbiology. 2007;153:3360–9. doi: 10.1099/mic.0.2007/010850-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guidry TV, Olsen M, Kil KS, Hunter RL, Jr, Geng YJ, Actor JK. Failure of CD1D−/− mice to elicit hypersensitive granulomas to mycobacterial cord factor trehalose 6,6′-dimycolate. J Interferon Cytokine Res. 2004;24:362–71. doi: 10.1089/107999004323142222. [DOI] [PubMed] [Google Scholar]
- 54.Pagel W, Pagel M. Zur Histochemie der Lungentuberkulose, mit besonderer Berücksichtigung der Fettsubstanzen und Lipoide [Fat and lipoid content to tuberculous tissue. Histochemical investigation] Virchows Arch path Anat. 1925;256:629–40. [Google Scholar]
- 55.Osler W. The principles and practice of medicine. New York: D. Appleton and Company; 1892. Chapter 26, Tuberculosis; pp. 184–255. [Google Scholar]
- 56.Mays TJ. Pulmonary consumption, pneumonia and allied diseaeses of the lungs: their etiology, pathology and treatment – with a chapter on physical diagnosis. New York: E.B. Treat & Company; 1901. Pathology of pulmonary consumption; pp. 247–88. [Google Scholar]
- 57.Dannenberg AM., Jr . Pathogenisis of human pulmonary tuberculosis. Insights from the rabbit model. Washington, DC: ASM press; 2006. [Google Scholar]
- 58.Creighton C. Bovine tuberculosis in man: an account of the pathology of suspected cases. London: MacMillan and Co; 1881. [Google Scholar]
- 59.Kashyap S, Mohapatra PR, Saini V. Endobronchial tuberculosis. Indian J Chest Dis Allied Sci. 2003;45:247–56. [PubMed] [Google Scholar]
- 60.Tamura A, Hebisawa A, Fukushima K, Yotsumoto H, Mori M. Lipoid pneumonia in lung cancer: radiographic and pathological features. Jpn J Clin Oncol. 1998;28:492–6. doi: 10.1093/jjco/28.8.492. [DOI] [PubMed] [Google Scholar]
- 61.Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 2008;4:e1000204. doi: 10.1371/journal.ppat.1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.North RJ, Jung YJ. Immunity to tuberculosis. Annu Rev Immunol. 2004;22:599–623. doi: 10.1146/annurev.immunol.22.012703.104635. [DOI] [PubMed] [Google Scholar]
- 63.Mustafa T, Phyu S, Nilsen R, Jonsson R, Bjune G. In situ expression of cytokines and cellular phenotypes in the lungs of mice with slowly progressive primary tuberculosis. Scand J Immunol. 2000;51:548–56. doi: 10.1046/j.1365-3083.2000.00721.x. [DOI] [PubMed] [Google Scholar]
- 64.Laennec R. A treatise on diseases of the chest in which they are described according to their anatomical characters, and their diagnosis established on a new principle by means of acoustick instruments. T&G Underwood; London: 1821. [Reprinted 1979 by The Classics of Medicine Library. Birmingham, AL.] [Google Scholar]