

Abstract
Background and Purpose
Blood-stage Plasmodium parasites cause morbidity and mortality from malaria. Parasite resistance to drugs makes development of new chemotherapies an urgency. Aminoacyl-tRNA synthetases have been validated as antimalarial drug targets. We explored long-term effects of borrelidin and mupirocin in lethal P. yoelii murine malaria.
Experimental Approach
Long-term (up to 340 days) immunological responses to borrelidin or mupirocin were measured after an initial 4 day suppressive test. Prophylaxis and cure were evaluated and the inhibitory effect on the parasites analysed.
Key Results
Borrelidin protected against lethal malaria at 0.25 mg·kg−1·day−1. Antimalarial activity of borrelidin correlated with accumulation of trophozoites in peripheral blood. All infected mice treated with borrelidin survived and subsequently developed immunity protecting them from re-infection on further challenges, 75 and 340 days after the initial infection. This long-term immunity in borrelidin-treated mice resulted in negligible parasitaemia after re-infections and marked increases in total serum levels of antiparasite IgGs with augmented avidity. Long-term memory IgGs mainly reacted against high and low molecular weight parasite antigens. Immunofluorescence microscopy showed that circulating IgGs bound predominantly to late intracellular stage parasites, mainly schizonts.
Conclusions and Implications
Low borrelidin doses protected mice from lethal malaria infections and induced protective immune responses after treatment. Development of combination therapies with borrelidin and selective modifications of the borrelidin molecule to specifically inhibit plasmodial threonyl tRNA synthetase should improve therapeutic strategies for malaria.
Keywords: borrelidin, Plasmodium, malaria, treatment, humoral response, aminoacyl-tRNA synthetases
Introduction
Among all parasite diseases, malaria causes the highest morbidity and mortality in the world. In countries where malaria is endemic, there were about 216 million cases in 2010 causing nearly 655 000 deaths, mostly in children under 5 years old and in pregnant women. Global plans to control, eliminate and ultimately eradicate malaria have mainly focused on prophylaxis, chemotherapy, vaccine development and vector control programmes. Unfortunately, these strategies are being hampered by the emergence of parasite resistance to old and newly introduced drugs (Bloland, 2001), so new long-acting antimalarial drugs are urgently needed for combination therapies. Parasite load, the innate host resistance to the infection (Stevenson and Riley, 2004), the naturally acquired immunity and the capacity of the parasite to evade the host immune response (Doolan et al., 2009) are all known to play important roles in the course of infection and in the outcome of the treatment. Although the acquisition of immunity against Plasmodium falciparum after a single infection can be generated, it is incomplete, non-sterilizing and transient, requiring repeated infections to be retained, and it is compromised in pregnant women and almost non-existent in children (Doolan et al., 2009). This condition provides clinical protection against new infections by maintaining a low-grade (Druilhe and Perignon, 1997) and generally asymptomatic parasitaemia in adults (Collins and Jeffery, 1999). Because the naturally acquired immunity (NAI) is an efficient resource against severe disease or lethality in continuously malaria-exposed adults (Doolan et al., 2009), efforts directed towards prophylactic interventions based on facilitating an efficient immunological response would eventually help to control malaria (Achtman et al., 2005). To this end, experimental inoculations of very small doses of intact sporozoites in volunteers during chloroquine treatment have shown to confer higher and longer levels of protection than vaccination with radiation-attenuated sporozoites (Hoffman et al., 2002; Roestenberg et al., 2009; 2011). More than 30 years ago, the same combination of sporozoites and chloroquine was reported to be successful for the immunological protection of mice (Beaudoin et al., 1977). These studies highlight the potential of inoculation with parasites combined with antimalarial treatment to favour the native exposure of antigens for the development of protective immunity (Borrmann and Matuschewski, 2011; Sauerwein et al., 2010). However, it seems that this type of immunoprotection cannot be induced in untreated malarial infections, because the high parasitaemia level reached could impair the development of protective immunity (Ocana-Morgner et al., 2003; Wilson et al., 2006). Taken together, these data support the development of new antimalarial strategies based on sustaining NAI through drug treatment.
We have therefore focused on inhibitors of aminoacyl-tRNA synthetases (ARS), essential enzymes for cell viability that have been identified and validated as antimalarial drug targets (Schimmel et al., 1998; Hurdle et al., 2005; Istvan et al., 2011; Hoepfner et al., 2012). Mupirocin is an inhibitor of isoleucyl tRNA synthetase (IleRS) (Hughes and Mellows, 1978) while borrelidin is an inhibitor of prokaryotic threonyl tRNA synthetase (ThrRS) (Hutter et al., 1966) and yeast cyclin-dependent kinase Cdc28/Cln2 (Tsuchiya et al., 2001), as well as an activator of eukaryotic caspase-3 and caspase-8 (Kawamura et al., 2003). Several pharmacological activities have been reported for borrelidin: antibiotic (Berger et al., 1949), angiogenesis inhibitor (Wakabayashi et al., 1997), anti-metastatic (Funahashi et al., 1999), antimitotic (Tsuchiya et al., 2001), antiviral (Dickinson et al., 1965), herbicidal and insecticidal (Dorgerloh et al., 1988), and antitumoural (Habibi et al., 2012). Both antibiotics, borrelidin and mupirocin, display substantial in vitro inhibitory activity against P. falciparum with an IC50 value in the nanomolar range (Otoguro et al., 2003; Istvan et al., 2011). In addition, recent data suggest that while mupirocin inhibited apicoplast-specific translation inducing ‘delayed-death’, the effects of borrelidin were not restricted to an organelle-specific phenotype and promoted an immediate arrest of parasite growth (Jackson et al., 2012). On the other hand, borrelidin has also antimalarial activity in vivo, against P. berghei and P. yoelii ssp. when administered subcutaneously and orally, although its effect has been only studied during the first 4 days post-infection (pi) during a primary contact (Otoguro et al., 2003).
Here, we have analysed the effect of treatment with either borrelidin or mupirocin, administered during exposure of mice to lethal blood-stage malaria, focusing on the immune response acquired. These effects were compared with those of the standard antimalarial drug chloroquine, a rapid parasiticide widely used in the past for the treatment of human malaria.
Methods
Rodent parasites and animals
All animal care and experimental procedures carried out at the Universidad Complutense de Madrid complied with Spanish (R.D. 32/2007) and European Union legislation (2010/63/CE) and were approved by the Animal Experimentation Committee of this institution. The experiments here described involving animals are reported following the ARRIVE guidelines for pharmacological studies (Kilkenny et al., 2010). The number of animals was calculated using Statgraphics Centurion 16.1.18 software (Statpoint Technologies, Inc, Warrenton, VA, USA) to provide about 80% of statistical power with 95% confidence level, and always following the 3Rs principles. A total of 120 animals were used in the experiments described here. The rodent malaria parasite P. yoelii 17XL (Py17XL) was kindly provided by Dr Virgilio Do Rosario (Instituto de Higiene e Medicina Tropical, Universidade Nova de Lisboa) and stored in liquid nitrogen after serial blood passages in mice. Inbred BALB/cAnNHsd pathogen-free female mice, aged 6–8 weeks and 16–18 g of weight were purchased from Harlan Laboratories (Udine, Italy) and housed at random in airy racks containing Lignocel® three-fourths bedding (Rettenmaier & Sohne, Rosenberg, Germany) and kept under constant standard conditions of light (12:12 h light : dark cycles), temperature (22–24°C) and humidity (around 50%) at the Animal House of the Universidad Complutense de Madrid. All mice were fed a commercial diet (2018 Teklad Global 18% Protein Rodent Diet, Harlan Laboratories) ad libitum.
In vivo antimalarial activity
Firstly, the in vivo antimalarial activity of borrelidin (Fluorochem, Hadfield, Derbyshire, UK), mupirocin (GlaxoSmithKline, Brentford, Middlesex, UK) and chloroquine (Sigma-Aldrich, St. Louis, MO, USA) was assessed using a 4 day suppressive test as previously described (Peters and Robinson, 1999). Briefly, mice were inoculated with 2 × 107 Py17XL-infected red blood cells (RBCs) from infected mice by i.p. injection between 10:00 and 12:00 h. The dose used for in vivo drug treatment was calculated based on the in vitro IC50 of each drug considering the toxicity and solubility data of each compound. Then, mice were treated daily for 4 days by i.p. injection – using a 30 G one-half needle under an approximately 10–15° angle – in the lower quadrant of the abdomen off midline. Borrelidin (0.25 mg·kg−1·day−1; n = 10), mupirocin (2.5 mg·kg−1·day−1; n = 10) or chloroquine in two doses (1 mg·kg−1·day−1; n = 5 or 30 mg·kg−1·day−1; n = 10), were used, starting 2 h after the infection. The tested drugs were prepared at appropriate doses in aqueous vehicle containing 7% Tween-80 and 3% ethanol. Control animals received aqueous vehicle by the same route (n = 5). Mice for each group were selected at random and treatments were carried out at the animal room. Parasitaemia was monitored daily (between 9:00 and 10:00 h) by microscopic examination of Wright's-stained thin-blood smears using the Plasmoscore 1.3 software (Burnet Institute, Melbourne, Australia; Proudfoot et al., 2008). To assess immunity against re-infection, cured mice were challenged with the same parasite dose 75 and 340 days after the primary infection and parasitaemia was monitored for a further 30 days. Three independent experiments were performed.
To study cure of the disesase in mice with established parasitaemia, borrelidin (0.25 mg·kg−1·day−1) and chloroquine (30 mg·kg−1·day−1) were administered daily for 4 days, beginning when blood parasitaemia had reached 10% (from day 3 pi to day 6 pi). Data from two independent experiments with n = 4 mice per group.
In vitro determination of the type of antimalarial action
Drug assays were performed using P. falciparum strain Dd2 (clone MRA-150; Malaria Research and Reference Reagent Resource Center: http://www.mr4.org), maintained in continuous culture following the protocol previously described by Radfar et al. (2009). To determine the type of antimalarial activity, we used the previously described procedures (Bahamontes-Rosa et al., 2011) for drug exposure duration and parasite culture intervals. Briefly, parasites in the ring stage were seeded at 1% parasitaemia and 2% haematocrit and exposed for 48 h to borrelidin or chloroquine at 20 times the IC50 value (25 nM and 3 μM, respectively) previously described (Moneriz et al., 2009; Jackson et al., 2012). Parasites were then harvested and washed three times with 10 mL washing medium RPMI 1640 (Sigma-Aldrich) supplemented with 100 μM hypoxanthine (Sigma-Aldrich), 25 mM HEPES (Sigma-Aldrich) and 12.5 μg·mL−1 gentamicine (Sigma-Aldrich) to completely remove the drugs. Then they were cultured in culture medium (Radfar et al., 2009) without drug for a further 8 days. After drug withdrawal, culture medium was changed daily. Parasitaemia and life cycle stages were monitored by microscopic examination of Wright's-stained thin-blood smears using Plasmoscore software. Four independent experiments were performed.
Characterization of specific P. yoelii 17XL antibodies in mice serum
Extracting parasite proteins from infected whole blood
Py17XL protein lysates were extracted from the erythrocytes of infected mice showing >50% parasitaemia. Mice were anaesthetized with isoflurane, as recommended by the local Animal Experimentation Committee, and whole blood was collected from the aorta into tubes containing EDTA 0.1 M as anticoagulant and kept at −80°C until protein extraction. Protein isolation began with erythrocyte lysis using 10 vol of saponin 0.1% (w/v) in PBS. After centrifugation (320× g, 5 min, 4°C) and washing twice in cold PBS, the pellet was treated with 2–3 vol of extraction buffer (50 mM Tris-HCl, pH 8.0; 50 mM NaCl; 0.5% Mega 10) containing protease inhibitor cocktail (Roche, Indianapolis, IN, USA) and subjected to four freeze–thaw cycles. Finally, lysates were centrifuged (7800× g, 10 min, 4°C) and total Py17XL protein samples stored at −20°C until use. Protein concentration was determined by the Bradford protein assay (Bio-Rad, Hercules, CA, USA).
IgG concentrations
Specific anti-Py17XL antibodies in sera from mice were quantified using mouse IgG ELISA detection kits following the manufacturer's instructions (Bethyl Laboratories, Montgomery, TX, USA). Briefly, microtiter plates were coated overnight with 100 μL per well of Py17XL protein extracts from infected RBCs (described above) at 5 μg·mL−1 in carbonate-bicarbonate buffered solution (Sigma). For specific IgG antibody quantification, diluted mouse serum was incubated for 1 h at room temperature (dilutions 1:150–1:800). IgG binding was detected with goat anti-mouse IgG conjugated with HRP at a 1:50 000 dilution. The enzymic reaction was developed using 3,3′,5,5'tetramethyl benzidine as enzyme substrate. Absorbance readings of reaction products were obtained at 652 nm in a Varian Cary 50 Bio spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). Sera from naïve mice, uninfected drug-treated mice and untreated infected mice were used as negative controls. Purified myeloma-derived mouse IgG (Bethyl Laboratories) was used to generate a sigmoid logistic four-parameter standard curve.
IgG avidity
To test antibody avidity (AI), five different concentrations (0, 1, 2, 3 and 4 M) of the chaotropic agent NaSCN (Sigma-Aldrich) were used to independently disrupt antigen-antibody binding during the ELISA protocol described above, as previously described (Pullen et al., 1986). Py17XL protein extracts from infected RBCs (described above) were used as antigen. After the serum incubation step, wells were washed three times using PBS plus 0.05% Tween-20. Next, each NaSCN concentration was added to a different well. Plates were allowed to stand at room temperature for 15 min and extensively washed (×6) with PBS containing 0.05% Tween-20. Subsequent steps were performed as described in the ELISA protocol. After incubation with various NaSCN concentrations, spectrophotometric readings at 652 nm were translated into percentage immunoglobulin binding with respect to values obtained at 0 M NaSCN. The avidity index is given as the NaSCN concentration value that produced a 50% reduction in immunoglobulin binding.
Western blot analysis
Samples of parasite proteins (10 μg) were fractionated on 10% SDS-PAGE (Bio-Rad) and transferred onto nitrocellulose membranes following standard procedures. Blots were blocked for 2 h in 5% non-fat skimmed milk in PBS and then incubated with mouse serum antibodies (1:10 000) overnight. Anti-mouse IgG HRP linked (Amersham Bioscience, Buckinghamshire, UK) secondary antibody was incubated for 1 h at room temperature at a 1:5000 dilution and the antigen-antibody reaction visualized using the SuperSignal chemiluminescent substrate (Pierce) and exposure to X-ray film. Sera from naïve mice, uninfected drug-treated mice and untreated infected mice were used as negative controls.
Immunofluorescence assay
To identify the specificity of IgG antibodies for intraerythrocyte stage parasites, thin-blood smears were prepared using iRBCs from mice at 40% parasitaemia, and subsequently fixed in freshly prepared 90% acetone 10% methanol for 2 min. The parasites were blocked with 3% BSA and 10% goat serum in PBS for 1 h at room temperature, and subsequently incubated with a 1:2500 dilution of mouse serum in the same conditions. Finally, smears were incubated with goat anti-mouse IgG labelled with Alexa Fluor 488 (1:400; Invitrogen, Carlsbad, CA) and DAPI (0.3 μM; Invitrogen) for 1 h at room temperature, and then mounted according to standard procedures. Two different controls were established following the same procedures, but substituting the primary antibody with the same volume of IgG serum from uninfected mice or with PBS. Also, because the acetone–methanol fixation method used in these immunofluorescence assays permeabilizes RBC membranes, additional staining controls were prepared to determine whether IgG also recognized surface antigens on the infected RBCs. Thus, thin-blood smears were fixed in 4% paraformaldehyde and permeabilized or not with 0.1% Triton X-100 (Sigma-Aldrich).
Labelling was detected by confocal immunofluorescence microscopy using a Leica CTR 6500 fluorescence microscope (Leica Microsystems, Wetzlar, Germany). Alexa Fluor was monitored by excitation with the 488 nm wavelength laser and DAPI were excited at wavelengths of 405 nm.
Statistical analysis
Data are presented as means ± SEM. Groups were compared using non-parametric Mann–Whitney test. The statistical significance was set at P ≤ 0.05.
Materials
Borrelidin (Fluorochem), mupirocin (GlaxoSmithKline, Brentford, Middlesex, UK), chloroquine (Sigma-Aldrich); NaSCN (Sigma-Aldrich), Triton x-100 (Sigma-Aldrich); AlexaFluor 488 (Invitrogen) and DAPI (Invitrogen).
Results
In vivo antimalarial activity of borrelidin and mupirocin against lethal P. yoelii 17XL infection
Drugs were administered to mice for the first four days following a primary contact with Py17XL infection. As shown in Figure 1, borrelidin was the only antibiotic successful at curing lethal malaria infection in mice, a result comparable to that observed with the positive control of the higher dose of chloroquine (30 mg·kg−1: chloroquine-30). In contrast, individuals treated with mupirocin or low chloroquine dosage (1 mg·kg−1) were unable to halt parasite growth, which was detectable from day 2 pi and subsequently died by days 4.2 ± 0.1 and 5 ± 0.3 pi, respectively, as did untreated mice (death on day 4.6 ± 0.3 pi). All mice treated with borrelidin and the standard chloroquine (30 mg·kg−1) dose regimes showed decreased parasite growth during the 4 days of its administration (<1% parasitaemia in peripheral blood; P < 0.0001 between parasitaemia of untreated and both, chloroquine-30 and borrelidin treated mice at day 3 pi). The ED90 value for borrelidin was around 0.25 mg·kg−1 against the lethal P. yoelii strain 17XL, slightly lower than for other P. yoelii strains (Otoguro et al., 2003). Different effects on the control of parasitaemia were observed after withdrawal of borrelidin or chloroquine-30 treatments at day 4 pi. Whereas in borrelidin-treated mice, parasitaemia values started to rise during the subsequent days up to a maximum of 35% attained on day 11 pi, the parasite growth was notably reduced in chloroquine–30-treated mice, which reached a peak below 10% at day 10 pi (P < 0.0001 between parasitaemia of chloroquine–30- and borrelidin-treated mice at day 10 pi). Thereafter, a progressive decrease was observed until no parasites could be microscopically detected at day 12 pi in chloroquine–30-treated mice and significantly later in borrelidin-treated mice around day 20 pi (P = 0.0003).
Figure 1.

Survival and parasitemia course in infected mice subjected to 4 day suppressive antimalarial treatment. BALB/c mice infected with 2 × 107 Plasmodium yoelii 17XL infected RBCs were treated for 4 days with vehicle (n = 5),1 mg·kg−1·day−1 chloroquine (n = 5), 30 mg·kg−1·day−1 chloroquine (n = 10), 25 mg·kg−1·day−1 borrelidin (n = 10) or 2.5 mg·kg−1·day−1 mupirocin (n = 10). (A) Survival and (B) parasitaemia percentages are shown for each group as mean ± SEM. Arrows indicate the four i.p. injections of drug/vehicle. Data from three independent experiments. *P < 0.05, significant differences between indicated groups.
Parasite stages distribution in treated P. falciparum cultures
The two antimalarial drugs that were able in vivo to cure lethal malaria in mice were also assayed in vitro on the drug-resistant P. falciparum strain Dd2. Following the approaches used in previous drug activity studies, the P. falciparum cultures were exposed to borrelidin and chloroquine for 48 h (one complete parasite life cycle) and then cultured for four subsequent life cycles (8 additional days) (Figure 2) (Goodman et al., 2007; Bahamontes-Rosa et al., 2011). The concentration used was 20 times the corresponding IC50 values of borrelidin and chloroquine for P. falciparum Dd2.
Figure 2.
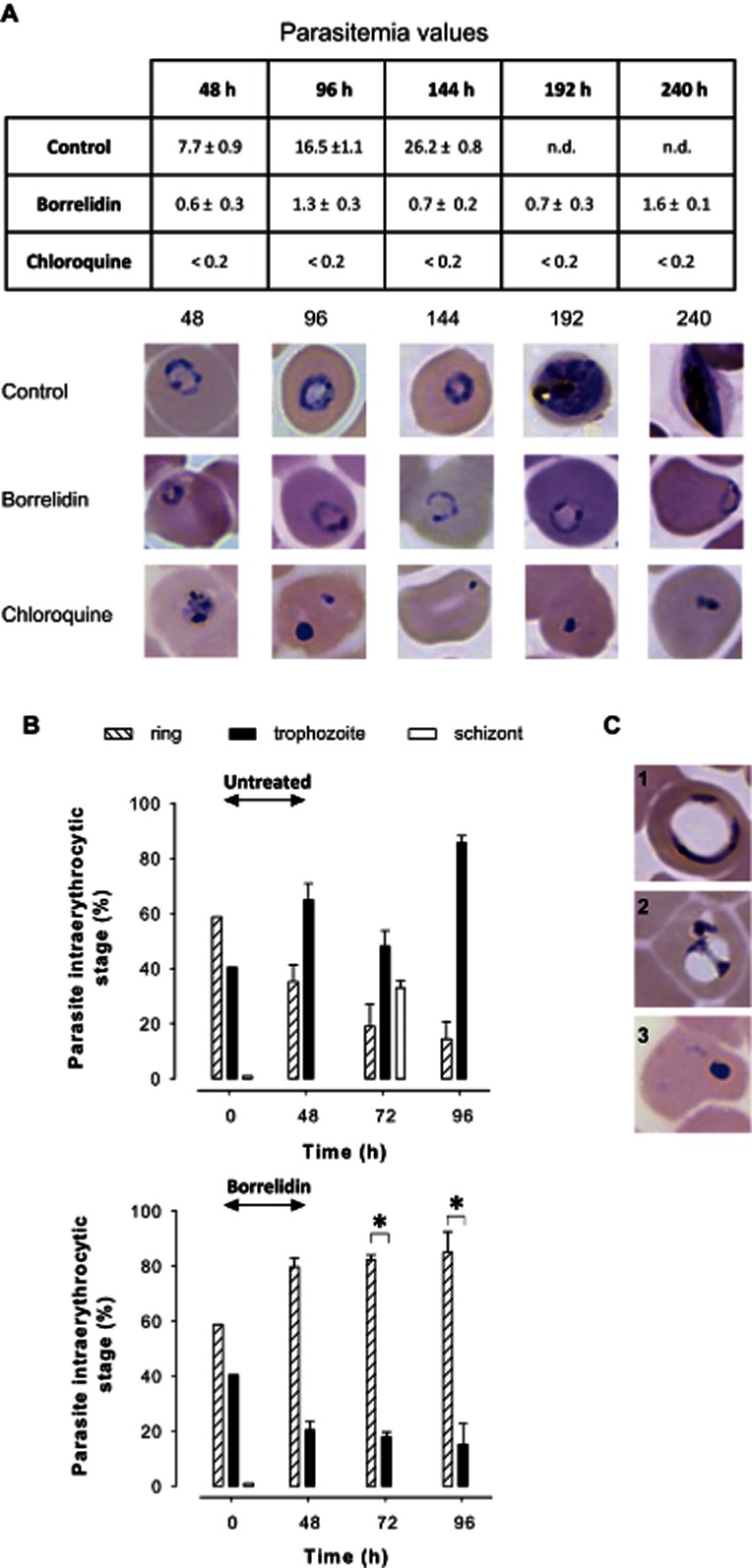
In vitro antimalarial activity in a 240 h growth assay. P.falciparum Dd2 parasites were treated with borrelidin (25 nM) or chloroquine (3 μM) for 48 h and then were further cultured in medium in the absence of the drugs. (A) Variation of parasitaemia and parasites images and (B) percentages of parasite intraerythrocytic stages show the morphology and evolution of the parasites at the different time points over 96 h with respect to the control. Bars show the percentages of ring, trophozoite and schizont stage parasites in infected RBCs. (C) Representative images of altered borrelidin-treated parasites. Results are expressed as the mean ± SEM in two independent experiments. n.d. = not determined parasitaemia in control cultures due to the saturation of parasites after two cycles that made growth not comparable to the drug-treated cultures. Four independent experiments were conducted. *P < 0.05.
Microscopic observation of the treated cultures (Figure 2A) revealed a reduced parasite growth after 48 h treatment with borrelidin (19-fold) and chloroquine (>100-fold), compared with control cultures (P = 0.034; P = 0.016, respectively) with an abundance of shrunk forms of the parasitised cell. The parasites remaining after treatment with borrelidin struggled throughout the 4 following days to recover their viability, which was only partially regained after 240 h (four parasite life cycles; P = 0.05). Analysis of the distribution of intraerythrocytic stages, after antibiotic withdrawal, in surviving parasites showed a high percentage of ring forms (>75%; P = 0.028 after 72 and 96 h) suggesting a specific effect of borrelidin on mature stages (Figure 2B). Remarkably, the antibiotic treatment also induced an altered cellular development in a fraction of surviving early-stage parasites, which seems to lead to death in the following schizogonic cycles (72–144 h) (Figure 2C). Control cultures followed a normal growth during the first three life cycles. Afterwards, the high parasite growth did not allow new invasion cycles without the addition of new erythrocytes and consequently the conditions in cycles 4 and 5 were not comparable.
Distribution of parasite stages in borrelidin treated mice
To follow potential changes in the typical asynchronic P. yoelii infection, analysis of the distribution of parasite stages was performed in the infected RBCs from the different groups of treated mice from days 0 to 10 pi by microscopic examination of thin-blood smears. Control mice and mupirocin-treated mice displayed identical P. yoelii asynchronicity (Figure 3A,B). Both groups showed a high proportion of ring-stage parasites during the first 2 days of increasing parasitaemia while mature forms (trophozoite/schizonts) were predominant in the last 2 days before death. This observation is in agreement with the 22–25 h required to complete the P. yoelii infection cycle in the erythrocyte. In the first 2 days, when parasitaemia was not too high, healthy schizonts containing merozoites were able to continue invading any intact RBCs that were still available. However, when erythrocytes were in short supply because of the high parasitaemia level (more than 80%), merozoites could not easily find red cells to generate new rings and therefore the remaining mature forms, including schizonts, were the predominant observed forms (P = 0.01 schizont vs. rings at day 5 pi). Similar results were found in mice treated with sub-therapeutic chloroquine doses (data not shown). The borrelidin-treated mice showed a different picture (Figure 3C). Thus, at day 3 pi, mice treated with borrelidin showed a significant dominance of trophozoite-stage parasites (P = 0.002) that was double that of the corresponding forms in the untreated (P = 0.004) or in the mupirocin-treated (P = 0.004) groups. Conversely, at this time, ring-stage parasites in borrelidin treated animals decreased by around 2.5-fold compared with untreated and mupirocin-treated mice (P = 0.06). In agreement with the above figures, borrelidin-treated vs. untreated or mupirocin-treated mice also showed differences in the percentage of schizont-stage parasites. In the borrelidin treated mice, trophozoites persisted as the most abundant form until day 10 pi (P < 0.05 trophozoites vs. schizonts at days 7, 8, 9 and 10 pi; and trophozoites vs. rings at day 7 and 10 pi), when parasitaemia levels peaked.
Figure 3.
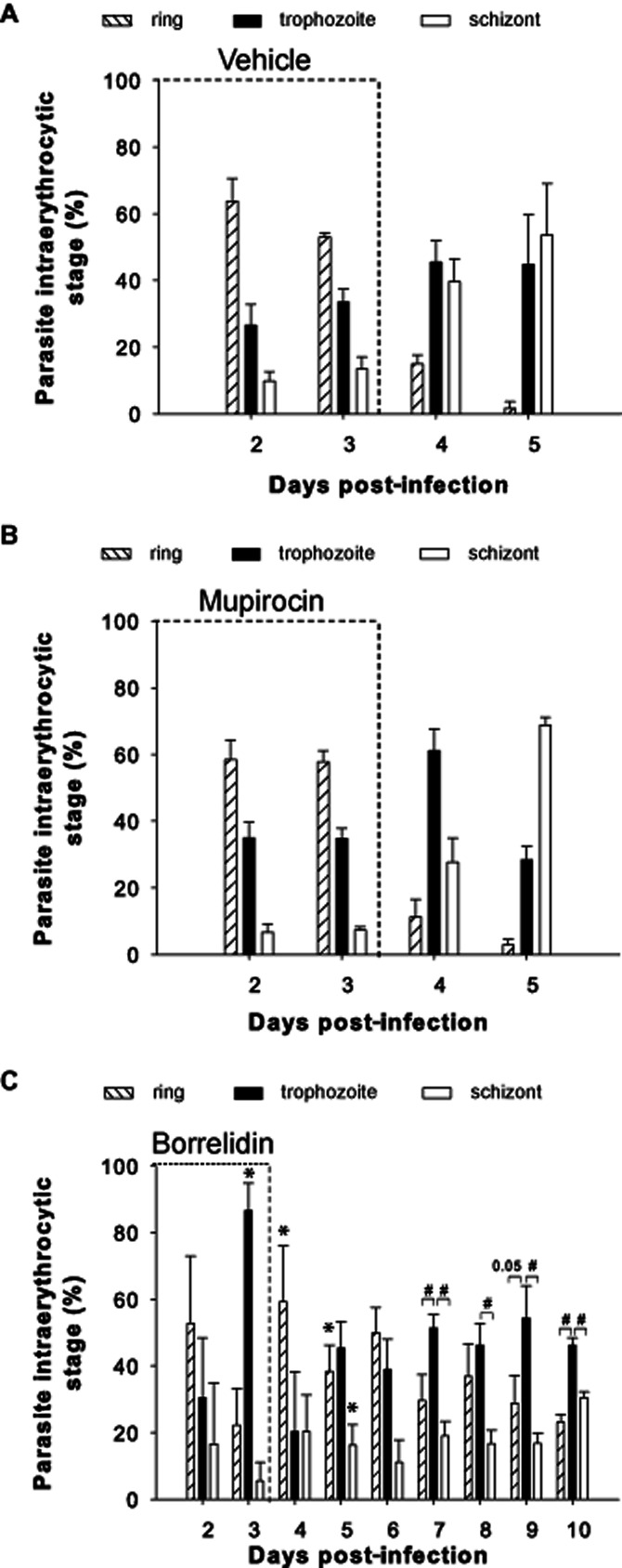
Distribution of intraerythrocytic parasite stages in blood samples from treated and untreated mice. Bars show the percentages of ring, trophozoite and schizont stage parasites in infected iRBCs from days 2 to 5 pi in (A) vehicle-injected (n = 5) and (B) mupirocin-treated mice (n = 10) or (C) to day 10 pi in borrelidin-treated mice (n = 10). Data were obtained by microscopy inspection of Wright's-stained thin blood smears. Results, expressed as the mean ± SEM, represent the percentages of cells of each parasite stage among a total of 500 erythrocytes per slide. Three independent experiments were performed. *P < 0.05, significant differences versus same parasite form in vehicle-injected mice; #P < 0.05, significant differences between parasite forms in the borrelidin-treated group.
Long-term immunity in cured mice
To ascertain whether the borrelidin treatment influenced the development of protective malaria immunity to re-infection, cured mice were re-infected on day 75 pi and parasitaemia was monitored for the following 30 days. The mice cured with the chloroquine-30 regime were also re-infected and used for comparison. As shown in Figure 4A, all the borrelidin-treated animals showed long-term full protection against the second lethal infection that was lethal to vehicle controls. In fact, blood parasitaemia levels (lower than 0.01% in most mice) were barely perceptible (Figure 4B). A lower protection was obtained in chloroquine–30-treated animals. After the secondary challenge, parasite growth was also transient and maintained at extremely low rates (<0.01%) with the exception of one out of 10 mice, which did not show microscopic parasitaaemia during the primary infection. This mouse was unable to control re-infection and died on day 84 pi. Interestingly, parasitaemia in this mouse began to be detected on day 79 pi (0.2%) whereas control mice of the same age exhibited a mean value of 75% parasitaaemia by this day. Mice cured by either borrelidin or chloroquine–30 from this re-infection, were challenged again on day 340 pi and all of them showed full protection (Figure 4A,B).
Figure 4.

Survival and parasitaemia course of the borrelidin and chloroquine-treated mice in response to a second and third re-infection. Borrelidin and chloroquine-treated mice (n = 10) surviving primary infection were re-infected twice on days 75 and 340 pi with the same doses of P. yoelii 17XL and (A) survival and (B) parasitaemia were then daily monitored for 30 days pi. Data from naïve infected mice (n = 5), chloroquine 30-cured, chloroquine 30-dead mice and borrelidin-treated miceare shown. Results are expressed as mean ± SEM.
Protective humoural immune response acquired in cured mice
To explore the humoural immune response developed after borrelidin or chloroquine-30 treatments, we determined the concentrations of specific IgGs in sera obtained once the blood parasitaemia had been cleared after each infection on days 21, 85 and 350 pi. The low specific IgG levels detected in borrelidin-treated mice after the first infection was significantly increased (P = 0.004) after the second challenge, and these higher levels persisted after the third antigenic challenge (P = 0.024) (Figure 5A). Similar results were obtained in the chloroquine-30 group (Figure 5A). Parasite-specific IgG antibodies in sera from naïve mice taken before first infection (day 0) and from untreated dead mice taken on day 4 pi, were undetectable.
Figure 5.

Parasite-specific IgG responses in borrelidin and chloroquine 30-treated mice. (A) Anti-P. yoelii IgG concentration in sera obtained from the differently treated mice after total clearance of the parasite from the first (21 dpi), second (85 dpi) and third (350 dpi) infections. n = 6 samples of each experiment were measured in duplicate by ELISA. (B) Anti-P. yoelii IgG avidity values of sera (n = 3–6) after total clearance of the parasite from the first (21 pi), second (85 pi) and third (350 pi) infections. Data are shown as mean ± SEM. Avidity of n = 3 samples of each experiment were determined in duplicate using NaSCN in ELISA. (C) Time course immunoblot analysis of anti-P. yoelii IgG in sera from borrelidin-treated mice. Total protein extracts (10 μg) from P. yoelii were separated on 10% SDS-PAGE, transferred to nitrocellulose membranes and developed with sera collected during the three infections. Arrows indicate the time of infection/re-infection. MW = molecular weight markers.
Qualitative traits of humoural response during consecutive infections were studied by measuring the antibody avidities, defined as the strength with which an antibody binds to an antigen (Goldblatt et al., 1998). Avidity was determined by ELISA after treatment with several concentrations of NaSCN, a chaotropic agent that disrupts antigen–antibody interactions (Figure 5B). Thus, low avidity antibody binding is disrupted at lower NaSCN concentrations than high, avidity binding. The avidity index (AI) corresponds to the molar concentration of NaSCN at which 50% of the bound antibodies was eluted. As shown in Figure 5B, in borrelidin-treated animals the AI increased significantly from the primary infection (AI = 0.88 M) up to twofold in the second (AI = 1.83 M; P = 0.036) and 1.6-fold in the third challenges (AI = 1.44 M). From the second to third challenges, a small, but significant decrease in the AI was detected (P = 0.036). A similar trend was observed in the chloroquine-30 group (Figure 5B). Clear avidity maturation occurred during the second infection (AI = 1.69 M; twofold increase), but the avidity fell somewhat also in the third infection (AI = 1.02 M on day 350 pi). Thus, although the avidity of the specific IgG response substantially increased after the second infection, if the animal remains without parasite boosting for a long period of time, this response can decay slightly, regardless of the treatment applied.
Immunoblot analysis of total P. yoelii proteins was performed using the sera from different infection time-points. The profile of immunodetected parasite proteins in cured mice after both borrelidin and chloroquine-30 treatments revealed a progressive increase in specific IgG levels, whose signal was boosted after each infection (days 21, 85 and 350 in Figure 5C). Moreover, IgG antibodies recognized an ever-wider range of parasite antigens as the number of re-infections increased. Strongest signals were detected in the immunogenic profiles in the molecular weight ranges 22–36, 64–98 and 140–150 kDa. Antibodies against parasite proteins in the 36–64 kDa range only appeared after re-infections and were less stable without parasite exposure, as shown by their marked decrease after the second challenge on day 340 pi. In contrast, a reduced variety of antigenic proteins were recognized with the serum from the chloroquine–30-treated mouse that died after secondary infection (Figure 5C), indicating the development of an immune response enough to delay the parasite growth, but not to guarantee survival (Figure 4). Serum from naïve mice, taken before first infection, showed an non-specific reaction with some high molecular weight proteins of infected RBCs. Similar results were obtained with sera from uninfected borrelidin-treated mice and untreated deceased mice (data not shown).
Specificity of protective IgG response against intraerythrocytic parasite stages
To identify the intraerythrocytic parasite stages bound by the specific IgGs presented in the sera of protected mice, immunofluorescence microscopy analysis was performed on blood smears of infected RBCs (Figure 6). Mature stages were identified by DAPI fluorescence of the nucleus (in blue) because during parasite growth, the nucleus divides and the resulting nuclei are transferred to merozoites before their release (Matteelli et al., 1997). The images obtained showed that specific IgGs (in green) from borrelidin and chloroquine–30-cured mice from day 85 pi preferentially bind to late parasite stages and 100% of the schizonts and merozoites are recognized by these antibodies (Figure 6B,C, images 2, 3 and 2', 3') Early parasite stages exhibited a weak immunostaining (Figure 6B,C, images 1 and 1'). The binding of IgG antibodies to infected RBCs was observed only when the infected RBC membranes were permeabilized (data not shown). Preimmune sera under the same experimental conditions did not show any signal (Figure 6B).
Figure 6.

Immunofluorescence microscopy analysis of IgG specificity against blood-stage P. yoelii infection. (A) Thin-blood smears showing high parasitaemia from untreated P. yoelii infected mice. (B–D) Fixed thin-blood smears showing high parasitaemia from untreated P. yoelii infected mice were stained with DAPI (blue) to identify parasite DNA and incubated with different sera. (B) No detectable signal was obtained with preimmune serum from non-infected mice as control. (C) Borrelidin-treated mice serum (1:2500) or (D) chloroquine–30-treated mice serum (1:2500) from day 85 pi was detected by Alexa Fluor 488-labeled anti-mouse IgG (green) (1:400). Double staining of DAPI and Alexa Fluor 488-labeled anti-mouse IgG shows the co-localization of late stage intraerythrocyte parasites with antibody binding. Images are representative of n = 3 duplicate samples taken from experiments shown in Figure 4. Scale bars, panels C-D: 10 μm. Images 1 and 1' are representative of early parasite stages; Images 2 and 2' show the presence of stained schizonts; Images 3 and 3' correspond to P. yoelii stained invasive forms (merozoites). Scale bars, panels 1–3 and 1–3': 2 μm.
Curative properties of borrelidin against lethal blood-stage malaria
As the drug treatment, which follows a classic 4 day suppression test begins just 2 h after infection, the results reflect the prophylactic properties of the drug. Thus, to study curative effects, a 4 day drug treatment of mice starting later, when mice exhibited around 10% of parasitaemia (day 3 pi) was carried out. Under these conditions, borrelidin was slower at decreasing parasitaemia rates than chloroquine (Figure 7B) and 25% (2/8) of treated mice did not survive (Figure 7A), although their parasitaemia levels were less severe than in untreated mice. Parasitaemia peaks in both groups were similar to those achieved after the 4 day prophylactic treatment (Figure 1B). All surviving mice were re-infected on day 135 pi and successfully overcame the infection (data not shown).
Figure 7.
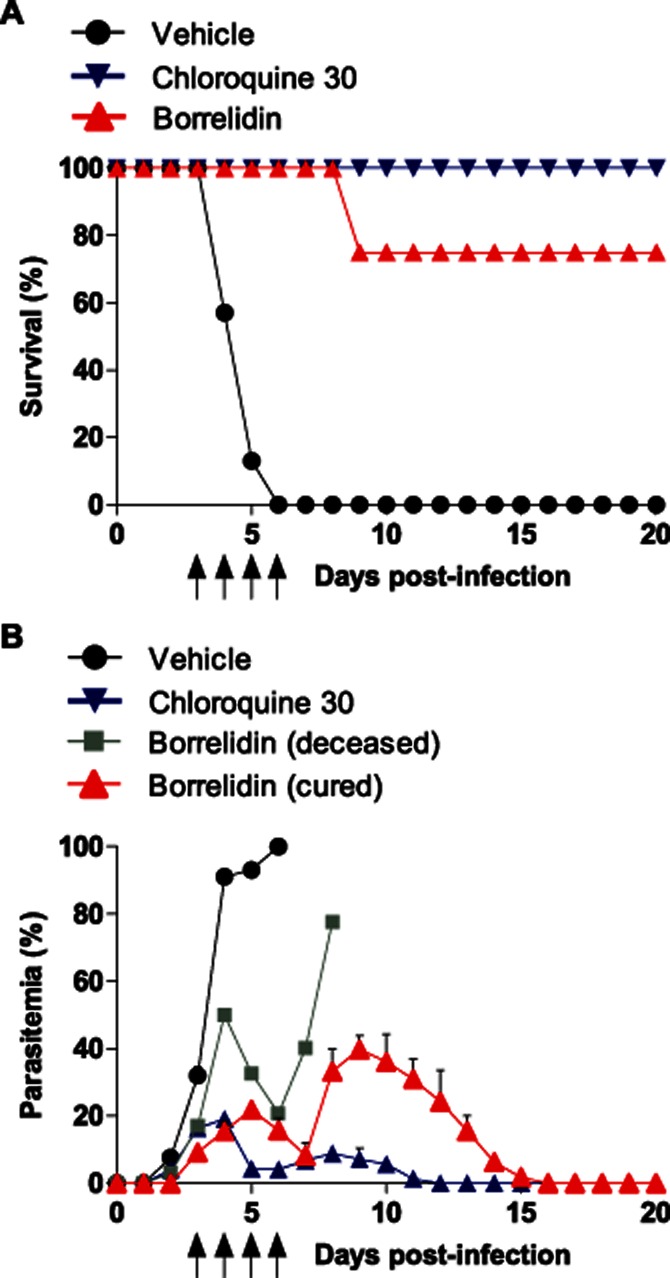
Survival and parasitaemia course in infected mice with 10% parasitaemia subjected to antimalarial treatment. BALB/c mice infected with 2 × 107 P. yoelii 17XL infected RBCs developed 10% parasitaemia (monitored by microscopic examination) before being treated for 4 days with vehicle, 30 mg·kg−1·day−1 chloroquine or 0.25 mg·kg−1·day−1 borrelidin. Data from dead borrelidin-treated mice are also shown. Mean (A) survival and (B) parasitaemia percentages are shown for each group. One representative experiment is shown (n = 4). Arrows indicate the four ip injections of drug/vehicle.
Discussion and conclusions
Our results demonstrated that prophylactic treatment with borrelidin during the exposure to live blood-stages of lethal P. yoelii 17XL was enough to halt the infection and protect mice from death. In contrast, mupirocin was unable to control primary malaria infection in vivo which could be attributed to its rapid hydrolysis in blood plasma, its binding to serum and the decrease of biological activity at pH near 8 (Thomas et al., 2010) and thus, now it is clinically restricted to topical use (Sutherland et al., 1985). Remarkably, a borrelidin concentration 120-fold lower compared with chloroquine showed the same efficacy in terms of survival after primary infection. Both borrelidin and chloroquine induced a rapid inhibition of P. falciparum growth in vitro within the first 48 h, confirming a ‘not delayed-death’ effect (Jackson et al., 2012), usually attributed to antibiotics that inhibit prokaryote translation (Barthel et al., 2008). These results are also consistent with previous data describing the stage specificity of borrelidin to the trophozoite and schizont stages (Ishiyama et al., 2011; Jackson et al., 2012) probably because the ARS maximal expression happens in mature life cycle stages (Jackson et al., 2012). Remarkably, although we used borrelidin at a fivefold higher concentration and for twice the duration used in previous studies (Ishiyama et al., 2011; Jackson et al., 2012), a fraction of P. falciparum parasites at ring and young trophozoite stages were capable of surviving and resume the multiplication in the fifth cycle. Therefore, the effect of borrelidin on ring blood-stage parasites would better fit with a static activity as they suffered growth arrest that recovered after drug withdrawal, in contrast to mature stages that would be susceptible to a cidal activity, leading to a rapid death (Bahamontes-Rosa et al., 2011). However, because aberrant parasite forms were also found in the two life cycles following antibiotic withdrawal, the normal development of a fraction of surviving early-stage parasites can also be affected. This altered development is also observed after other antibiotic treatments (Barthel et al., 2008). Results derived from in vivo assays may also support a static activity for borrelidin, which is a typical effect of ARS inhibitors (Critchley et al., 2005). The examination of intraerythrocytic parasite stages during borrelidin treatment in mice during Py17XL infection reflected an accumulation of trophozoites in peripheral blood at the end of the treatment (day 3 pi). This accumulation could be attributed not only to the in vitro effect of borrelidin on mature stages, which prevent the appearance of new rings (Ishiyama et al., 2011), but also on the development of rings and young trophozoites, which instead of completing the cycle from day 2 to 3 pi were delayed in our in vivo assays and only grew until the trophozoite stage.
Besides direct antimalarial activity, our results showed that borrelidin treatment sustained protective humoural response during a primary infection. All borrelidin-treated mice were capable of controlling parasitaaemia and subsequently they developed an efficient immune response with the production of specific antibodies that completely eliminated parasites after re-infection. Static compounds seem to allow the host immune system to participate in the battle against infection by increasing the period of antigen presentation (Scholar and Pratt, 1939), which is particularly important in developing immunity to malaria (Urban et al., 2005; Amante et al., 2011). Currently, some of the antimalarial drugs in use such as atovaquone or pyrimethamine have static activity (Bahamontes-Rosa et al., 2011) and their use, combined with other compounds, is recommended by the World Health Organization (World Health Organization, 2010). Treatment with the antibiotic borrelidin allowed a robust humoural response that totally prevented subsequent lethal infections in all animals. Chloroquine cidal activity led to a protective humoural response in 90% of mice (9/10) after first challenge similar to the borrelidin-treated mice group, but 10% of mice (1/10) died during re-infection. Thus, antimalarial treatments or doses that may provide rapid elimination of parasites in blood would eventually reduce the residence time of native parasite antigen for the efficient presentation to the immune cells. This could be the case of the one chloroquine–30-treated mouse that died in one of the re-infection experiments, which in turn diminished malaria parasite recognition by the raised antibodies, as shown in its corresponding immunoblot.
Experimental inoculations of malaria in humans and mice have demonstrated that a drug controlled exposure to blood- or liver-stage parasites can result in protection (Pombo et al., 2002; Roestenberg et al., 2009; Friesen et al., 2010; Sauerwein et al., 2010). In mice, treatments that maintain the chronicity of the infection show a high surviving rate after re-infection, whereas mice receiving radical treatments that completely abolish parasite multiplication, die in a second challenge (Long et al., 2002). Moreover, the parasite level seems to be a very important factor because very low levels of blood-stage P. falciparum in volunteers were not capable of eliciting antibodies, but induced efficient cell-mediated immunity (Pombo et al., 2002). Any efficient immune response to malaria is complex and involves several cell and humoural factors. Cell-mediated immune mechanisms are fundamental to the control of the first wave of infective Plasmodium parasites (Achtman et al., 2005). Conversely, the particular importance of antibodies in malaria immunity has been shown by serum transfer experiments in humans (Cohen et al., 1961) and mice (Jayawardena et al., 1978).
Thus, the generation of immunological memory in the borrelidin and chloroquine–30-treated mice was supported by the robust antibody response in the re-infections, the presence of switched specific antibodies during 9 months of barely detectable parasitaemia after a second and third re-infection (Kinyanjui et al., 2004; Achtman et al., 2007; Weiss et al., 2009) and by the increase in avidity of specific IgGs after re-infections (Berek, 1993). The decrease of antigen-IgG binding strength observed after the third infection was more evident in chloroquine than borrelidin-treated mice and could reflect a progressive loss of immune response after the absence of parasite contact during 9 months, as described in humans (Linares et al., 2011). These data are also in agreement with non-lethal Plasmodium re-infections in mice that do not increase AI, in the long term (Bull et al., 2002). In addition, in both treated mouse groups, the repertoire of Py17XL antigens recognized by the specific IgGs raised was also amplified after each re-infection, as observed for acquired malaria immunity in humans, which is likely to depend on the accumulation of a wide repertoire of antigenic specificities over a long time (Kinyanjui et al., 2004) and which parallels with a gradual gaining of clinical immunity (see Bull et al., 2002). After both treatments, mice showed IgG binding to merozoite antigens and internal antigens of Py17XL infected RBCs, as identified recently in a similar experimental set (Kamali et al., 2012). Because internal antigens are only exposed in disrupted target cells, they are detected as secreted antigens, which seem to induce antibody responses more efficiently than membrane and cytoplasmic antigens due to an enhanced ability to reach the lymph node (Boyle et al., 1997). However, antibodies to intracellular proteins are usually considered markers of past infection and could only indicate an increased parasite killing. There are some exceptions of human and rodent antibodies with reactivity to intracellular antigens of Plasmodium or other parasites that can induce humoural protection (Vedi et al., 2008; Crompton et al., 2010). Thus, given that the immunogenic antigens encoded by the parasite are largely unknown (Langhorne et al., 2008), we cannot exclude the possibility that some intracellular Plasmodium antigens released from the infected RBCs might cooperate in immune protection.
Interestingly, IgGs binding to antigens in the medium MW range (36–64 KDa) vanished after infection in all mice. (Mota et al., 2001; Pombo et al., 2002; Elliott et al., 2005) Similarly, human antibody patterns in seasonal malaria transmission show strong preference towards high MW antigens (Thelu et al., 1991) and IgGs from mice suffering non-lethal malaria infections recognize more trophozoite- or schizont- than ring-infected erythrocytes (Mota et al., 1998).
Finally, although prophylactic administration of borrelidin turned out to be more effective in protecting mice from lethal malaria than a therapeutic dosage applied when the acute infection was already established, we expect that improvement of borrelidin combination therapies and chemical modifications of the borrelidin polyketide molecule, would eventually generate better therapeutic strategies and more selective analogues for inhibiting ThrRS from Plasmodium species, respectively.
In conclusion, our results provide new insights into the potential use of borrelidin as an antimalarial drug and contribute to the validation of ThrRS as a target for prophylaxis or therapy against malaria. We showed that treatment with a low borrelidin dose had parasite–stage-specific actions and induced a robust long-term protective response in 100% of treated animals.
Acknowledgments
We thank Dr Jesús Sánchez-Nogueiro for accession and technical support in immunofluorescence imaging, and Ana Burton for reading and commenting on the manuscript. This work was supported by the Spanish Ministry of Innovation and Science (grant BIO2010-17039) and by the Programme of Consolidate Research Teams from UCM-Comunidad de Madrid (Research Team 920267). I.G.A. holds a fellowship awarded by the Spanish Ministry of Innovation and Science under grant BIO2007-67885. The work of N.C and L.R.d.P. was supported by grants BIO2009-09776 from the Spanish Ministry of Innovation and Science, and the EU-FP7 collaborative project Mephitis (HEALTH-F3-2009-223024).
Glossary
- ARS
aminoacyl-tRNA synthetases
- IleRS
isoleucyl t-RNA synthetase
- NAI
naturally acquired immunity
- pi
post-infection
- Py17XL
P. yoelii 17XL
- ThrRS
threonyl t-RNA synthetase
Conflict of interest
None.
References
- Achtman AH, Bull PC, Stephens R, Langhorne J. Longevity of the immune response and memory to blood-stage malaria infection. In: Langhorne J, editor. Immunology and Immunopathogenesis of Malaria. 1st edn. Berlin-Heidelberg: Springer; 2005. pp. 71–102. [DOI] [PubMed] [Google Scholar]
- Achtman AH, Stephens R, Cadman ET, Harrison V, Langhorne J. Malaria-specific antibody responses and parasite persistence after infection of mice with Plasmodium chabaudi chabaudi. Parasite Immunol. 2007;29:435–444. doi: 10.1111/j.1365-3024.2007.00960.x. [DOI] [PubMed] [Google Scholar]
- Amante FH, Engwerda CR, Good MF. Experimental asexual blood stage malaria immunity. Curr Protoc Immunol. 2011;93:19.14.01–19.14.26. doi: 10.1002/0471142735.im1904s93. [DOI] [PubMed] [Google Scholar]
- Bahamontes-Rosa N, Rodriguez-Alejandre A, Gonzalez-del-Rio R, Garcia-Bustos JF, Mendoza-Losana A. A new molecular approach for cidal vs static antimalarial determination by quantifying mRNA levels. Mol Biochem Parasitol. 2011;181:171–177. doi: 10.1016/j.molbiopara.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Barthel D, Schlitzer M, Pradel G. Telithromycin and quinupristin-dalfopristin induce delayed death in Plasmodium falciparum. Antimicrob Agents Chemother. 2008;52:774–777. doi: 10.1128/AAC.00892-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoin RL, Strome CP, Mitchell F, Tubergen TA. Plasmodium berghei: immunization of mice against the ANKA strain using the unaltered sporozoite as an antigen. Exp Parasitol. 1977;42:1–5. doi: 10.1016/0014-4894(77)90054-6. [DOI] [PubMed] [Google Scholar]
- Berek C. Somatic mutation and memory. Curr Opin Immunol. 1993;5:218–222. doi: 10.1016/0952-7915(93)90007-f. [DOI] [PubMed] [Google Scholar]
- Berger J, Jampolsky LM, Goldberg MW. Borrelidin, a new antibiotic with antiborrelia activity and penicillin enhancement properties. Arch Biochem. 1949;22:476–478. [PubMed] [Google Scholar]
- Bloland PB. Drug Resistance in Malaria. Geneva: World Health Organization; 2001. [Google Scholar]
- Borrmann S, Matuschewski K. Protective immunity against malaria by ‘natural immunization’: a question of dose, parasite diversity, or both? Curr Opin Immunol. 2011;23:500–508. doi: 10.1016/j.coi.2011.05.009. [DOI] [PubMed] [Google Scholar]
- Boyle JS, Koniaras C, Lew AM. Influence of cellular location of expressed antigen on the efficacy of DNA vaccination: cytotoxic T lymphocyte and antibody responses are suboptimal when antigen is cytoplasmic after intramuscular DNA immunization. Int Immunol. 1997;9:1897–1906. doi: 10.1093/intimm/9.12.1897. [DOI] [PubMed] [Google Scholar]
- Bull PC, Lowe BS, Kaleli N, Njuga F, Kortok M, Ross A, et al. Plasmodium falciparum infections are associated with agglutinating antibodies to parasite-infected erythrocyte surface antigens among healthy Kenyan children. J Infect Dis. 2002;185:1688–1691. doi: 10.1086/340420. [DOI] [PubMed] [Google Scholar]
- Cohen S, McGregor IA, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature. 1961;192:733–737. doi: 10.1038/192733a0. [DOI] [PubMed] [Google Scholar]
- Collins WE, Jeffery GM. A retrospective examination of secondary sporozoite- and trophozoite-induced infections with Plasmodium falciparum: development of parasitologic and clinical immunity following secondary infection. Am J Trop Med Hyg. 1999;61:20–35. doi: 10.4269/tropmed.1999.61-020. [DOI] [PubMed] [Google Scholar]
- Critchley IA, Young CL, Stone KC, Ochsner UA, Guiles J, Tarasow T, et al. Antibacterial activity of REP8839, a new antibiotic for topical use. Antimicrob Agents Chemother. 2005;49:4247–4252. doi: 10.1128/AAC.49.10.4247-4252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton PD, Kayala MA, Traore B, Kayentao K, Ongoiba A, Weiss GE, et al. A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc Natl Acad Sci U S A. 2010;107:6958–6963. doi: 10.1073/pnas.1001323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson L, Griffiths AJ, Mason CG, Mills RF. Anti-viral activity of two antibiotics isolated from a species of Streptomyces. Nature. 1965;206:265–268. doi: 10.1038/206265a0. [DOI] [PubMed] [Google Scholar]
- Doolan DL, Dobano C, Baird JK. Acquired immunity to malaria. Clin Microbiol Rev. 2009;22:13–36. doi: 10.1128/CMR.00025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorgerloh M, Kretschmer A, Schmidt RR, Steffens R, Zoebelein G, Tietjen K, et al. Borrelidin insecticide and herbicide, and its preparation by fermentation. German Patent. 1988;11 (1988) DE 3607287. [Google Scholar]
- Druilhe P, Perignon JL. A hypothesis about the chronicity of malaria infection. Parasitol Today. 1997;13:353–357. doi: 10.1016/s0169-4758(97)01095-8. [DOI] [PubMed] [Google Scholar]
- Elliott SR, Kuns RD, Good MF. Heterologous immunity in the absence of variant-specific antibodies after exposure to subpatent infection with blood-stage malaria. Infect Immun. 2005;73:2478–2485. doi: 10.1128/IAI.73.4.2478-2485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen J, Silvie O, Putrianti ED, Hafalla JC, Matuschewski K, Borrmann S. Natural immunization against malaria: causal prophylaxis with antibiotics. Sci Transl Med. 2010;2:40ra49. doi: 10.1126/scitranslmed.3001058. [DOI] [PubMed] [Google Scholar]
- Funahashi Y, Wakabayashi T, Semba T, Sonoda J, Kitoh K, Yoshimatsu K. Establishment of a quantitative mouse dorsal air sac model and its application to evaluate a new angiogenesis inhibitor. Oncol Res. 1999;11:319–329. [PubMed] [Google Scholar]
- Goldblatt D, Vaz AR, Miller E. Antibody avidity as a surrogate marker of successful priming by Haemophilus influenzae type b conjugate vaccines following infant immunization. J Infect Dis. 1998;177:1112–1115. doi: 10.1086/517407. [DOI] [PubMed] [Google Scholar]
- Goodman CD, Su V, McFadden GI. The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol Biochem Parasitol. 2007;152:181–191. doi: 10.1016/j.molbiopara.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Habibi D, Ogloff N, Jalili RB, Yost A, Weng AP, Ghahary A, et al. Borrelidin, a small molecule nitrile-containing macrolide inhibitor of threonyl-tRNA synthetase, is a potent inducer of apoptosis in acute lymphoblastic leukemia. Invest New Drugs. 2012;30:1361–1370. doi: 10.1007/s10637-011-9700-y. [DOI] [PubMed] [Google Scholar]
- Hoepfner D, McNamara CW, Lim CS, Studer C, Riedl R, Aust T, et al. Selective and specific inhibition of the Plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe. 2012;11:654–663. doi: 10.1016/j.chom.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman SL, Goh LM, Luke TC, Schneider I, Le TP, Doolan DL, et al. Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites. J Infect Dis. 2002;185:1155–1164. doi: 10.1086/339409. [DOI] [PubMed] [Google Scholar]
- Hughes J, Mellows G. Inhibition of isoleucyl-transfer ribonucleic acid synthetase in Escherichia coli by pseudomonic acid. Biochem J. 1978;176:305–318. doi: 10.1042/bj1760305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurdle JG, O'Neill AJ, Chopra I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob Agents Chemother. 2005;49:4821–4833. doi: 10.1128/AAC.49.12.4821-4833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter R, Poralla K, Zachau HG, Zahner H. Metabolic products of microorganisms. 5l. On the mechanism of action of borrelidin-inhibition of the threonine incorporation in sRNA. Biochem Z. 1966;344:190–196. [PubMed] [Google Scholar]
- Ishiyama A, Iwatsuki M, Namatame M, Nishihara-Tsukashima A, Sunazuka T, Takahashi Y, et al. Borrelidin, a potent antimalarial: stage-specific inhibition profile of synchronized cultures of Plasmodium falciparum. J Antibiot. 2011;64:381–384. doi: 10.1038/ja.2011.6. [DOI] [PubMed] [Google Scholar]
- Istvan ES, Dharia NV, Bopp SE, Gluzman I, Winzeler EA, Goldberg DE. Validation of isoleucine utilization targets in Plasmodium falciparum. Proc Natl Acad Sci U S A. 2011;108:1627–1632. doi: 10.1073/pnas.1011560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson KE, Pham JS, Kwek M, De Silva NS, Allen SM, Goodman CD, et al. Dual targeting of aminoacyl-tRNA synthetases to the apicoplast and cytosol in Plasmodium falciparum. Int J Parasitol. 2012;42:177–186. doi: 10.1016/j.ijpara.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Jayawardena AN, Targett GA, Leuchars E, Davies AJ. The immunological response of CBA mice to P. yoelii. II. The passive transfer of immunity with serum and cells. Immunology. 1978;34:157–165. [PMC free article] [PubMed] [Google Scholar]
- Kamali AN, Marin-Garcia P, Azcarate IG, Diez A, Puyet A, Bautista JM. Plasmodium yoelii blood-stage antigens newly identified by immunoaffinity using purified IgG antibodies from malaria-resistant mice. Immunobiology. 2012;217:823–830. doi: 10.1016/j.imbio.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Kawamura T, Liu D, Towle MJ, Kageyama R, Tsukahara N, Wakabayashi T, et al. Anti-angiogenesis effects of borrelidin are mediated through distinct pathways: threonyl-tRNA synthetase and caspases are independently involved in suppression of proliferation and induction of apoptosis in endothelial cells. J Antibiot. 2003;56:709–715. doi: 10.7164/antibiotics.56.709. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinyanjui SM, Mwangi T, Bull PC, Newbold CI, Marsh K. Protection against clinical malaria by heterologous immunoglobulin G antibodies against malaria-infected erythrocyte variant surface antigens requires interaction with asymptomatic infections. J Infect Dis. 2004;190:1527–1533. doi: 10.1086/424675. [DOI] [PubMed] [Google Scholar]
- Langhorne J, Ndungu FM, Sponaas AM, Marsh K. Immunity to malaria: more questions than answers. Nat Immunol. 2008;9:725–732. doi: 10.1038/ni.f.205. [DOI] [PubMed] [Google Scholar]
- Linares M, Albizua E, Mendez D, Rubio JM, Martinez-Serna A, Martinez MA, et al. Malaria hidden in a patient with diffuse large B-cell lymphoma and sickle cell trait. J Clin Microbiol. 2011;49:4401–4404. doi: 10.1128/JCM.00911-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long TT, Nakazawa S, Huaman MC, Kanbara H. Influence of antimalarial treatment on acquisition of immunity in Plasmodium berghei NK65 malaria. Clin Diagn Lab Immun. 2002;9:933–934. doi: 10.1128/CDLI.9.4.933-934.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteelli A, Castelli F, Caligaris S. Life cycle of malaria parasites. In: Carosi G, Castelli F, editors. Handbook of Malaria Infection in the Tropics. 1st edn. Bologna: Associazione Italiana “Amici di R. Follereau”; 1997. pp. 17–23. [Google Scholar]
- Moneriz C, Marin-Garcia P, Bautista JM, Diez A, Puyet A. Haemoglobin interference and increased sensitivity of fluorimetric assays for quantification of low-parasitaemia Plasmodium infected erythrocytes. Malar J. 2009;8:279. doi: 10.1186/1475-2875-8-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mota MM, Brown KN, Holder AA, Jarra W. Acute Plasmodium chabaudi chabaudi malaria infection induces antibodies which bind to the surfaces of parasitized erythrocytes and promote their phagocytosis by macrophages in vitro. Infect Immun. 1998;66:4080–4086. doi: 10.1128/iai.66.9.4080-4086.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mota MM, Brown KN, Do Rosario VE, Holder AA, Jarra W. Antibody recognition of rodent malaria parasite antigens exposed at the infected erythrocyte surface: specificity of immunity generated in hyperimmune mice. Infect Immun. 2001;69:2535–2541. doi: 10.1128/IAI.69.4.2535-2541.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocana-Morgner C, Mota MM, Rodriguez A. Malaria blood stage suppression of liver stage immunity by dendritic cells. J Exp Med. 2003;197:143–151. doi: 10.1084/jem.20021072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otoguro K, Ui H, Ishiyama A, Kobayashi M, Togashi H, Takahashi Y, et al. In vitro and in vivo antimalarial activities of a non-glycosidic 18-membered macrolide antibiotic, borrelidin, against drug-resistant strains of Plasmodia. J Antibiot (Tokyo) 2003;56:727–729. doi: 10.7164/antibiotics.56.727. [DOI] [PubMed] [Google Scholar]
- Peters W, Robinson B. Malaria. In: Zak O, Sande M, editors. Handbook of Animal Models of Infection. 1st edn. San Diego, CA: Academic Press; 1999. pp. 757–773. [Google Scholar]
- Pombo DJ, Lawrence G, Hirunpetcharat C, Rzepczyk C, Bryden M, Cloonan N, et al. Immunity to malaria after administration of ultra-low doses of red cells infected with Plasmodium falciparum. Lancet. 2002;360:610–617. doi: 10.1016/S0140-6736(02)09784-2. [DOI] [PubMed] [Google Scholar]
- Proudfoot O, Drew N, Scholzen A, Xiang S, Plebanski M. Investigation of a novel approach to scoring Giemsa-stained malaria-infected thin blood films. Malar J. 2008;7:62. doi: 10.1186/1475-2875-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen GR, Fitzgerald MG, Hosking CS. Antibody avidity determination by ELISA using thiocyanate elution. J Immunol Methods. 1986;86:83–87. doi: 10.1016/0022-1759(86)90268-1. [DOI] [PubMed] [Google Scholar]
- Radfar A, Mendez D, Moneriz C, Linares M, Marin-Garcia P, Puyet A, et al. Synchronous culture of Plasmodium falciparum at high parasitemia levels. Nat Protoc. 2009;4:1899–1915. doi: 10.1038/nprot.2009.198. [DOI] [PubMed] [Google Scholar]
- Roestenberg M, McCall M, Hopman J, Wiersma J, Luty AJ, van Gemert GJ, et al. Protection against a malaria challenge by sporozoite inoculation. N Engl J Med. 2009;361:468–477. doi: 10.1056/NEJMoa0805832. [DOI] [PubMed] [Google Scholar]
- Roestenberg M, Teirlinck AC, McCall MB, Teelen K, Makamdop KN, Wiersma J, et al. Long-term protection against malaria after experimental sporozoite inoculation: an open-label follow-up study. Lancet. 2011;377:1770–1776. doi: 10.1016/S0140-6736(11)60360-7. [DOI] [PubMed] [Google Scholar]
- Sauerwein RW, Bijker EM, Richie TL. Empowering malaria vaccination by drug administration. Curr Opin Immunol. 2010;22:367–373. doi: 10.1016/j.coi.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Schimmel P, Tao J, Hill J. Aminoacyl tRNA synthetases as targets for new anti-infectives. FASEB J. 1998;12:1599–1609. [PubMed] [Google Scholar]
- Scholar EC, Pratt WB. The Antimicrobial Drugs. 2nd edn. New York: Oxford University Press; 1939. [Google Scholar]
- Stevenson MM, Riley EM. Innate immunity to malaria. Nat Rev. 2004;4:169–180. doi: 10.1038/nri1311. [DOI] [PubMed] [Google Scholar]
- Sutherland R, Boon RJ, Griffin KE, Masters PJ, Slocombe B, White AR. Antibacterial activity of mupirocin (pseudomonic acid), a new antibiotic for topical use. Antimicrob Agents Chemother. 1985;27:495–498. doi: 10.1128/aac.27.4.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelu J, Sheick-Zakiuddin I, Boudin C, Peyron F, Picot S, Ambroise-Thomas P. Development of natural immunity in Plasmodium falciparum malaria: study of antibody response by Western immunoblotting. J Clin Microbiol. 1991;29:510–518. doi: 10.1128/jcm.29.3.510-518.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CM, Hothersall J, Willis CL, Simpson TJ. Resistance to and synthesis of the antibiotic mupirocin. Nat Rev Microbiol. 2010;8:281–289. doi: 10.1038/nrmicro2278. [DOI] [PubMed] [Google Scholar]
- Tsuchiya E, Yukawa M, Miyakawa T, Kimura KI, Takahashi H. Borrelidin inhibits a cyclin-dependent kinase (CDK), Cdc28/Cln2, of Saccharomyces cerevisiae. J Antibiot. 2001;54:84–90. doi: 10.7164/antibiotics.54.84. [DOI] [PubMed] [Google Scholar]
- Urban B, Ing R, Stevenson M. Early interactions between blood-stage plasmodium parasites and the immune system. Curr Top Microbiol Immunol. 2005;297:25–70. doi: 10.1007/3-540-29967-x_2. [DOI] [PubMed] [Google Scholar]
- Vedi S, Dangi A, Hajela K, Misra-Bhattacharya S. Vaccination with 73 kDa recombinant heavy chain myosin generates high level of protection against Brugia malayi challenge in jird and mastomys models. Vaccine. 2008;26:5997–6005. doi: 10.1016/j.vaccine.2008.08.073. [DOI] [PubMed] [Google Scholar]
- Wakabayashi T, Kageyama R, Naruse N, Tsukahara N, Funahashi Y, Kitoh K, et al. Borrelidin is an angiogenesis inhibitor; disruption of angiogenic capillary vessels in a rat aorta matrix culture model. J Antibiot (Tokyo) 1997;50:671–676. doi: 10.7164/antibiotics.50.671. [DOI] [PubMed] [Google Scholar]
- Weiss GE, Crompton PD, Li S, Walsh LA, Moir S, Traore B, et al. Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J Immunol. 2009;183:2176–2182. doi: 10.4049/jimmunol.0901297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NS, Behrens GM, Lundie RJ, Smith CM, Waithman J, Young L, et al. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol. 2006;7:165–172. doi: 10.1038/ni1300. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Guidelines for the Treatment of Malaria. Geneva: World Health Organization; 2010. [Google Scholar]