

Abstract
Background and Purpose
Nitrite (NO2−) has recently been shown to represent a potential source of NO, in particular under hypoxic conditions. The aim of the current study was to compare the haemodynamic effects of NO2− in healthy volunteers and patients with stable congestive heart failure (CHF).
Experimental Approach
The acute haemodynamic effects of brachial artery infusion of NO2− (0.31 to 7.8 μmol·min−1) was assessed in normal subjects (n = 20) and CHF patients (n = 21).
Key Results
NO2− infusion was well tolerated in all subjects. Forearm blood flow (FBF) increased markedly in CHF patients at NO2− infusion rates which induced no changes in normal subjects (anova: F = 5.5; P = 0.02). Unstressed venous volume (UVV) increased even with the lowest NO2− infusion rate in all subjects (indicating venodilation), with CHF patients being relatively hyporesponsive compared with normal subjects (anova: F = 6.2; P = 0.01). There were no differences in venous blood pH or oxygen concentration between groups or during NO2− infusion. Venous plasma NO2− concentrations were lower in CHF patients at baseline, and rose substantially less with NO2− infusion, without incremental oxidative generation of nitrate, consistent with accelerated clearance in these patients. Plasma protein-bound NO concentrations were lower in CHF patients than normal subjects at baseline. This difference was attenuated during NO2− infusion. Prolonged NO2− exposure in vivo did not induce oxidative stress, nor did it induce tolerance in vitro.
Conclusions and Implications
The findings of arterial hyper-responsiveness to infused NO2− in CHF patients, with evidence of accelerated transvascular NO2− clearance (presumably with concomitant NO release) suggests that NO2− effects may be accentuated in such patients. These findings provide a stimulus for the clinical exploration of NO2− as a therapeutic modality in CHF.
Keywords: sodium nitrite, vascular effects, heart failure
Introduction
Nitrite (NO2−), present in plasma at submicromolar concentrations, has been regarded in the past as a relatively inert product of NO catabolism and dietary nitrate/ NO2− ingestion. NO2− is also generated as a component of organic nitrate metabolism. It is now clear that NO2− can indeed exert vasodilator effects, possibly via reduction to NO (Cosby et al., 2003; Crawford et al., 2006).
Although the mechanism(s) of bioactivation remain incompletely understood, a number of reductases have been shown to ‘reactivate’ NO from NO2−, independent of endothelial NOS (eNOS) activity (Baker et al., 2007; Webb et al., 2008). This process appears to be markedly potentiated in hypoxia, although once again, it is not clear what mechanism(s) underlie this (Maher et al., 2008). The implication is that exogenous administration of NO2− represents a means of selective release of NO (and hence vasodilatation) to hypoxic (and presumably ischaemic) tissues, with minimal risk of inducing the ‘steal’ phenomenon and no dependence on intact endothelial function. Thus, NO2− represents a potential treatment for conditions such as limb and myocardial ischaemia, and congestive heart failure (CHF).
A desirable characteristic of a vasodilator agent for potential use in the management of acutely decompensated heart failure is that it exerts marked venodilator, rather than arteriolar dilating effects. Salutary consequences of selective venodilatation include relief of congestive symptoms without significant risk of precipitating symptomatic hypotension. Specifically, venodilator agents ameliorate the phenomenon of diastolic ventricular interaction, thus increasing cardiac output and thereby organ perfusion (Dupuis et al., 1990; Atherton et al., 1997; Williams and Frenneaux, 2006).
In this regard, organic nitrates such as glyceryl trinitrate (GTN) are frequently utilized in the management of acute heart failure with pulmonary congestion. Although organic nitrate-based therapy exerts prominent venodilator effects (Muir and Nolan, 1991; Manyari et al., 1993) and appears to have a number of advantages over a diuretic-based treatment regimen in such patients, there is no evidence that NO release from GTN is hypoxia selective. Furthermore, therapy with GTN and other organic nitrates not only suffers from the problem of attenuation of effect during prolonged therapy (nitrate tolerance and pseudo-tolerance) (Daiber et al., 2005; Munzel et al., 2005), but also exhibits the phenomenon of NO resistance (de novo hyporesponsiveness to all sources of NO) in the presence of CHF, despite infusion at very high rates (Chirkov et al., 1999; 2001; Anderson et al., 2004). Moreover, the arteriolar vasodilation induced by organic nitrates can lead to deleterious reductions in systemic blood pressure, often accompanied by throbbing headache.
We have previously described the local forearm vascular responses to infused NO2− in normal subjects, documenting a marked venodilation and modest arteriolar vasodilation under normoxic conditions. However, during hypoxia, there was selective potentiation of arterial vasodilation (Maher et al., 2008).
In the current study, we have compared vasomotor responsiveness to NO2− in stable CHF patients and in normal subjects, relating response to concomitant plasma NO2− concentrations. We theorized that CHF patients might largely circumvent the problem of NO resistance at the arterial level by virtue of the potentiating effect of tissue hypoxia on NO2− bioactivation. As such, the primary null hypothesis was that the vasomotor effects of NO2− would not vary between patients with CHF and normal subjects. The secondary hypothesis was that the pharmacokinetics of NO2− would not differ significantly between the two groups. Additional experiments were performed to determine whether prolonged NO2− exposure might result in incremental oxidative stress and/or induce the development of tolerance.
Methods
Subject selection
The study involved a comparison between patients with stable New York Heart Association Class II–III CHF (n = 21) and healthy volunteers (n = 20). CHF patients were recruited from an ‘advanced heart failure and cardiomyopathy’ outpatient clinic. Among CHF patients, contraindications to study entry were long-acting nitrate therapy, symptomatic hypotension and clinically significant hepatic or renal dysfunction. None of the normal subjects had any known coronary risk factors, and none was taking cardioactive medications or vitamin supplements. The study was approved by the Local Research Ethics Committee and all patients gave written informed consent. The study conformed to the principles of the Declaration of Helsinki. Subjects had consumed a light breakfast and abstained from caffeine drink intake for at least 6 h. Pre-study dietary nitrate/NO2− intake was not modified.
Experimental protocol
Instrumentation
Subjects rested supine in a dedicated vascular laboratory and brachial artery cannulation was performed as previously described (Maher et al., 2008).
Haemodynamic assessment
The principal haemodynamic investigations performed were serial determination of unstressed forearm venous volume (UVV) as an index of venodilator response, and forearm blood flow (FBF) as an index of arteriolar response to infused NO2−. Forearm venous volume (FVV) was assessed utilizing radionuclide plethysmography as previously described (Schmitt et al., 2002) and FBF was measured utilizing strain-gauge-plethysmography, as previously described (Gunaruwan et al., 2002). Results were expressed relative to baseline values and those in the infused arm, corrected for changes in the non-infused arm.
NO2− infusion
Figure 1 is a schematic of the overall experimental design. After determination of baseline data, NO2− was infused into the non-dominant brachial artery. Infusion rates were 0.31 μmol·min−1 for 30 min, thereafter increasing to 0.78 μmol·min−1, 3.1 μmol·min−1 and 7.8 μmol·min−1, each for further 30-min infusion periods. Changes in haemodynamic parameters were measured 5, 12 and 20 min after initiation of each infusion rate.
Figure 1.
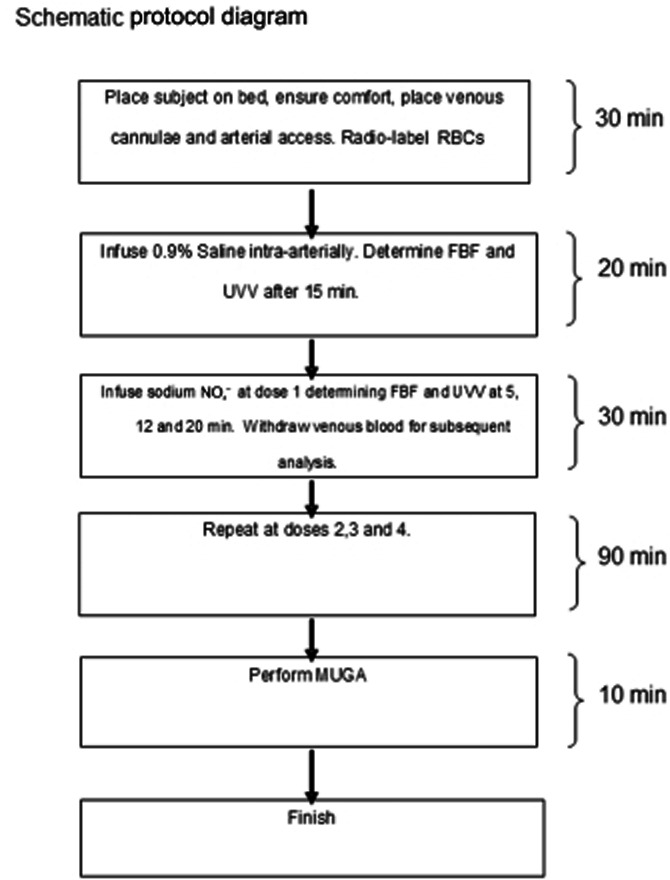
Schematic diagram of study protocol.
Blood sampling/analysis
Blood was withdrawn via venous cannulae in both arms at baseline and after the conclusion of each infusion. Blood gas analysis was performed for pH, oxygen and methaemoglobin concentrations (Bayer Rapidlab 865, Siemens, Erlangen, Germany). Blood for determination of venous plasma NO2−, nitrate and protein-bound NO (RXNO) concentrations was taken into EDTA tubes and immediately centrifuged (200 rpm for 10 min at 4°C). Samples were stored at −80°C prior to assay. Plasma NO2−, nitrate and RXNO content were determined via ozone-based chemiluminescence (Pinder et al., 2009) or HPLC (Rassaf et al., 2002) as previously described.
Assessment of NO2− clearance in human plasma in vitro
To ensure that NO2− clearance in vitro did not vary between normal subjects and patients, experiments were performed in which fresh venous blood (EDTA) from normal and CHF subjects was spiked with sodium NO2− in vitro to final concentrations of 2 and 20 μM; after spiking samples were incubated under gentle agitation at 37°C with aliquots being removed after 1, 2, 5, 10, 20 and 60 min prior to addition of N-ethylmaleimide (10 mM), centrifugation and assay.
Reagents
Sodium NO2− was purchased from Martindale Pharmaceuticals, UK. HPLC-grade NO2−-free water (Fisher Scientific, Loughborough, UK) was utilized for extractions and dilutions.
In vitro tolerance induction experiments
In vitro studies were performed to address the possibility that prolonged infusion of NO2− might induce tolerance to itself and/or cross-tolerance to GTN. Segments of saphenous veins discarded after bypass grafting were collected from patients undergoing non-emergent coronary artery bypass grafting who had not received long-acting nitrates for at least 24 h, placed in ice-cold Krebs solution, cleaned and cut into 2–3 mm segments. For vascular reactivity studies, venous segments were suspended under tension in 15-mL organ baths containing Krebs solution at 37°C. Resting tension was set at 1 g, as previously described (Sage et al., 2000) and segments were equilibrated for 60 min before being constricted with 120 mmol·L−1 KCl; vessel segments in which constriction was less than 1 g were discarded. After a further 30 min of washout, the segments were pre-constricted with phenylephrine to produce 70% of maximal tension. Once contractile response had reached a plateau, each segment was exposed to increasing concentrations of NaNO2 (4 × 10−9 to 1.2 × 10−2 M) and GTN (4 × 10−9 to 1.2 × 10−5 M) in order to obtain cumulative vasodilator concentration-response curves. The order of administration of NaNO2 and GTN was randomized. A washout period of 30 min was allowed between vasodilator response curves.
Tolerance induction experiments were performed via incubation of venous segments in 10−2 M NaNO2 under resting tension for 45 min. After a further washout period of 30 min, NaNO2 and GTN concentration-response curves were repeated, again in random order. In each experiment, control vessels were utilized in order to exclude spontaneous changes in responsiveness to either vasodilator.
Vascular relaxation responses to NaNO2 and GTN were compared before and after prolonged NaNO2 exposure, via curve fitting of individual concentration-response data to obtain EC50 values for each curve. In the case of NaNO2 administration, which yielded bi-sigmoidal concentration-response curves, EC50 values were calculated from the low-affinity, high-capacity component of the response. As EC50 data were normally distributed, these were compared via paired t-tests.
Assessment of NO2−infusion upon levels of oxidative stress
Heart failure patients (n = 15) were exposed to saline infusion (20 min), followed by two incremental doses of NO2− (7.84 nM and 7.84 μM; 20 min i.v. infusion for each dose) under normoxic conditions. The patients were then exposed to 12% hypoxia for 20 min and infused with 7.84 μM NO2−. At the end of each infusion, blood samples were taken for plasma 8-isoprostane analysis.
Assessment of oxidative stress
Total plasma 8-iso prostaglandin F2α (8-isoprostane) was measured using a commercial 8-isoprostane EIA assay (Cayman Chemical Company, Ann Arbor, MI, USA). Briefly, plasma samples were collected in vacutainers containing EDTA that was supplemented with 0.005% BHT to prevent spontaneous oxidative formation of 8-isoprostane. Total 8-isoprostane was determined by first hydrolysing the samples, followed by affinity sorbent/column purification step. Total 8-isoprostane content was then measured according to the assay kit protocol. The assay of both free and bound isoprostanes was used as a substantial proportion of 8-isoprostanes, which are esterified in lipids, would not be detected by measurement of free isoprostane alone.
Analysis of results
The current studies had >80% power to detect 20% differences in both FBF and UVV responses between groups at P < 0.05 level.
Clinical characteristics of normal subjects and CHF patients were compared utilizing non-paired t-tests (two-sided) for normally distributed parameters, and Wilcoxon tests for skewed data. Categorical data were compared using a Fisher's exact test. Serial changes in FBF and UVV, pH and venous O2 saturation, and concentrations of NO2− and RX NO in both the infused and non-infused forearms were compared in normal and CHF subjects by two-way anova with repeated measures, utilizing Dunnett's t-test to assess for significance of changes at individual time points. Logit transformation of data was utilized to detect possible disparity of concentration-response relationships between groups of subjects. All results are expressed as mean ± SEM unless otherwise stated. P-value of <0.05 was taken as statistically significant.
Results
Subject/patient characteristics
The characteristics of the patients and healthy controls are detailed in Table 1 and the drug therapy in patients in Table 2. It should be noted that although no patient had calculated creatinine clearance values less than 30 mL·min−1, mean plasma creatinine levels were marginally abnormal in the CHF group, typical of populations with heart failure.
Table 1.
Demographic data for both groups of subjects
| Demographic and clinical features | CHF patients (mean ± SEM) | Healthy controls (mean ± SEM) | P-value |
|---|---|---|---|
| Age (year) | 62.9 ± 2.7 | 57.6 ± 1.2 | 0.08 |
| Sex, M/F (%) | 18/3 (86/14) | 15/5 (75/25) | 0.70 |
| Body mass index (kg·m−2) | 30.0 ± 1.2 | 26.8 ± 0.6 | 0.05 |
| Serum cholesterol (mmol·L−1) | 5.3 ± 0.4 | 5.4 ± 0.2 | 0.69 |
| Plasma glucose (mmol·L−1) | 5.8 ± 0.4 | 4.7 ± 0.1 | 0.05 |
| Serum creatinine (μmol·L−1) | 125 ± 6.7 | 103.4 ± 5.2 | 0.03 |
| Heart rate | 74 ± 2 | 61.5 ± 1.9 | 0.01 |
| Blood pressure (mmHg) | |||
| Systolic | 117 ± 3.7 | 128 ± 2.0 | 0.03 |
| Diastolic | 74.8 ± 1.9 | 73.4 ± 1.4 | 0.73 |
| Left ventricular ejection fraction | 34.8 ± 2.5 | 56.6 ± 1.7 | 0.001 |
| Aetiology (ischaemic/DCM) | 10/11 | N/A |
Table 2.
Summary of the medication taken by the CHF patients
| Medication | % of patients receiving |
|---|---|
| Beta blockers | 76% |
| ACE inhibitors/ARB | 95% |
| Aldosterone antagonists | 57% |
| Loop diuretics | 100% |
| Perhexiline | 29% |
| Warfarin | 57% |
| Antiplatelets | 48% |
| ‘Statins’ | 52% |
| Digoxin | 19% |
Haemodynamic effects of NO2− infusion
Sodium NO2− infusion was well tolerated. Mean arterial blood pressure and heart rate did not vary significantly during the experiment: mean arterial pressure values for normal subjects were 88 ± 4 mmHg and 88 ± 15 at baseline and peak NO2− infusion respectively. For CHF patients, the mean arterial pressure values were 73 ± 4 and 70 ± 8 mmHg at baseline and peak NO2− infusion, respectively, while the corresponding values for heart rate were 62 ± 5 and 65 ± 4 beats·min−1 in normal subjects and 66 ± 4 and 69 ± 4 beats·min−1 in CHF patients. Maximal venous methaemoglobin concentrations in the infused arm were less than 2% of total haemoglobin at all NO2− infusion rates.
FBF changes are shown in Figure 2. In the non-infused forearm, both in normal subjects and CHF patients, there was significant vasoconstriction during the course of the study (anova: F = 2.6; P = 0.04), which was attenuated at the highest NO2− infusion rate, raising the possibility of the onset of NO2− induced vasodilatation due to recirculation (inset, Figure 2). In the infused arm (main graph, Figure 2), the relationship between infusion rate and effect varied markedly between groups. For normal subjects, there was a progressive increase in FBF with infusion rates of 3.14 μmol·min−1 and higher. In CHF patients, vasodilator responses were induced already at the lowest infusion rates of NO2−, but with similar responses at doses >3.14 μmol·min−1. Logit transformation of concentration-response data confirmed that the gradients of the linearized relationships were significantly steeper (P = 0.017) in normal subjects than in CHF patients; partitioned anova methodology was therefore utilized for analysis of these data. At lower, but not higher, infusion rates, there were considerably greater FBF responses (F = 5.5; P = 0.02) than those in normal subjects, in whom the threshold for increases in FBF was NO2− infusion rate of 3.1 μmol·min−1.
Figure 2.
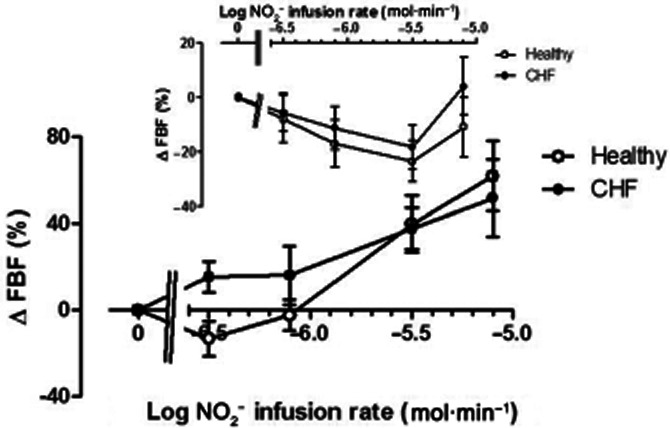
Changes in FBF in the non-infused arm (inset) and infused arm in normal subjects (open symbols) and CHF patients (closed symbols). In the non-infused arm, FBF decreased significantly (F = 2.6; P = 0.04); in the infused arm there was a selective increase in FBF associated with lower NO2− infusion rates (F = 5.5; P = 0.02). * = P < 0.05 versus normal subjects.
UVV changes are summarized in Figure 3. In the non-infused arm (inset), both in normal subjects and patients, there was venoconstriction (F = 9.6; P < 0.0001), which was more marked in normal subjects (F = 15.4; P < 0.001). In the infused arm (main graph, Figure 3), both groups exhibited evidence of increases in UVV commencing with the lowest infusion rate of sodium NO2− infusion, with a monophasic and progressive dose-related increase in UVV. However, overall responses in CHF patients were substantially lower than those in normal subjects (F = 6.3; P = 0.01).
Figure 3.
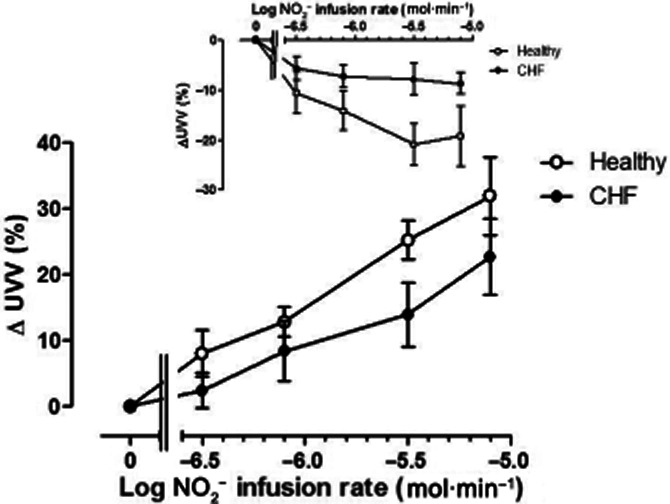
Changes in unstressed venous volume (UVV) in the non-infused arm (inset) and infused arm in normal subjects (open symbols) and CHF patients (closed symbols). In the non-infused arm, there was a decrease in UVV, which was more marked in normal subjects (F = 15.4; P < 0.001). In the infused arm, the increase in UVV with NO2− infusion was attenuated in CHF patients (F = 6.2; P = 0.01). * = P < 0.05 versus normal subjects.
Changes in venous pH and O2 saturations
pH in venous blood did not vary significantly between normal subjects and CHF patients, nor did it fluctuate significantly (Figure 4) during NO2− infusion in either arm. Venous oxygen saturation also did not vary significantly between patient groups (P = 0.27 and P = 0.35 for changes in the CHF group versus healthy controls in the non-infused and infused arms respectively). With NO2− infusion, there was a trend to lower venous oxygen saturation with time in the non-infused arm (P = 0.07), which may be explained by prolonged immobility and consecutive decreases in blood flow with time. In comparison in the infused arm, there was no significant change in venous oxygen saturation with time (P = 0.53). At peak dose, venous oxygen saturations were 79.5% ± 7.2 and 83.4 ± 1.8 in the CHF and healthy volunteers respectively.
Figure 4.
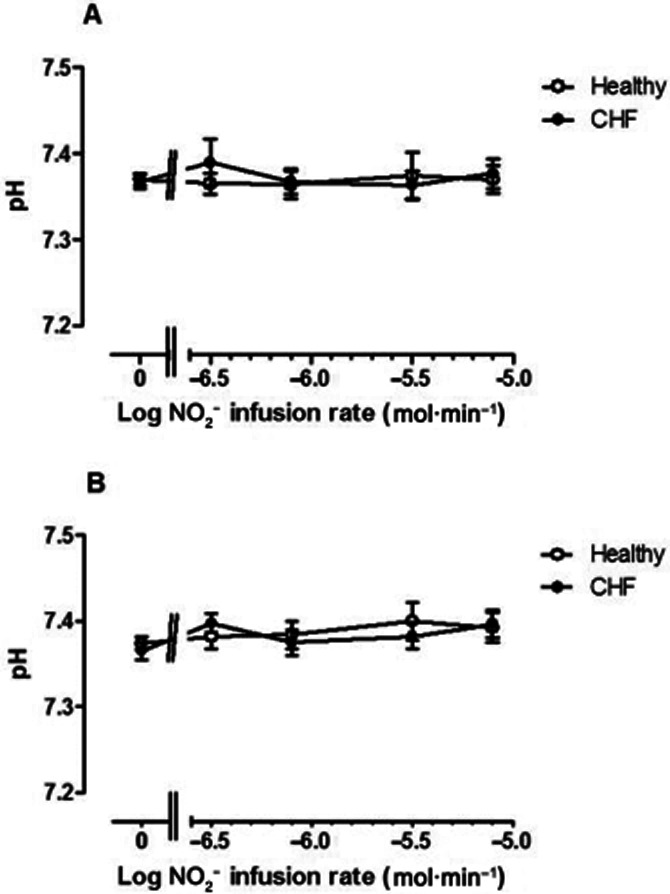
pH fluctuations in venous blood from (A) non-infused arm and (B) infused arm in normal subjects (open symbols) and CHF patients (closed symbols). pH did not vary significantly either with NO2− infusion rate or between groups.
Plasma NO2− and nitrate concentrations
Changes in NO2− concentrations in venous blood during NO2− infusion are summarized in Figure 5A and B. Resting plasma NO2− concentrations did not differ statistically between groups (0.60 ± 0.0.07 vs. 0.41 ± 0.08 μM in control vs. CHF patients: P = 0.08). NO2− concentrations did not change much in venous blood in the non-infused arm in both normal subjects and CHF patients (Figure 5A), but revealed a trend towards greater levels (F = 4.3; P = 0.04) in the normal subject group.
Figure 5.
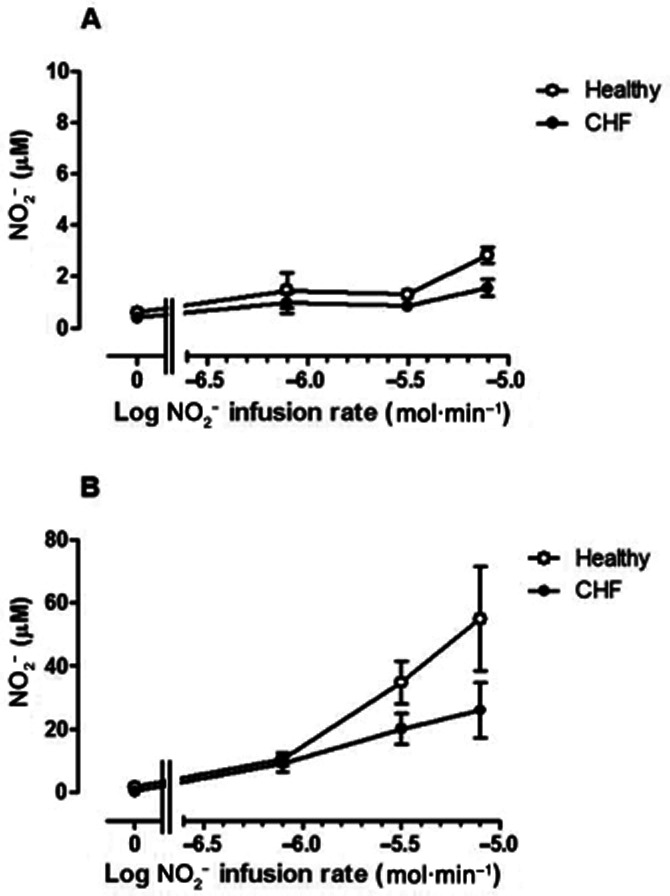
Venous plasma NO2− concentration (μM) in (A) non-infused arm and (B) infused arm in normal subjects (open symbols) and CHF patients (closed symbols). Baseline NO2− concentrations were marginally lower in CHF patients than in normal subjects (P = 0.08). NO2− concentrations rose significantly (F = 32.7; P < 0.0001) with NO2− infusion in the infused arm: this increase was attenuated significantly (F = 10.6; P = 0.002) in CHF patients. * = P < 0.05 versus normal subjects.
In the infused arm (Figure 5B), NO2− concentrations increased approximately 70-fold during NO2− infusion (F = 32.7; P < 0.0001). In normal subjects, venous NO2− concentrations increased from 1.2 ± 0.10 to 54.9 ± 16.5 μM, while in CHF patients, the increase was from 0.39 ± 0.07 to 29.0 ± 9.3 μM. Thus, the increase in venous NO2− levels was greater in normal subjects than in CHF patients (F = 10.6; P = 0.002). The increase in venous NO2− concentrations per 10-fold increase in NO2− infusion rate was disproportionately small: approximately sevenfold for normal subjects and 3–4-fold for CHF patients.
Consistent with previous observations (Usui et al., 1998) basal nitrate levels in the venous effluent were found to be higher in the CHF patients compared to healthy volunteers (Figure 6B; P = 0.0034). Venous nitrate levels rose similarly (P = 0.96 and P = 0.67 for the non-infused and infused arms, respectively) in the two groups of subjects (Figure 6A and B) suggesting that the degree of oxidation of NO2− to nitrate did not differ between groups.
Figure 6.
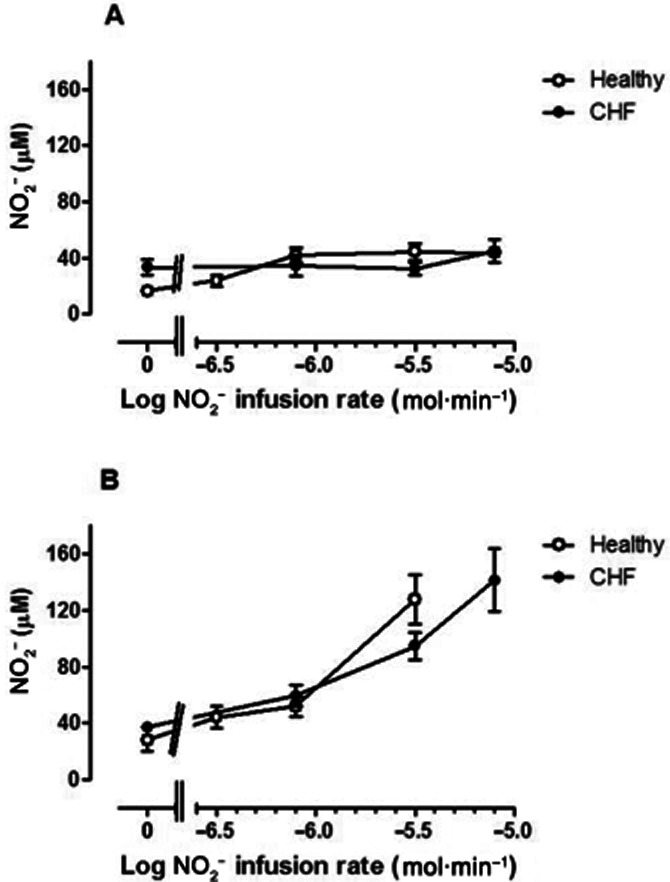
Venous plasma nitrate concentrations in (A) non-infused arm and (B) infused arm in normal subjects (open symbols) and CHF patients (closed symbols). Baseline nitrate concentrations were greater (P = 0.003) in CHF subjects. During NO2− infusion, nitrate concentrations increased (F = 16.2 P < 0.0001) in the infused arm, but without significant difference between normal subjects and CHF patients.
RXNO concentrations
In the non-infused arm (Figure 7A), RXNO concentrations were significantly greater in normal subjects than in patients with CHF (F = 8.6; P = 0.04). In the infused arm, this difference was attenuated during NO2− infusion, becoming non-significant. Furthermore, RXNO tended to increase with the highest NO2− infusion rate.
Figure 7.
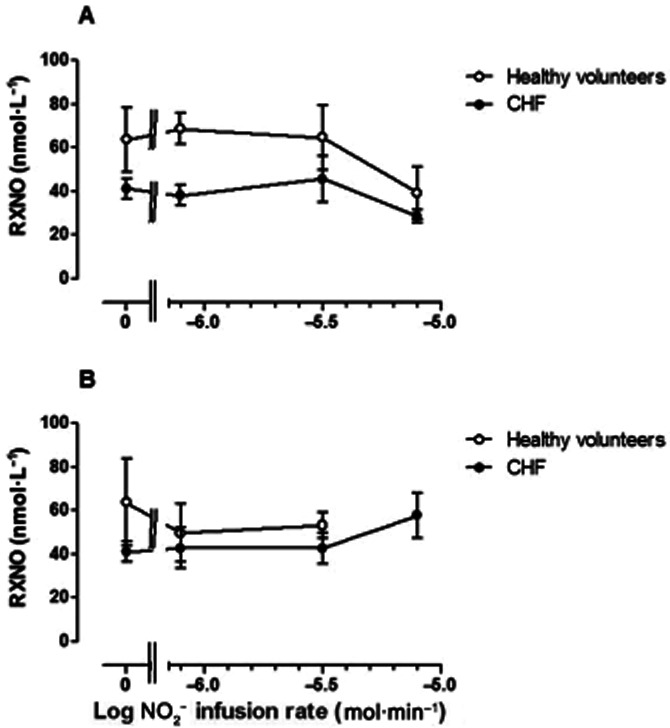
Venous plasma RXNO (nmol·L−1) in (A) non-infused arm and (B) infused arm in normal subjects (open symbols)and CHF patients (closed symbols). RXNO concentrations did not vary significantly with NO2− infusion in either arm (F = 0.44 P = 0.99), but were significantly lower in CHF patients than in normal subjects in the non-infused arm (P = 0.04), while this difference was attenuated in the infused arm.
Assessment of in vitro NO2− clearance in human plasma
Following spiking of whole blood with NO2− in vitro, plasma NO2− decayed similarly (P = 0.66) in the two groups, with an apparent half-life of 5–6 min, irrespective of the initial concentration of NO2− achieved, as summarized in Figure 8. Although basal levels of nitrate were higher in heart failure patients, the accompanying rate of nitrate formation was similar (P = 0.99) in the two groups (not shown). Fitting attempts revealed that the reaction obeyed neither first- nor second-order kinetics suggesting the involvement of multiple processes including uptake, oxidation, reduction and redistribution between plasma and erythrocytes.
Figure 8.
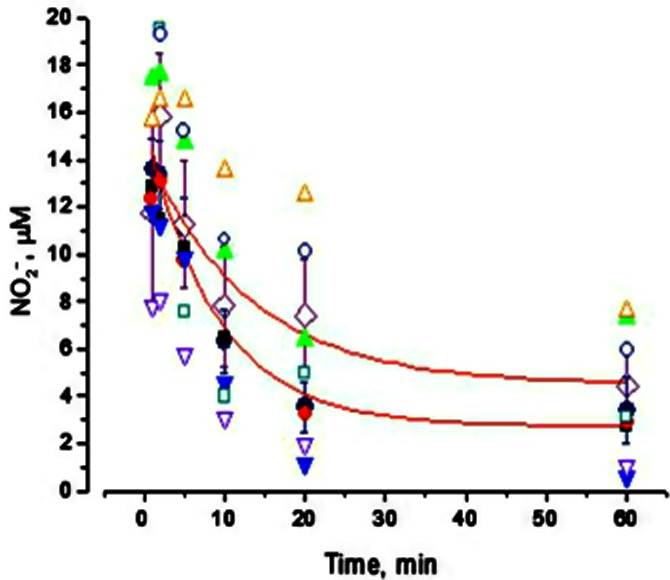
Clearance of NO2− in vitro from human plasma. Closed symbols represent data from normal subjects; open symbols represent data from CHF patients.
In vitro tolerance/cross-tolerance study
In pre-constricted saphenous vein rings in vitro, NO2− induced vasorelaxation with a bi-sigmoidal concentration-response characteristic (Figure 9). After exposure to very high concentrations of NO2− (10−2 M) for 45 min followed by 30 min washout, there was no significant shift in the NO2− concentration-response curve (log EC50 for the low-affinity components −3.7 ± 0.10 vs. −4.0 ± 0.08 M (n = 7; P = NS, for before and after attempted tolerance induction respectively). Furthermore, there was no cross-tolerance to GTN (log EC50 −7.9 ± 0.09 vs. −7.9 ± 0.07 M; n = 7, P = NS for before and after NO2− tolerance induction respectively).
Figure 9.
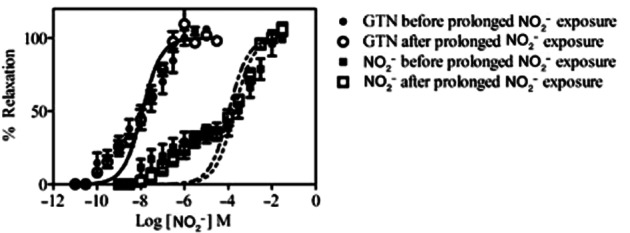
In vitro tolerance/cross-tolerance induction study. Concentration-response curves are shown for NO2− and GTN, indicating percentage relaxation of saphenous vein rings, before and after prolonged incubation with NO2−.
Assessment of NO2− infusion on oxidative stress
Plasma total isoprostane levels (Figure 10) did not increase significantly with NO2− infusion in both normoxia and hypoxia [8.99 ± 1.71, 7.07 ± 0.93, 11.11 ± 2.11 and 9.09 ± 1.48 at baseline, ‘low-dose NO2−’, ‘higher dose NO2−’ and during hypoxia respectively (P = 0.44)].
Figure 10.
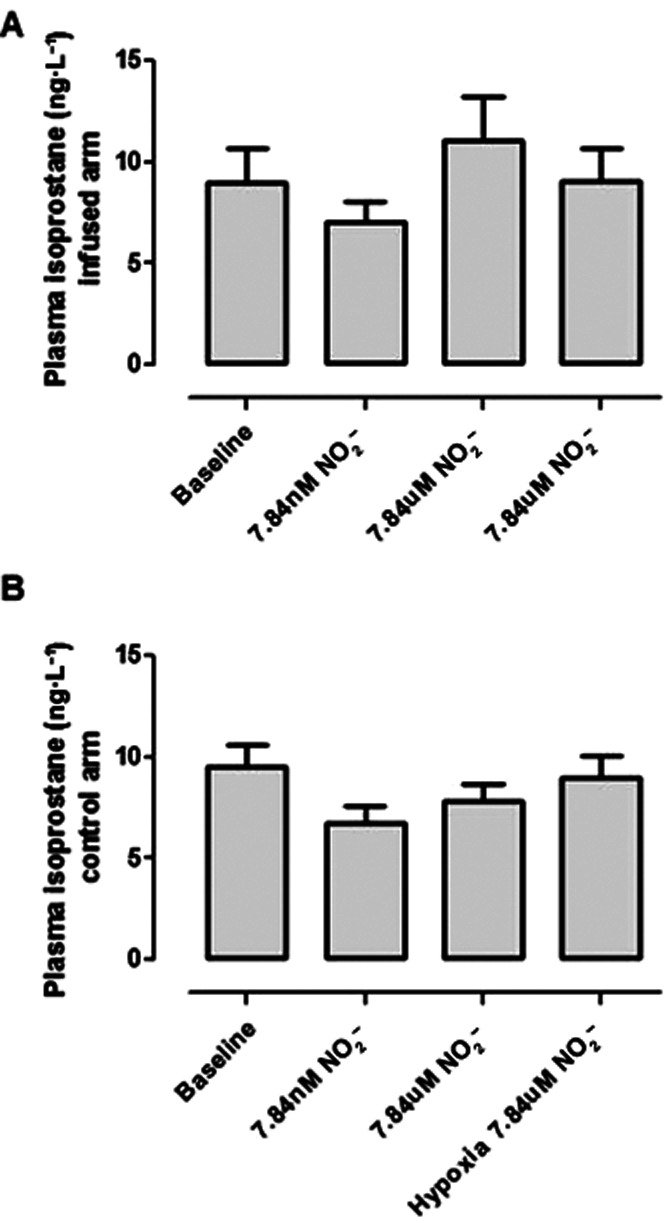
Plasma 8-isoprostane concentrations following NO2− infusion in heart failure patients. Values did not vary significantly between infused (A) and non-infused (B) arms.
Discussion
Previous studies have suggested that infused NO2− exerts vasomotor responses that are similar to those of the organic nitrates, with marked venodilatation, and moderate dose-dependent arteriolar dilatation, but in the case of NO2−, these effects are augmented by hypoxia or exercise. (Cosby et al., 2003; Maher et al., 2008). On the other hand, infused NO2− represents in many ways a particularly attractive agent for treatment of CHF complicated by fluid overload. Not only is it a potent venodilator, but its effects appear to be (somewhat surprisingly) devoid of tolerance development (Dejam et al., 2007). Furthermore, NO2− provides a ‘needs-based’ vasodilator effect, with effects accentuated by hypoxia in many systems (Shiva et al., 2007; Ingram et al., 2010; Milsom et al., 2010). The precise mechanism(s) underlying this accentuated effect remain incompletely understood.
In the present study, effects of NO2− infusion were compared in normal subjects and patients with stable CHF. The previously documented (Maher et al., 2008) selective venodilator effects of NO2− at low infusion rates were again documented in normal subjects: the lowest two infusion rates of NO2− induced significant increases in UVV, without increasing FBF. On the other hand, in CHF patients marked and selective hyper-responsiveness of forearm resistance vessels to lower rates of NO2− infusion was observed, with substantially reduced venodilator responses in CHF patients. The latter finding may represent NO resistance at the level of the capacitance bed. Alternatively, it may be a consequence of increased clearance of NO2− across the vascular bed, resulting in lower concentrations in the capacitance vasculature at any given infusion rate (see below). Thus, the main implication of the current findings is that in the presence of CHF, the spectrum of vasomotor responses to NO2− is altered, with enhancement of arteriolar dilatation, but attenuation of venodilator responses.
The finding that CHF patients had lower RXNO and marginally lower NO2− concentrations than normal subjects is consistent with previous findings in subjects with endothelial dysfunction (Lauer et al., 2001; Kleinbongard et al., 2003; Heiss et al., 2006). Steady-state NO2− concentrations (in the absence of dietary or i.v. NO2− supplementation) may be regarded as being indicative of NO generation and therefore indicative of diminished eNOS activity in CHF (Lauer et al., 2001), whereas RXNO concentrations reflect the generation of S-nitrosoproteins from either NO or NO2− (Bryan et al., 2005), as well as a measure of prevalent redox stress (Ng et al., 2004). With sodium NO2− infusion, venous NO2− concentrations increased, but not in proportion to the increases in infusion rates, suggesting some selective loss of NO2− at higher infusion rates. These changes were noted both in the infused and non-infused arms, indicating that the non-infused arm cannot be regarded as a simple ‘control’ site: indeed the FBF changes (Figure 2, inset) suggest the onset of some dilator effect with the highest NO2− infusion rate. Furthermore, NO2− levels increased far less markedly with increasing sodium NO2− infusion rates in CHF patients compared to normal subjects (Figure 5B) indicating greater rates of NO2− clearance in these patients. Although NO2− may be reduced to NO (bioactivation), a major clearance mechanism for NO2− is oxidation to nitrate. Therefore, venous nitrate concentrations were measured in both infused and non-infused arms, revealing that, in the presence of predominant oxidation of NO2− to nitrate, there was nevertheless a similar increase in plasma nitrate levels in the two groups upon infusion of NO2−, making enhanced clearance through oxidation to nitrate less likely to account for these differences. Similarly, our in vitro results suggest that there is no systematic difference in the rate of NO2− disappearance in the two groups, thus excluding sampling artefacts as a possible explanation for the differences in NO2− concentrations between normal subjects and CHF patients.
As regards the additional potential formation of S-nitrosoproteins via NO2− metabolism, venous RXNO concentrations tended to increase in the infused arm in the CHF patients (coupled with a relative decrease in RXNO concentrations in the controls), attenuating the difference between baseline levels in the two groups. These findings are also consistent with selective bioactivation of NO2− in the CHF patients, and might have been accounted for by the presence of tissue hypoxia and/or acidosis in these patients, given the previously described incremental bioactivation of NO2− in the presence of hypoxia (Maher et al., 2008). However, neither venous blood pH nor oxygen concentrations differed significantly between groups. There was a trivially (non-significantly) lower venous oxygen saturation in the infused arm of the heart failure patients, but this seems unlikely to provide an explanation for the difference in clearance. It therefore appears either that a component of subcellular/microvascular hypoxia was present in the CHF patients without detection in venous samples (a possible but somewhat unlikely event) or that these results indicate that, apart from hypoxia, NO2− bioactivation to NO can be induced by factors other than hypoxia, such as changes in redox state. This issue is worthy of further investigation.
Taken together, these results show that CHF patients are hyper-responsive to the arteriolar dilating effects of infused sodium NO2−, presumably due to increased release of NO (despite the absence of arterial hypoxaemia). This finding may imply the need for some caution in the use of infused NO2− preparations in patients with decompensated CHF, for fear of precipitating falls in systolic blood pressure. On the other hand, symptomatic hypotension was not observed in the currently studied patient cohort. However, CHF patients were hyporesponsive both to the effects of infused sodium NO2− on venous capacitance and, to a lesser extent, to the effects on FBF at high infusion rates. These findings may imply the existence of NO resistance, as previously documented in both arteries and platelets of such patients (Anderson et al., 2004). Although platelet NO resistance exhibits partial responsiveness to ACE inhibitor therapy (Chirkov et al., 2004), which was utilized in the majority of the CHF group, its interaction with vascular NO resistance is less certain. Alternatively, the apparent resistance of the capacitance vessels may in whole or in part be due to a lower concentration of NO2− in venous blood due to the increased transvascular clearance. This would potentially result in higher doses being required when treating heart failure patients acutely. As net responsiveness to NO2− reflects both accelerated bioactivation and diminished tissue responses to NO, it is likely that individual responses vary markedly, according to each of these component factors.
Nitrate therapy is associated with the development of tolerance and increased oxidative stress. It is now accepted that a central component of nitrate tolerance induction is progressive failure of enzymatic bioactivation (Sage et al., 2000), and although the nature of the relevant nitrate reductase remains controversial (D'Souza et al., 2011), it is clear that GTN inhibits aldehyde dehydrogenases (ALDH; Towell et al., 1986; Chen et al., 2002; D'Souza et al., 2011). In particular, the possibility that inhibition of the predominantly mitochondrial form of ALDH may contribute to nitrate tolerance development (and possibly also to accentuation of redox stress) has been the subject of considerable recent investigation (Chen et al., 2002; Mackenzie et al., 2005). Given that the bioactivation of NO2− is catalysed by a large number of NO2− reductases, which may include ALDH (Feelisch et al., 2008), there is a theoretical potential both for the induction of tolerance after prolonged administration of NO2− and for the associated induction of redox stress. We therefore evaluated the potential for NO2− tolerance induction utilizing cross-tolerance to GTN, a relevant issue given that this may theoretically develop after prolonged exposure to the agent (Henry et al., 1990). Under the conditions of this experiment, NO2− did not induce tolerance to itself (consistent with the findings of Dejam et al., 2007) or to GTN. Furthermore, prolonged systemic administration of NO2− in patients with CHF was not associated with aggravation of redox stress, as measured by plasma concentrations of total isoprostanes. On the other hand, it is possible that oxidative modification of proteins may have occurred.
A number of potential limitations and caveats apply to the current findings. Most importantly, the biochemical basis of the observed arterial hyper-responsiveness to NO2− was not determined, and indeed, the possible relevance of tissue hypoxia to this phenomenon cannot be completely excluded with the methodology utilized in this study. Furthermore, changes in tissue redox stress were not documented: this remains a major priority for future investigations. While the observation of attenuation of the increase in plasma NO2− in association with increasing rates of sodium NO2− infusion implies accelerated transvascular clearance of NO2− in such patients, the release of NO was not measured and nitrate generation data were incomplete for normal subjects. Therefore, it cannot be absolutely certain that there was a close relationship between such accelerated clearance and incremental rates of NO generation. Indeed, there is recent evidence that NO2− may exhibit vasoactivity independent of bioconversion and that NO2− may exert a component of its effects independent of the formation of NO or NO adduct (Bryan et al., 2005; Shiva and Gladwin, 2009). Quantitation of heme-nitrosyl adducts in red blood cells and/or identification of specific NO adducts in plasma might have provided an additional, more direct index of NO formation, However, as observed by Dejam et al. (2007), increases in concentrations of such adducts are likely to parallel those of plasma NO2− concentrations. Furthermore, the precise proportion of NO2− clearance via oxidation to nitrate cannot be assessed completely from the current results, although it was possible to exclude the possibility that transvascular nitrate generation occurred selectively in CHF patients given the similar rise in nitrate levels.
Oxidative stress is associated with many chronic diseases and oxidative damage markers can be measured in the plasma. However, plasma measurement of oxidative stress remains a challenging area of research because of the highly reactive nature of these molecules. Several studies have focused on measuring either the total antioxidant capacity of plasma or specific measures of free radical-mediated damage such as F2-isoprostane or oxidized-LDL. While we acknowledge that other oxidative stress markers could have been assessed in the current study, we opted to quantify total 8-isoprostane (8-iso prostaglandin F2α). Isoprostanes are among the most reliable markers of oxidative stress that can be assessed in translational studies by specific immunoassay-based techniques. Moreover, these products are not only by-products of oxidative stress, but also effector molecules involved in pathophysiology. 8-isoprostane is a potent pulmonary and renal vasoconstrictor and has been implicated as a causative mediator of hepato-renal syndrome and pulmonary oxygen toxicity.
Finally, the findings of the current investigation, while relevant to the potential therapeutic administration of NO2−, may not be indicative of physiological modulation of NO2− at far lower concentrations.
While NO2− infusion has the potential to lead to the generation of methaemoglobin (a possible concern), we did not observe a significant increase in venous methaemoglobin levels in this study.
We noted a significant vasoconstriction in the non-infused arm during the study. In theory, this could have been secondary to hypotension; however, no significant drop in blood pressure was observed; in fact, there appeared to be a trend towards an increase. It has been our experience in prolonged intrabrachial infusion studies with other agents that forearm blood flow falls in the contralateral arm over time despite no significant changes in blood pressure. We suspect this result from the effects of slight discomfort and often a full bladder during these long studies. It is because of such changes in flow in the non-infused arm during prolonged studies that bilateral plethysmography with correction for the non-infused arm is recommended for intrabrachial infusion studies (Benjamin et al., 1995).
With regards to the medication received by the CHF patients, while there are data to suggest that ACE inhibitors and angiotensin receptor blockers improve NO production/endothelial function, we could not find any evidence to suggest any impact upon NO2− conversion. The possibility that the medication taken by the CHF patients may account for some of the differences observed remains a limitation of this study.
In the current study, NO2− was infused intra-arterially in order to document peripheral vasomotor responsiveness with minimal changes in systemic blood pressure. This circumstance differs both from the potential mode of treatment to increase NO2− effect in chronic heart failure (e.g. via administration of dietary sources of nitrate) and from the i.v. infusion of NO2− as a component of management of acute heart failure. Nevertheless, the findings of the current investigation extend the question of the potential clinical utility of NO2− as a component of the management of patients with both acute and chronic heart failure. The major residual issue to be explored is the extent of change in vascular responsiveness to be seen in such patients, especially in decompensated CHF, where significant tissue hypoxia is more likely to be present, with resultant potentiation of NO2− bioactivation, perhaps counterbalanced in part by the phenomenon of tissue resistance to NO (Chirkov and Horowitz, 2007). Despite the occurrence of NO resistance, treatment with organic nitrates is at least as effective as diuretic/morphine-based acute pharmacotherapy for decompensated heart failure with acute pulmonary oedema (Cotter et al., 1998; Sharon et al., 2000). However, nitrate therapy is often associated with the unpleasant side effect of headache. In this study, none of the subjects who received i.v. NO2− suffered from this symptom, potentially providing a distinct advantage over traditional nitrate therapies. A comparison with NO2−-based therapy now seems indicated.
Acknowledgments
We thank Rebekah Weaver and Ulasini Naidoo for technical and nursing support, Bill Thomson, Joe O'Brien and the radiopharmacy team for their medical physics and radiopharmaceutical support. We also acknowledge Janis Weeks (Medical Microbiology, Cardiff) for conducting the plasma nitrite/protein NO measurements and for her expertise in the biochemical aspects of this study.
Glossary
- ALDH
aldehyde dehydrogenases
- FBF
forearm blood flow
- FVV
forearm venous volume
- GTN
glyceryl trinitrate
- RXNO
protein-bound NO
- UVV
unstressed venous volume
Conflict of interest
Professor Frenneaux has received a research grant from Medtronic. He has an ownership interest in a ‘method of use’ patent held for Perhexiline in Chronic Heart Failure. He has also served on the advisory board or as a consultant to Medtronic and Biotronik.
References
- Anderson RA, Ellis GR, Chirkov YY, Holmes AS, Payne N, Blackman DJ, et al. Determinants of platelet responsiveness to nitric oxide in patients with chronic heart failure. Eur J Heart Fail. 2004;6:47–54. doi: 10.1016/S1388-9842(03)00038-2. [DOI] [PubMed] [Google Scholar]
- Atherton JJ, Moore TD, Lele SS, Thomson HL, Galbraith AJ, Belenkie I, et al. Diastolic ventricular interaction in chronic heart failure. Lancet. 1997;349:1720–1724. doi: 10.1016/S0140-6736(96)05109-4. [DOI] [PubMed] [Google Scholar]
- Baker JE, Su J, Fu X, Hsu A, Gross GJ, Tweddell JS, et al. Nitrite confers protection against myocardial infarction: role of xanthine oxidoreductase, NADPH oxidase and K(ATP) channels. J Mol Cell Cardiol. 2007;43:437–444. doi: 10.1016/j.yjmcc.2007.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin N, Calver A, Collier J, Robinson B, Vallance P, Webb D. Measuring forearm blood flow and interpreting the responses to drugs and mediators. Hypertension. 1995;25:918–923. doi: 10.1161/01.hyp.25.5.918. [DOI] [PubMed] [Google Scholar]
- Bryan NS, Fernandez BO, Bauer SM, Garcia-Saura MF, Milsom AB, Rassaf T, et al. Nitrite is a signaling molecule and regulator of gene expression in mammalian tissues. Nat Chem Biol. 2005;1:290–297. doi: 10.1038/nchembio734. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci U S A. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirkov YY, Horowitz JD. Impaired tissue responsiveness to organic nitrates and nitric oxide: a new therapeutic frontier? Pharmacol Ther. 2007;116:287–305. doi: 10.1016/j.pharmthera.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Chirkov YY, Holmes AS, Chirkova LP, Horowitz JD. Nitrate resistance in platelets from patients with stable angina pectoris. Circulation. 1999;100:129–134. doi: 10.1161/01.cir.100.2.129. [DOI] [PubMed] [Google Scholar]
- Chirkov YY, Holmes AS, Willoughby SR, Stewart S, Wuttke RD, Sage PR, et al. Stable angina and acute coronary syndromes are associated with nitric oxide resistance in platelets. J Am Coll Cardiol. 2001;37:1851–1857. doi: 10.1016/s0735-1097(01)01238-4. [DOI] [PubMed] [Google Scholar]
- Chirkov YY, Holmes AS, Martelli JD, Horowitz JD. Effect of perindopril on platelet nitric oxide resistance in patients with chronic heart failure secondary to ischemic left ventricular dysfunction. Am J Cardiol. 2004;93:1438–1440. doi: 10.1016/j.amjcard.2004.02.052. A10. [DOI] [PubMed] [Google Scholar]
- Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- Cotter G, Metzkor E, Kaluski E, Faigenberg Z, Miller R, Simovitz A, et al. Randomised trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary oedema. Lancet. 1998;351:389–393. doi: 10.1016/S0140-6736(97)08417-1. [DOI] [PubMed] [Google Scholar]
- Crawford JH, Isbell TS, Huang Z, Shiva S, Chacko BK, Schechter AN, et al. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2006;107:566–574. doi: 10.1182/blood-2005-07-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza Y, Dowlatshahi S, Bennett BM. Changes in aldehyde dehydrogenase 2 expression in rat blood vessels during glyceryl trinitrate tolerance development and reversal. Br J Pharmacol. 2011;164:632–643. doi: 10.1111/j.1476-5381.2011.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A, Mulsch A, Hink U, Mollnau H, Warnholtz A, Oelze M, et al. The oxidative stress concept of nitrate tolerance and the antioxidant properties of hydralazine. Am J Cardiol. 2005;96:25i–36i. doi: 10.1016/j.amjcard.2005.07.030. [DOI] [PubMed] [Google Scholar]
- Dejam A, Hunter CJ, Tremonti C, Pluta RM, Hon YY, Grimes G, et al. Nitrite infusion in humans and nonhuman primates: endocrine effects, pharmacokinetics, and tolerance formation. Circulation. 2007;116:1821–1831. doi: 10.1161/CIRCULATIONAHA.107.712133. [DOI] [PubMed] [Google Scholar]
- Dupuis J, Lalonde G, Lebeau R, Bichet D, Rouleau JL. Sustained beneficial effect of a seventy-two hour intravenous infusion of nitroglycerin in patients with severe chronic congestive heart failure. Am Heart J. 1990;120:625–637. doi: 10.1016/0002-8703(90)90021-o. [DOI] [PubMed] [Google Scholar]
- Feelisch M, Fernandez BO, Bryan NS, Garcia-Saura MF, Bauer S, Whitlock DR, et al. Tissue processing of nitrite in hypoxia: an intricate interplay of nitric oxide-generating and -scavenging systems. J Biol Chem. 2008;283:33927–33934. doi: 10.1074/jbc.M806654200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaruwan P, Schmitt M, Taylor J, Lee L, Struthers A, Frenneaux MP. Lack of rapid aldosterone effects on forearm resistance vasculature in health. J Renin Angiotensin Aldosterone Syst. 2002;3:123–125. doi: 10.3317/jraas.2002.013. [DOI] [PubMed] [Google Scholar]
- Heiss C, Lauer T, Dejam A, Kleinbongard P, Hamada S, Rassaf T, et al. Plasma nitroso compounds are decreased in patients with endothelial dysfunction. J Am Coll Cardiol. 2006;47:573–579. doi: 10.1016/j.jacc.2005.06.089. [DOI] [PubMed] [Google Scholar]
- Henry PJ, Horowitz JD, Louis WJ. Nitrate tolerance induced by nicorandil or nitroglycerin is associated with minimal loss of nicorandil vasodilator activity. J Cardiovasc Pharmacol. 1990;15:365–370. doi: 10.1097/00005344-199003000-00004. [DOI] [PubMed] [Google Scholar]
- Ingram TE, Pinder AG, Bailey DM, Fraser AG, James PE. Low-dose sodium nitrite vasodilates hypoxic human pulmonary vasculature by a means that is not dependent on a simultaneous elevation in plasma nitrite. Am J Physiol Heart Circ Physiol. 2010;298:H331–H339. doi: 10.1152/ajpheart.00583.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinbongard P, Dejam A, Lauer T, Rassaf T, Schindler A, Picker O, et al. Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic Biol Med. 2003;35:790–796. doi: 10.1016/s0891-5849(03)00406-4. [DOI] [PubMed] [Google Scholar]
- Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M, et al. Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc Natl Acad Sci U S A. 2001;98:12814–12819. doi: 10.1073/pnas.221381098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IS, Maki-Petaja KM, McEniery CM, Bao YP, Wallace SM, Cheriyan J, et al. Aldehyde dehydrogenase 2 plays a role in the bioactivation of nitroglycerin in humans. Arterioscler Thromb Vasc Biol. 2005;25:1891–1895. doi: 10.1161/01.ATV.0000179599.71086.89. [DOI] [PubMed] [Google Scholar]
- Maher AR, Milsom AB, Gunaruwan P, Abozguia K, Ahmed I, Weaver RA, et al. Hypoxic modulation of exogenous nitrite-induced vasodilation in humans. Circulation. 2008;117:670–677. doi: 10.1161/CIRCULATIONAHA.107.719591. [DOI] [PubMed] [Google Scholar]
- Manyari DE, Wang Z, Cohen J, Tyberg JV. Assessment of the human splanchnic venous volume-pressure relation using radionuclide plethysmography. Effect of nitroglycerin. Circulation. 1993;87:1142–1151. doi: 10.1161/01.cir.87.4.1142. [DOI] [PubMed] [Google Scholar]
- Milsom AB, Patel NS, Mazzon E, Tripatara P, Storey A, Mota-Filipe H, et al. Role for endothelial nitric oxide synthase in nitrite-induced protection against renal ischemia-reperfusion injury in mice. Nitric Oxide. 2010;22:141–148. doi: 10.1016/j.niox.2009.10.010. [DOI] [PubMed] [Google Scholar]
- Muir AL, Nolan J. Modulation of venous tone in heart failure. Am Heart J. 1991;121(6 Pt 2):1948–1950. doi: 10.1016/0002-8703(91)90830-b. [DOI] [PubMed] [Google Scholar]
- Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–628. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- Ng ES, Jourd'heuil D, McCord JM, Hernandez D, Yasui M, Knight D, et al. Enhanced S-nitroso-albumin formation from inhaled NO during ischemia/reperfusion. Circ Res. 2004;94:559–565. doi: 10.1161/01.RES.0000117771.63140.D6. [DOI] [PubMed] [Google Scholar]
- Pinder AG, Rogers SC, Khalatbari A, Ingram TE, James PE. The measurement of nitric oxide and its metabolites in biological samples by ozone-based chemiluminescence. Methods Mol Biol. 2009;476:10–27. doi: 10.1007/978-1-59745-129-1_2. [DOI] [PubMed] [Google Scholar]
- Rassaf T, Bryan NS, Kelm M, Feelisch M. Concomitant presence of N-nitroso and S-nitroso proteins in human plasma. Free Radic Biol Med. 2002;33:1590–1596. doi: 10.1016/s0891-5849(02)01183-8. [DOI] [PubMed] [Google Scholar]
- Sage PR, de la Lande IS, Stafford I, Bennett CL, Phillipov G, Stubberfield J, et al. Nitroglycerin tolerance in human vessels: evidence for impaired nitroglycerin bioconversion. Circulation. 2000;102:2810–2815. doi: 10.1161/01.cir.102.23.2810. [DOI] [PubMed] [Google Scholar]
- Schmitt M, Blackman DJ, Middleton GW, Cockcroft JR, Frenneaux MP. Assessment of venous capacitance. Radionuclide plethysmography: methodology and research applications. Br J Clin Pharmacol. 2002;54:565–576. doi: 10.1046/j.1365-2125.2002.t01-7-01689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon A, Shpirer I, Kaluski E, Moshkovitz Y, Milovanov O, Polak R, et al. High-dose intravenous isosorbide-dinitrate is safer and better than Bi-PAP ventilation combined with conventional treatment for severe pulmonary edema. J Am Coll Cardiol. 2000;36:832–837. doi: 10.1016/s0735-1097(00)00785-3. [DOI] [PubMed] [Google Scholar]
- Shiva S, Gladwin MT. Nitrite mediates cytoprotection after ischemia/reperfusion by modulating mitochondrial function. Basic Res Cardiol. 2009;104:113–119. doi: 10.1007/s00395-009-0009-3. [DOI] [PubMed] [Google Scholar]
- Shiva S, Huang Z, Grubina R, Sun J, Ringwood LA, MacArthur PH, et al. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ Res. 2007;100:654–661. doi: 10.1161/01.RES.0000260171.52224.6b. [DOI] [PubMed] [Google Scholar]
- Towell JF, Barboriak JJ, Townsend WF, Kalbfleisch JH, Wang RI. Erythrocyte aldehyde dehydrogenase: assay of a potential biochemical marker of alcohol abuse. Clin Chem. 1986;32:734–738. [PubMed] [Google Scholar]
- Usui M, Matsuoka H, Miyazaki H, Ueda S, Okuda S, Imaizumi T. Increased endogenous nitric oxide synthase inhibitor in patients with congestive heart failure. Life Sci. 1998;62:2425–2430. doi: 10.1016/s0024-3205(98)00225-2. [DOI] [PubMed] [Google Scholar]
- Webb AJ, Milsom AB, Rathod KS, Chu WL, Qureshi S, Lovell MJ, et al. Mechanisms underlying erythrocyte and endothelial nitrite reduction to nitric oxide in hypoxia: role for xanthine oxidoreductase and endothelial nitric oxide synthase. Circ Res. 2008;103:957–964. doi: 10.1161/CIRCRESAHA.108.175810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L, Frenneaux MP. Diastolic ventricular interaction: from physiology to clinical practice. Nat Clin Pract Cardiovasc Med. 2006;3:368–376. doi: 10.1038/ncpcardio0584. [DOI] [PubMed] [Google Scholar]