

Abstract
Background and Purpose
Nutrient sensing in the gut is believed to be accomplished through activation of GPCRs expressed on enteroendocrine cells. In particular, L-cells located predominantly in distal regions of the gut secrete glucagon-like peptide 1 (GLP-1) and peptide tyrosine-tyrosine (PYY) upon stimulation by nutrients and bile acids (BA). The study was designed to address the mechanism of hormone secretion in L-cells stimulated by the BA receptor G protein-coupled bile acid receptor 1 (GPBAR1).
Experimental Approach
A novel, selective, orally bioavailable, and potent GPBAR1 agonist, RO5527239, was synthesized in order to investigate L-cell secretion in vitro and in vivo in mice and monkey. In analogy to BA, RO5527239 was conjugated with taurine to reduce p.o. bioavailability yet retaining its potency. Using RO5527239 and tauro-RO5527239, the acute secretion effects on L-cells were addressed via different routes of administration.
Key Results
GPBAR1 signalling triggers the co-secretion of PYY and GLP-1, and leads to improved glucose tolerance. The strong correlation of plasma drug exposure and plasma PYY levels suggests activation of GPBAR1 from systemically accessible compartments. In contrast to the orally bioavailable agonist RO5527239, we show that tauro-RO5527239 triggers PYY release only when applied intravenously. Compared to mice, a slower and more sustained PYY secretion was observed in monkeys.
Conclusion and Implications
Selective GPBAR1 activation elicits a strong secretagogue effect on L-cells, which primarily requires systemic exposure. We suggest that GPBAR1 is a key player in the intestinal proximal-distal loop that mediates the early phase of nutrient-evoked L-cell secretion effects.
Keywords: bile acid, glucagon-like peptide 1, cyclic adenosine monophosphate, GLP-1 secretion, incretin, glucose tolerance
Introduction
The gut is the largest endocrine organ due to the location of chemosensory cells that secrete multiple regulatory peptide hormones and bioactive amines. A major morphological characteristic of chemosensory cells is that they are scattered along the epithelial lining with direct access to the luminal content. Among enterocytes and brush border epithelial cells, enteroendocrine cells (EEC) are the primary chemosensory cells representing less than 1% of the entire epithelial population. Over 20 different EEC types have been classified according to the content of their secretory granules (reviewed in Steinert and Beglinger, 2011). Of particular interest are L-cells, which co-secrete therapeutically relevant peptides by sensing the arrival of nutrients such as carbohydrates, fats and proteins. These peptides include peptide tyrosine-tyrosine (PYY) and preproglucagon-derived peptides such as glucagon-like peptide 1 (GLP-1), oxyntomodulin and GLP-2.
GLP-1 is a potent antihyperglycaemic hormone, which induces glucose-dependent stimulation of insulin secretion, suppresses glucagon secretion, and stimulates the proliferation and differentiation of insulin-secreting beta cells. In addition, GLP-1 suppresses appetite and thus reduces body weight. Current GLP-1-based therapies target the treatment of type 2 diabetes by either mimicking GLP-1 or slowing its degradation through inhibition of the endogenous enzyme dipeptidyl peptidase 4 (reviewed in Drucker, 2001; Drucker and Nauck, 2006). GLP-2 is an important intestinotrophic growth factor and mediator of intestinal adaptation and is under therapeutic evaluation for the treatment of gut disorders such as short bowel syndrome (Wallis et al., 2009). Actions of oxyntomodulin overlap with those of GLP-1 and glucagon, leading to a decrease in food intake and increased energy expenditure giving rise to body weight reduction (Wynne et al., 2006; Kosinski et al., 2012). Finally, PYY has been shown to reduce appetite (Batterham et al., 2002). Interestingly, co-administration of oxyntomodulin and PYY exerts additive effects on food intake in overweight and obese humans (Field et al., 2010), suggesting that mechanisms triggering the secretion of several bioactive peptides from L-cells may offer a new avenue for the treatment of type 2 diabetes, obesity and intestinal disorders.
In addition to nutrients, bile acids (BA) have been shown to stimulate peptide secretion from L-cells. Intracolonic infusion of deoxycholic acid (DCA) was first shown to increase plasma PYY and glucagon immunoreactivity in humans (Adrian et al., 1993). The luminal induced secretory mechanism also translates to rodents because luminal perfusion of bile into perfused rat colon preparations was found to release GLP-1 (Plaisancie et al., 1996). Since L-cell number and hormone content are highest in the distal parts in the intestine, increases in plasma GLP-1 and PYY levels were also observed after intrarectal infusion of taurocholic acid in obese, type 2 diabetic volunteers (Adrian et al., 2012). Delivery of BA to distal parts of the intestine can also be achieved by anionic exchange resins (AER), which are non-absorbable polymers with BA sequestering properties. Treatment with AER increased plasma GLP-1 levels in humans (Garg et al., 2011) and rodents (Chen et al., 2010; Shang et al., 2010), which was associated with improved glycaemic control. Recently, the BA-responsive receptor, G protein-coupled bile acid receptor 1 (GPBAR1; TGR5, GPR131, M-BAR) or GPBA (receptor nomenclature as in Alexander et al., 2011), has been shown to be essential for the AER-mediated increase in GLP-1 release in the distal intestine in mice (Harach et al., 2012).
GPBAR1 is a GPCR, which is activated by BA, resulting in stimulation of Gαs proteins and downstream cAMP signalling pathways (Maruyama et al., 2002; Kawamata et al., 2003). GPBAR1 has been shown to be expressed in immortalized EEC lines, such as mouse STC-1 (Katsuma et al., 2005; Thomas et al., 2009) and GLUTag (Parker et al., 2012) and human NCI-H716 cells (Maruyama et al., 2002), and its activation results in enhanced GLP-1 secretion. Stimulation of GLUTag cells by different BA was also shown to trigger the secretion of GLP-2. The GPBAR-mediated secretory effects were also observed in primary cultures of the adult mouse colon and small intestine (Parker et al., 2012). Besides the established cAMP signalling, additional downstream pathways were proposed for the secretory effect such as elevation of intracellular Ca2+ concentrations and closure of ATP-sensitive potassium (KATP) channels (Thomas et al., 2009). GPBAR1 stimulation could also enhance the calcium and secretory responses to glucose (Parker et al., 2012). In vivo, BA-dependent improvement of glucose homeostasis was shown to be mediated by GPBAR1-induced GLP-1 secretion using GPBAR1-overexpressing and knockout mice (Thomas et al., 2009).
In addition to the gut, GPBAR1 is functionally expressed in non-parenchymal cells of the liver such as sinusoidal (Keitel et al., 2007) and Kupffer cells (Keitel et al., 2008), cholangiocytes of the bile duct (Keitel et al., 2010) and in epithelial cells and smooth muscle cells of the gallbladder, where GPBAR1 activation promotes chloride and fluid secretion (Keitel et al., 2009) or gall bladder relaxation and filling (Lavoie et al., 2010; Li et al., 2011) respectively. GPBAR1 activation in skeletal muscle cells and brown adipose tissues contribute to the prevention of body weight gain during high-fat feeding (Watanabe et al., 2006; Thomas et al., 2009) and expression in resting CD14-positive cells (Kawamata et al., 2003) inhibits inflammatory responses in the liver (Keitel et al., 2008; Wang et al., 2011), in the gut (Cipriani et al., 2013) and attenuates atherosclerosis (Pols et al., 2011). In summary, GPBAR1 modulates several metabolic functions by both luminal stimulation in the gut and activation of systemically accessible tissues. Even though available data clearly suggest that luminal GPBAR1 activation of enteroendocrine L-cells is important for GLP-1 secretion, GPBAR1 activation of L-cells from the systemic side cannot be excluded from contributing to the secretory and overall antidiabetic effect. The aim of this study was to characterize pharmacologically a novel synthetic, small-molecule GPBAR1 agonist and to investigate its secretory effect in vitro using the enteroendocrine model cell line STC-1 and in vivo by specifically addressing the potential of GPBAR1-mediated L-cell activation during systemic drug exposure.
Experimental procedures
Animals
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). All animal care and experimental procedures were in accordance with national and international guidelines for animal care and were conducted according to the study protocol. In addition to authority approval, experiments performed in non-human primates (NHP) were reviewed by the F. Hoffmann-La Roche ethical commission. The F. Hoffmann-La Roche Pharma Research Basel test facility is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. During the course of the studies, no overt pharmacological or toxicological signs were observed. C57Bl/6 and db/db (B6.BKS(D)-Leprdb/J) male mice, were obtained from Charles River Laboratories Germany GmbH (Sulzfeld, Germany). Transgene GPBAR KI mice (GPBAR1_B6-Gpbar1tm1(GPBAR1)Ait_0109) were generated in-house, and then bred and supplied by Harlan Laboratories (Milan, Italy). Mice were housed with a 12/12 h light cycle in climate rooms in groups of 2–5 animals in standard Makrolon cages with sawdust and enrichment, and fed regular chow diet ad libitum before experimental procedures. Incompatible animals were housed individually. Cage sizes are defined according to national guidelines regarding number of animals and their body weight. NHP were housed with a 12/12 light cycle in socialized groups according to their needs and fed a typical NHP diet (cage sizes 50–100 m3).
Materials
RO5527239 ((R,E)-1-(4-(3-(hydroxyimino)-3-(2-methylpyridin-4-yl)-1-o-tolylpropyl)phenyl)piperidine-4-carboxylic acid), tauro-RO5527239 ((R,E)-2-(1-(4-(3-(hydroxyimino)-3-(2-methylpyridin-4-yl)-1-o-tolylpropyl)phenyl)piperidine-4-carboxamido)ethanesulfonic acid) (US Pat. Appl. US 20120010190) and sitagliptin were prepared at F. Hoffmann-La Roche AG. Krebs-Ringer, Forskolin, IBMX, phorbol 12-myristate 13-acetate (PMA) and fatty acid-free BSA were obtained from Sigma (Buchs, Switzerland).
Cell culture
CHO-DUKX-CRE-luci cells were cultured in DMEM 31331-028 (Invitrogen, Carlsbad, CA, USA) supplemented with GlutaMax, 10% FBS, hypoxanthine/thymidine (HT Supplement), 200 μg·mL−1 hygromycin and when transfected with pCIneo plasmids with 400 μg·mL−1 geneticin (G418). STC-1 cells were obtained from Dr. Douglas Hanahan (through Cold Spring Harbor Laboratory, NY, USA) and cultured in DMEM 31966 (Invitrogen, Carlsbad, CA, USA) supplemented with 15% horse serum and 2.5% fetal calf serum (FCS). Cells were incubated in a humidified atmosphere at 37°C with 5% CO2.
Stable GPBAR1 expression in mammalian cells
Human and mouse GPBAR1 receptor cDNA (GenBank: NM_001077191 and NM_174985, respectively) were inserted into pCIneo (Catalys, Wallisellen, Switzerland). CHO cells deficient in dihydrofolate reductase activity (CHO-dhfr-) harbouring a luciferase reporter gene under the control of 5 cAMP-responsive elements (CHO-DUKX-CRE-luci cells) were grown to 80% confluency in growth medium (DMEM supplemented with 1× HT, 10% FCS, 200 μg·mL−1 hygromycin) and were transfected with pCIneo-Gpbar1 using Lipofectamine plus (Invitrogen) according to the manufacturer's instructions. Clones were isolated in limited dilution conditions and identified by activities in the cAMP assay. Cells were stimulated for 30 min with 10 μmol·L−1 taurolithocholic acid (TLCA). Stable, clonal cell lines (CHO-DUKX-CRE-luci-hGpbar1, clone 165 and CHO-DUKX-CRE-luci-mGpbar1, clone 184) displaying the greatest induction of cAMP activity were identified as giving consistently good responses for up to at least 20 passages.
Transient transfection
CHO-DUKX cells were transfected using the Cell Line Nucleofector Solution V and an Amaxa Nucleofector II (Lonza, Cologne, Germany) with program A-033 according to the instruction of the manufacturer. 1 × 106 cells were transfected with the indicated amounts of plasmid and 3 × 104 cells were seeded per well in a 96-well plate. Transfections were controlled by using a pmaxGFP (Lonza) plasmid in parallel to each experiment. Cells were grown for 2 days in growth medium at 37°C and 5% CO2 prior to use in the cAMP assay.
Radioligand-binding assay
HEK 293 EBNA cells were transiently transfected with pCIneo-hGPBAR1, harvested 48 h later, followed by homogenization in 15 mmol·L−1 HEPES, 0.3 mmol·L−1 EDTA, 1 mmol·L−1 EGTA, 2 mmol·L−1 MgCl2, complete EDTA-free protease inhibitor (Roche Applied Science, Rotkreuz, Switzerland), pH 7.4 using a glass potter and centrifugation at 47 800 g at 4°C for 30 min. The pellet was then re-homogenized twice in the same buffer and centrifuged (47 800× g, 4°C, 30 min). The final pellet was then resuspended in 75 mmol·L−1 Tris, 0.3 mmol·L−1 EDTA, 1 mmol·L−1 EGTA, 12.5 mmol·L−1 MgCl2, 250 mmol·L−1 sucrose, pH 7.4 at a protein concentration of 1 to 3 mg·mL−1, aliquoted, frozen on dry ice and stored at −80°C. Saturation binding was performed with 0.1 to 100 nmol·L−1 [3H]RO5527239 (Chemistry Department of F. Hoffmann-La Roche AG) and 40 μg of membrane protein. TLCA (100 μmol·L−1) was used to define non-specific binding. About 50% of the total binding signal was specific. Assay buffer consisted of 50 mmol·L−1 Tris-HCl, 5 mmol·L−1 MgCl2, 2.5 mmol·L−1 EDTA and 0.1% ascorbic acid, pH 7.4. Assays were initiated by addition of membranes in a final volume of 250 μL per well. Assays were incubated for 3 h at room temperature (RT) and then vacuum-filtered and rinsed with wash buffer (50 mmol·L−1 Tris-HCl, 5 mmol·L−1 MgCl2, 2.5 mmol·L−1 EDTA and 100 mmol·L−1 NaCl, pH 7.4) on a Filtermate cell harvester (Perkin Elmer, Schwerzenbach, Switzerland) through GF/B filters pre-soaked in 0.3% polyethylenimine.
cAMP assay
For CHO cells, cAMP was measured using cAMP-Nano-TRF detection kit (Roche Applied Science, Mannheim, Germany). CHO-DUKX-CRE-luci-hGpbar1 and CHO-DUKX-CRE-luci-mGpbar1 cells were seeded 17–24 h prior to the experiment 5 × 104 or 3 × 104 cells per well, respectively, in a black 96-well plate with flat clear bottom (Corning, Wiesbaden, Germany) in growth medium and incubated in 5% CO2 at 37°C in a humidified incubator. The growth medium was exchanged with Krebs-Ringer bicarbonate buffer with 1 mmol·L−1 IBMX and incubated at 30°C for 60 min. Agonist was added to a final assay volume of 100 μL and incubated for 30 min at 30°C. The assay was stopped by the addition of 50 μL 3× lysis reagent and shaken for 2 h at RT. The time-resolved energy transfer was measured using a LF502 Nanoscan FLT (IOM, Berlin, Germany), equipped with a laser as excitation source. cAMP content was determined from the function of a standard curve spanning from 10 μmol·L−1 to 0.13 nmol·L−1 cAMP.
STC-1 cells (50 000 cells per well) were seeded 17–24 h prior to the experiment in a Poly D-Lysine-coated 96-well plate (BD Bioscience, Oxford, UK) in growth medium and incubated in 5% CO2 at 37°C in a humidified incubator for 24 h. Cells were washed once with Krebs-Ringer bicarbonate buffer containing 1 mmol·L−1 IBMX, stimulated and incubated herein for 15 min at 37°C. cAMP was measured using the Tropix cAMP-Screen immunoassay system (Applied Biosystems, Foster City, CA, USA) according to the instruction of the manufacturer.
GLP-1 secretion from STC-1 cells
Two days prior to the experiment, 3 × 104 STC-1 cells were seeded in a 96-well plate coated with Poly D-Lysine (BD Bioscience) in growth medium. The following day, growth medium was supplemented with 5 mmol·L−1 butyrate and incubated for 24 h. For the secretion experiment, cells were washed with Krebs-Ringer buffer (NaCl 115 mmol·L−1, KCl 4.7 mmol·L−1, CaCl2 2.56 mmol·L−1, KH2PO4 1.2 mmol·L−1, MgSO4 1.2 mmol·L−1, NaHCO3 20 mmol·L−1, HEPES 16 mmol·L−1) supplemented with 0.1% BSA (fatty acid free). Experiments were performed by incubating the cells with test reagents in the same solution for 2 h at 37°C. GLP-1 secreted into the supernatant was quantified using an immunoassay specific for active GLP-1(7–36; Millipore, St. Charles, MO, USA). Secretion was normalized to secretion measured upon stimulation with 1 μmol·L−1 PMA used as positive control in parallel in the same set of experiments.
Oral glucose tolerance test (oGTT) in humanized GPBAR1-KI and diabetic db/db mice
Efficacy after acute p.o. administration of RO5527239 was assessed in humanized GPBAR1-Knock-in male C57Bl/6 mice (GPBAR1-KI) expressing human GPBAR1 receptor instead of the murine counterpart (Supporting Information Fig. S1), and in male obese diabetic db/db mice [B6.BKS(D)-Leprdb/J]. GPBAR1-KI mice at an age of 12 weeks were randomized in two groups (n = 8 per treatment) and stratified for body weight. The evening before the oGTT, food was reduced to 1 g to have about 12 h of fasting. In the morning, animals were administered either vehicle (Klucel 2% Tween 0.1%, 10 mL·kg−1) or RO5527239 (30 mg·kg−1) via p.o. gavage 1 h before the p.o. glucose bolus (2 g·kg−1). In another study, obese diabetic db/db mice, at an age of 8 weeks, were randomized in four groups (n = 10 per treatment) and stratified for body weight and glucose 4 h after food was removed. Four days later and after 16 h fasting, animals were administered either vehicle or sitagliptin (10 mg·kg−1) via p.o. gavage 1 h before the p.o. glucose bolus, and challenged with vehicle or RO5527239 (30 mg·kg−1) concomitantly with the p.o. glucose bolus (1 g·kg−1). Blood glucose levels were determined at desired time points over a 2 h period using blood (5 μL) collected from tail nick using a handheld glucose meter (Accu-Chek, Roche, Switzerland). Active GLP-1 (7–36) and total GLP-1 (7–36 and 9–36) plasma GLP-1 levels were determined 2 h after glucose challenge. After decapitation, blood was collected in EDTA-coated tubes containing aprotinin (5 μL/100 μL blood; Sigma, Buchs, Switzerland). Plasma samples were obtained via centrifugation at 10 000× g for 20 min. Active and total plasma GLP-1 levels were determined with Luminex technology using Milliplex MapKit active GLP-1 beads (Millipore) and a mouse total GLP-1 assay (Mesoscale, Gaithersburg, MD, USA).
Pharmacokinetic (PK)/pharmacodynamic (PD) study in C57Bl/6 mice
C57Bl/6 male mice (8-week-old; 24–29 g; Charles River Laboratories Germany GmbH) received either one (n = 10, 1–5 h interval) or two consecutive (n = 10, 5–10 h interval) doses of RO5527239 by p.o. gavage (10 mg·kg−1 per dose as a microsuspension in Klucel, 2%/Tween 80, 0.1%). Animals were dosed in fed conditions and deprived of food throughout the study. Terminal blood samples (500 μL, n = 2 per time point) were taken at different time points post-dosing by decapitation and collected in pre-cooled EDTA coated tubes containing 25 μL aprotinin. The samples were kept on ice and immediately centrifuged at 4°C to obtain plasma. Samples were stored at −20°C until analysis. Total PYY levels in plasma were determined by radioimmunoassay (Millipore, St. Charles, MO, USA).
Comparison between systemic and p.o. administration of RO5527239 and tauro-RO5527239 was assessed in male GPBAR1-KI mice at the age of 24 and 32 weeks respectively. Mice were randomized in eight groups (n = 6 per group) and stratified for bodyweight. The evening before compound administration, animals received 1.8 g food each. Compound for p.o. administration was formulated as a microsuspension in equal parts of solutol/labrafil/oleic acid and administered in a volume of 10 mL·kg−1. Intravenous formulations were made as microsuspensions in NMP, 30%/Tris buffer, 70%) suitable for an administration volume of 2 mL·kg−1. 60 min after administration animals were sacrificed by decapitation and blood was collected for plasma total PYY and compound LC-MS/MS analysis (see below) in EDTA coated tubes containing 25 μL aprotinin.
PK/PD study in cynomolgus monkeys (NHP)
Male cynomolgus monkeys (n = 5; 8.7–15.6 kg, ages 6 to 14 years, Hoffmann-La Roche in-house colony) were selected based on their glucose/insulin parameters during an oGTT and categorized as mildly glucose intolerant in an evaluation conducted prior to the study. Subjects received an p.o. bolus of either vehicle or 10 mg·kg−1 of RO5527239 per p.o. gavage (microsuspension in Klucel, 2%/Tween 80, 0.1%) in a randomized crossover design. Animals were fasted for 8 h prior to compound administration and regular food, consisting of 70 g of pellets and vegetables, which was offered after sampling plasma 8 h post dose. Blood samples (1.5 mL) were taken at different time points post-dosing by cephalic vein puncture and collected in pre-cooled tubes containing EDTA and 75 μL aprotinin. The samples were kept on ice and immediately centrifuged at 4°C to obtain plasma. Samples were stored at −20°C until analysis. Total PYY levels in plasma were determined by radioimmunoassay (Millipore).
LC-MS/MS compound analysis
Quantification of compound levels in plasma was accomplished by means of LC-MS/MS analysis. Plasma (50 μL) was precipitated in three volumes of acetonitrile containing d6-midazolam (0.2 μg·mL−1) as internal standard, mixed and centrifuged (10 min, 4°C, 5850× g). The supernatant was diluted 20-fold with water containing 0.2% formic acid (v/v) and 1.0 μL was injected onto a Supelco Ascentis Express C18 2 cm × 2.1 mm × 2.7 μm operating with mobile phases A (0.2% formic acid in water containing 5% acetonitrile) and B (acetonitrile) at 0.45 mL·min−1 flow. The outlet of the column was coupled to AB Sciex QTRAP5500 mass spectrometer (AB Sciex, Brugg, Switzerland) with TurboIonSpray source. Detection was carried out using multiple reactions monitoring mode with positive ion detection focusing in the transition 458.1/308 for RO5527239, 565.1/415.1 for tauro-RO5527239 and 331.1/296 for d6-midazolam.
Statistical analysis
In vivo data were analysed by anova (alpha 0.05) followed by Dunnett's test, comparison versus vehicle (SAS/JMP software, SAS institute Inc., SAS Campus Drive, Cary, NC, USA). Student's t-test was applied for other comparisons. The dose-response curves in Figures 2 and 3 were created using a logistic equation y = Bottom + [(Top-Bottom)/(1 + ((LogEC50/x)∧nH))] using XLfit software (ID Business Solutions, Guildford, UK). All data are expressed as mean ± SEM.
Figure 2.
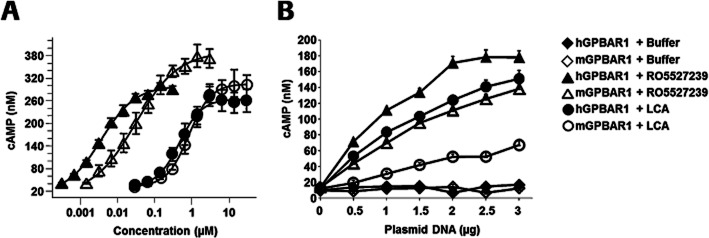
Agonist potency and efficacy of RO5527239 in comparison to LCA in recombinant CHO cells. (A) Intracellular cyclic AMP levels in the human (full symbols) and mouse (open symbols) CHO GPBAR1-expressing stable cell lines treated for 30 min with RO5527239 (triangle) and LCA (circle). Data are the means from three independent experiments, each of which is the mean of three samples. (B) Gene-dosing experiments with human (full symbols) and mouse (open symbols) GPBAR1 receptors in transiently transfected CHO cells and treated for 30 min with 1 μmol·L−1 RO5527239 (triangle) and 10 μmol·L−1 LCA (circle) or buffer (diamonds). Data represent mean cAMP levels ± SEM of three independent experiments made in triplicate.
Figure 3.
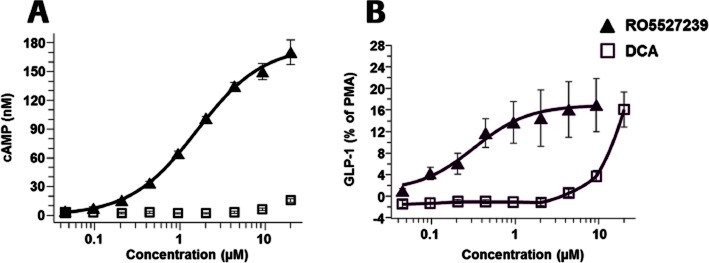
Agonist potency and efficacy of RO5527239 in comparison to DCA in STC-1 cells. (A) Effect of GPBAR1 agonists RO5527239 and the BA DCA on intracellular cAMP accumulation. STC-1 cells were stimulated with the indicated reagents and incubated for 15 min at 37°C in the presence of IBMX (1 mmol·L−1). (B) GPBAR1 activation promotes GLP-1 secretion from STC-1 cells in response to RO5527239 along with the BA DCA. STC-1 cells were incubated for 2 h at 37°C in Krebs-Ringer buffer containing various concentrations of RO5527239 and DCA. After incubation, the conditioned medium was collected and the concentration of GLP-1 was determined by enzyme immunoassay. Data represent the mean ± SEM of three independent experiments made in triplicate.
Results
Identification of RO5527239 and in vitro pharmacological characterization
A stable CHO-K1-derived cell line that expresses human GPBAR1 was generated and used to conduct a high-throughput screening campaign, leading to the identification of a number of small-molecule GPBAR1 agonists. In the course of a medicinal chemistry program, an iterative series of structural modifications within the chemical class of 1-hydroxyimino-3-phenyl-propanes were generated, leading to the discovery of RO5527239 ((R,E)-1-(4-(3-(hydroxyimino)-3-(2-methylpyridin-4-yl)-1-o-tolylpropyl)phenyl)piperidine-4-carboxylic acid) and the synthesis of its taurine conjugate (Figure 1A).
Figure 1.
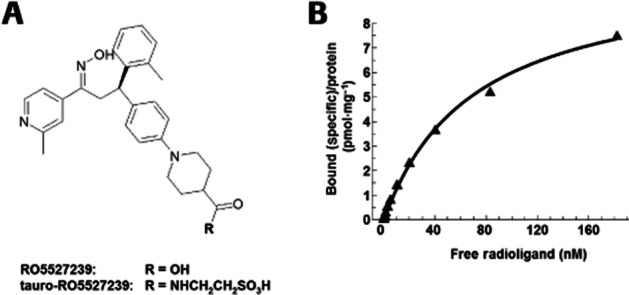
Structure of RO5527239 and binding properties to GPBAR1 receptors. (A) Chemical structure of the selective GPBAR1 agonists RO5527239 and tauro-RO5527239. (B) Saturation binding at human GPBAR1 shows high-affinity binding to membranes prepared from CHO-DUKX-CRE-luci-hGpbar1 cells and reveals a single high-affinity and saturable binding site with a Kd value of 70.4 nmol·L−1 and a Bmax value of 10.2 pmol·mg−1 protein. The binding site is in common with the BA LCA, which when used in excess displaced bound RO5527239. Data are the mean from two independent experiments, each of which is the mean of three samples.
In a saturation-binding experiment using membranes prepared from human GPBAR1-expressing CHO cells, [3H]RO5527239 competitively binds to GPBAR1 receptors in a mode similar to the endogenous ligand TLCA, which was used to displace the radioligand (Figure 1B). RO5527239 exhibits an affinity (Kd) of 70.4 nM at human GPBAR1 receptors. Determinations of cAMP accumulation were used as a measure of signalling through the Gs, adenylate cyclase pathway. In comparison to lithocholic acid (LCA), RO5527239 exhibits 127-fold higher potency at human GPBAR1 (EC50 = 3.6 vs. 457 nM) and 28-fold higher potency at mouse GPBAR1 (EC50 = 30 vs. 832 nM) in stably expressing CHO cells (Figure 2A). Maximal cAMP levels reached by RO5527239 stimulation were in a range similar (110%) to that achieved by LCA stimulation at human GPBAR1 receptors, and slightly higher than at mouse GPBAR1 (128%).
Gene-dosage experiments were performed to evaluate further RO5527239's pharmacological properties and mechanism of action at human and mouse GPBAR1 receptors. At maximal concentration, RO5527239 (1 μM) and LCA (10 μM) triggered a gene dosage-dependent increase in cAMP accumulation in cells expressing GPBAR1 receptors as opposed to non-transfected cells suggesting GPBAR1-specific actions. For the human GPBAR1 receptor, the efficacy upon RO5527239 and LCA stimulation appears to reach saturation at the same level, which is in agreement with Figure 2A dealing with stable expressing CHO cells and presumably higher expression levels compared to those achieved by transient transfections. For the mouse GPBAR1 receptor, RO5527239 exhibits higher efficacy than LCA, which is in agreement with the slightly higher efficacy of RO527239 as obtained with stable expressing CHO cells. Thus, in mice, RO5527239 might be more efficacious than the endogenous ligand LCA under conditions of low expression levels. Furthermore, the unchanged basal activity in non-stimulated cells independent of gene dosage suggests a lack of constitutive activity of both human and mouse GPBAR1 receptors (Figure 2B).
RO5527239 is highly selective for GPBAR1, as evaluated from radioligand binding and functional assays (CEREP, Celle l'Evescault, France; http://www.cerep.fr), which evaluated 97 target proteins (Supporting Information Table S1). The only activity at 10 μM observed (>50% inhibition) was glucocorticoid receptor binding, which was followed up and proved to be inactive in a functional transactivation assay (Supporting Information Table S1; Discoverx, Fremont, CA, USA). In addition, no activities were observed in a functional DPP4 activity assay (Supporting Information Table S1; as performed according to Lubbers et al., 2007) and cellular transactivation assays for FXR, LXRα, LXRβ, PPARα, PPARγ, PPARδ and RXRα (data not shown).
In accordance with several previous studies (Maruyama et al., 2002; Kawamata et al., 2003; Katsuma et al., 2005; Thomas et al., 2009), activation of GPBAR1 by RO5527239 and the BA DCA triggered a dose-dependent increase in intracellular cAMP levels in enteroendocrine STC-1 cells (Figure 3A). The compound RO5527239 displayed a potency (EC50) of 1.59 μM, which is 50-fold lower than that obtained from activation of recombinant mouse GPBAR1 receptors. The BA, DCA, displayed an EC50 of greater than 10 μM and a striking 14-fold lower efficacy compared to RO5527239 at 30 μM. The role of BA in inducing GLP-1 secretion through GPBAR1 by increases of intracellular cAMP concentrations was first demonstrated by Maruyama et al. (Maruyama et al., 2002) and confirmed by Thomas et al. (Thomas et al., 2009) in the same STC-1 cells. Accordingly, treatment with RO5527239 promotes a dose-dependent secretion of GLP-1 (EC50 321 nM), which is 33-fold lower compared to DCA (EC50 11 μM), reaching, however, the same efficacy (Figure 3B). This suggests that lower cAMP levels than those achieved by DCA are sufficient to trigger GLP-1 secretion in STC-1 cells. In summary, compared to BA, RO5527239 is a highly potent and efficacious GPBAR1 agonist capable of promoting a strong GLP-1 secretagogue effect in EECs.
RO5527239 improves glucose tolerance and promotes glucose-independent GLP-1 release
In the course of the high-throughput in vitro testing campaign, the majority of identified GPBAR1 agonists exhibited high potencies at human GPBAR1 compared to low or no activity at rodent GPBAR1 receptors. Therefore a humanized knock-in mouse (GPBAR1-KI) was generated that carries the human, instead of the mouse GPBAR1 gene (Supporting Information Fig. S1) and expressed GPBAR1 equivalently in the gall bladder, ileum, colon, kidney and spleen (Supporting Information Fig. S2). This GPBAR1-KI mouse was used for the initial assessment of GPBAR1 activation on glucose tolerance. RO5527239 exhibited good p.o. bioavailability in C57Bl/6 mice (Supporting Information Table S2), attaining micromolar plasma levels for more than 2 h at the doses used here. RO5527239 (30 mg·kg−1, orally) markedly improved p.o. glucose tolerance (Figure 4A). Based on the AUC, RO5527239 inhibited the glucose excursion by approximately 37%.
Figure 4.

RO5527239 improves glucose tolerance in mice and promotes GLP-1 secretion. (A) Oral glucose tolerance test (2 g·kg−1 glucose) in male 12-week-old GPBAR1-KI mice treated with vehicle (▵), or RO5527239 (30 mg·kg−1; ▴). Animals were first dosed with compound or vehicle 60 min before glucose bolus. Data represent the mean ± SEM. The inset represents the average delta AUC during 2 h (mean ± SD) (n = 8 per group). (B) Corresponding plasma total GLP-1 levels in GPBAR1-KI mice during an p.o. glucose challenge, 60 min after the p.o. administration of saline or RO5527239 (30 mg·kg−1). Data represent the mean ± SEM. (n = 8 per group). (C) Oral glucose tolerance test in male 8-week-old db/db mice using 1 g·kg−1 glucose treated concomitantly with vehicle (▵), RO5527239 (30 mg·kg−1; ▴), or preceded 60 min before by p.o. administration of sitagliptin (10 mg·kg−1; ○) alone, and in combination with RO5527239 (30 mg·kg−1; ♦). Data represent the mean ± SEM. (n = 8 per group). (D) Corresponding active GLP-1 plasma levels and (E) total GLP-1 plasma levels 2 h after glucose challenge. Data represent the mean ± SEM (n = 5 per group). Statistically significant: *P < 0.05; **P < 0.01, ***P < 0.001. Unpaired t-test (Figure 4A), anova followed by Dunnett's test (Figure 4C–E).
To determine whether the improved glucose tolerance was due to enhanced incretin release, we measured circulating total GLP-1 levels. Glucose administered alone did not increase total GLP-1 levels under these experimental conditions (Figure 4B), which would have been expected to occur within the first 30 min after the glucose challenge. Mice pretreated with RO5527239 had markedly elevated GLP-1 levels even before the glucose challenge (Figure 4B), suggesting that GPBAR1 activation promotes GLP-1 secretion in a glucose-independent manner. Furthermore, GLP-1 levels were sustained for more than 3 h after administration of RO5527239. Plasma levels of RO5527239 at the 3 h time point amounted to 8333 ng·mL−1. Taking into account 98% protein binding, free RO5527239 plasma levels are still 95 times above the human EC50 of 3.8 nM as measured in a recombinant cell line, or fit exactly to the EC50 as measured by GLP-1 release from the mouse STC-1 cell line. Therefore, the observed sustained secretion effect might to be due to sufficiently high RO5527239 levels in plasma. We then examined the effects of RO5527239 on glucose homeostasis in db/db mice, a rodent model of type 2 diabetes. RO5527239 significantly improved p.o. glucose tolerance in db/db mice (Figure 4C). The efficacy of RO5527239 (30 mg·kg−1) in reducing glucose excursion during the p.o. glucose tolerance test was comparable to that of the dipeptidyl peptidase 4 inhibitor (DPP4i), sitagliptin (10 mg·kg−1). A combination of RO5527239 with sitagliptin appears to further improve glucose tolerance, suggesting that GLP-1 that has been released upon GPBAR1 activation might have been stabilized by the DPP4i (Figure 4C). To prove this, active GLP-1 (7–36) levels were determined in plasma 1 h after glucose challenge and drug administration. As expected, RO5527239 significantly increased active GLP-1 levels in db/db mice, which were 7.9-fold higher compared to glucose-induced GLP-1 levels. Co-administration of DPP4i resulted in even more pronounced increases in active GLP-1 levels [9.5-fold increase vs glucose-induced GLP-1 (7–36) levels] as a consequence of its ability to prolong the half-life of plasma GLP-1 (7–36) (Figure 4D). The GLP-1 secretagogue effect upon GPBAR1 activation was further investigated by the determination of total plasma GLP-1 levels, constituting GLP-1 (7–36) and the DPP4-processed product GLP-1 (9–36). As expected glucose-induced total GLP-1 levels were identical during oGTT in vehicle- and DPP4i-treated mice (Figure 4E). RO5527239, however, triggered an eightfold increase in GLP-1 secretion compared to the vehicle during oGTT (Figure 4E). Interestingly, combination treatment of RO5527239 with DPP4i partially inhibited the effect by RO5527239 alone (P < 0.001). This is in agreement with previous data showing an inhibitory effect of DPP4 inhibition on GLP-1 release in dogs (Deacon et al., 2002) and humans (Bock et al., 2010). Taken together, these data demonstrate that RO5527239 acts by promoting GLP-1 release from intestinal L-cells, which subsequently results in enhanced glucose disposal.
Systemic RO5527239 levels promote L-cell secretion in mice
GLP-1 and PYY are secreted from human enteroendocrine L-cells in response to numerous stimuli, including bile salts (Adrian et al., 1993). Similarly, RO5527239 when administered orally to C57Bl/6 mice at a dose of 10 mg·kg−1 led to secretion of both PYY and GLP-1. The PK profile of the GPBAR1 agonist was found to peak at the same time as the PD response for both GLP-1 and PYY, and the overall PK/PD behaviour was consistent with a direct response-type mechanism (Figure 5A). The amounts of GLP-1 and PYY in the plasma directly correlate (R2 = 0.72), which indicates secretion of both peptides from enteroendocrine L-cells (Figure 5B). These data further confirm that GPBAR1 activation promotes peptide secretion from L-cells in a glucose-independent manner as first observed in GPBAR-KI mice expressing human GPBAR1 receptors (Figure 4B). Furthermore, the close correlation of the PD effect with the plasma PK profile suggests that GPBAR1 activation triggers L-cell secretion through systemic compound exposure, rather than through activation within the intestinal lumen.
Figure 5.
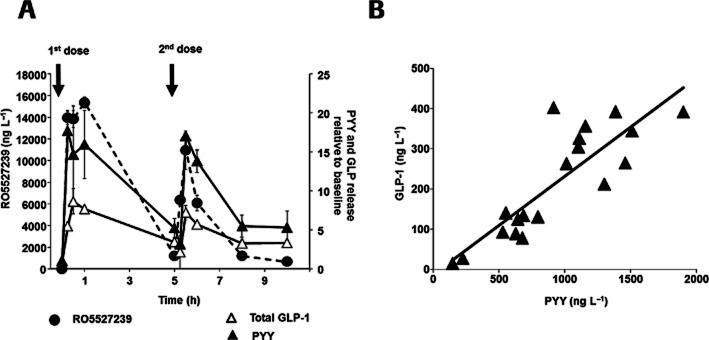
RO5527239 promotes GLP-1 and PYY secretion in mice. (A) Oral administration and repeated dosing 5 h later of RO5527239 (10 mg·kg−1) to male 8-week-old C57Bl/6 mice and corresponding plasma levels of RO5527239 (•), total GLP-1 (▵) and PYY (▴) expressed relative to baseline (t = 0). Data represent the mean ± SEM (n = 10 per group). (B) Correlation of mean plasma total GLP-1 and PYY levels determined from identical time points.
Peptide secretion is dependent on systemic and not intestinal luminal GPBAR1 activation
In order to confirm further peptide secretion by activation of systemically accessible GPBAR1 receptors, GPBAR1 agonists were tested comparing p.o. and i.v. bolus applications. Taurine conjugation of RO5527239, referred to as tauro-RO5527239 (Figure 1A), rendered the molecule poorly orally bioavailable, but retained its high potency at mouse (EC50: 19 ± 8 nM), and human (EC50: 1 ± 0.1 nM) GPBAR1 as determined in the cAMP assay. Administration of 10 mg·kg−1 of RO5527239 to GPBAR1-KI mice by either p.o. gavage or i.v. bolus yielded a significant and comparable PYY elevation irrespective of the route of administration (Figure 6A). RO5527239 plasma levels in the p.o. and i.v. groups were comparable, confirming systemic exposure as the major driver for the PD effect (Figure 6B). Similarly, tauro-RO5527239 was administered at a dose of 10 mg·kg−1 by either p.o. gavage or i.v. bolus and showed robust PYY elevation in the intravenously treated group but not in the orally treated group (Figure 6C). Plasma exposure of tauro-RO5527239 and the differences in the PD effect observed for the two routes of administration are in line with the required plasma exposure for PYY secretion (Figure 6D).
Figure 6.

Peptide secretion in mouse is dependent on systemic and not intestinal luminal GPBAR1 agonist levels. (A) Plasma PYY levels determined 1 h after administration of RO5527239 (10 mg·kg−1) by either p.o. gavage or i.v. bolus in male 24-week-old GPBAR1-KI mice. (B) RO5527239 plasma levels in the p.o. and i.v. groups. (C) Plasma PYY levels determined 1 h after administration of tauro-RO5527239 (10 mg·kg−1) by either p.o. gavage or i.v. bolus in male 32-week-old GPBAR1-KI mice. (D) tauro-RO5527239 plasma levels in the p.o. and i.v. groups. All data represent the mean ± SEM (n = 6 per group).
RO5527239 promotes L-cell secretion in non-human primates
A randomized crossover study in cynomolgus monkeys was performed in order to investigate the translational potential of GPBAR1-mediated peptide secretion from L-cells. Oral administration of RO5527239 to cynomolgus monkeys at a dose of 10 mg·kg−1 produced a significant, and compared to mice, a much more sustained elevation of circulating PYY levels (Figure 7A). RO5527239 and PYY exposure response profiles in the treated group did not run in parallel suggesting a slower and more sustained PYY secretion in monkeys as compared to mouse (Figure 7B). The PYY response starts with the first time point and peaks at a later time compared to plasma GPBAR1 agonist levels, consistent with an indirect response-type mechanism. The decline and reappearance of RO5527239 in the plasma is considered to be due to enterohepatic recycling of the compound.
Figure 7.
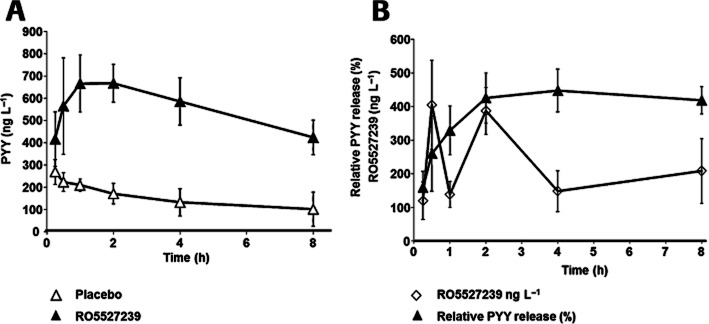
RO5527239 promotes PYY secretion in non-human primates. (A) Time course of plasma PYY levels in 6–14-year-old cynomolgus monkeys after single p.o. administration of RO5527239 (10 mg·kg−1; ▴) compared to the corresponding placebo (▵). Study was set up as a randomized crossover design. (B) Time course of relative plasma PYY levels expressed in percent to placebo (▴) and corresponding plasma levels of RO5527239 (◇). All data represent the mean ± SEM (n = 5 per group).
Discussion
In the present study, we provide the first evidence that systemic GPBAR1 activation is a major BA-mediated pathway to promote GLP-1 and PYY secretion from intestinal L-cells in rodents and non-human primates using a pharmacological approach. This involved the generation as well as in vitro and in vivo characterization of a novel, selective, orally bioavailable, and highly potent GPBAR1 agonist, RO5527239, and its less orally bioavailable taurine conjugate.
In vitro, RO5527239 exhibits high-affinity and saturable binding to a GPBAR1 site that overlaps with BA binding, and potently stimulates cAMP production, by activating both murine and human GPBAR1. Compared to LCA, the potency was 139-fold and 26-fold higher at human and mouse GPBAR1 receptors respectively. Compared to BA, the efficacy was equal at human GPBAR1 receptors but higher at the mouse GPBAR1 receptor. The high selectivity and bioavailability make RO5527239 a suitable tool for further exploring GPBAR1 functions in vivo. Acute treatment of mice with RO5527239 showed that selective stimulation of GPBAR1 improves glucose tolerance and promotes strong and sustained secretion of GLP-1. The anti-hyperglycaemic effect was confirmed in diabetic db/db mice and co-treatment with a DPP4 inhibitor suggested that GPBAR1-mediated GLP-1 secretion contributed to the additive anti-hyperglycaemic effect. These results are in line with a genetic approach showing elevated and blunted in vivo GLP-1 responses to BA in GPBAR-overexpressing and -knockout mice, respectively (Thomas et al., 2009), and with pharmacological approaches using GPBAR1 agonists with unrelated chemical structures (Thomas et al., 2009; Duan et al., 2012). With our pharmacological tools, RO5527239 and tauro-RO5527239, we addressed the contribution of luminal exposure in the gut and systemic exposure of the GPBAR1 agonist to induce the secretory effects in intestinal L-cells.
Previous data suggested a major contribution of direct luminal GPBAR1 activation on L-cells to stimulate secretion. Transcriptional analysis of primary L-cells revealed the presence of several GPCRs that are responsive to regulators such as the acylethanolamine receptor GPR119, the long-chain fatty acid receptors GPR40 and GPR120, the BA receptor GPBAR1 (Reimann et al., 2008) and the short-chain fatty acid receptor GPR41 (Samuel et al., 2008). In line with our data, GPBAR1 has been shown to be functionally expressed in immortalized EEC lines such as mouse STC-1 (Katsuma et al., 2005; Thomas et al., 2009), human NCI-H716 cells (Maruyama et al., 2002) and mouse GLUTag cells (Parker et al., 2012), where GPBAR1 activation was shown to involve rapid elevation of cAMP, and to enhance calcium and secretory responses to glucose (Parker et al., 2012). The secretory response upon GPBAR1 activation by BA or a synthetic GPBAR1 agonist could be recapitulated in primary murine intestinal culture (Parker et al., 2012). Ex vivo, this direct mechanism was initially investigated using isolated gut preparations by intraluminal perfusion of nutrients and BA that triggered the secretion of GLP-1 and PYY (Plaisancie et al., 1996). Using colonic explants from GPBAR1 knockout mice, the BA-induced GLP-1 release was confirmed to be mediated by GPBAR1 (Harach et al., 2012). In humans, intracolonic infusions of DCA, a major constituent of human bile, dose dependently promoted increases in plasma PYY and enteroglucagon concentrations in patients undergoing colonoscopy (Adrian et al., 1993; 2012) suggesting that GPBAR1-mediated stimulation of L-cells through the gut lumen translates to humans.
In contrast, our data suggest that intestinal L-cells can also be stimulated by BA through a mechanism other than through direct stimulation in the gut lumen, which we suggest contributes to the immediate phase of L-cell activation upon food intake. Firstly, plasma GLP-1 and PYY levels show a dynamic response concordant with the systemic GPBAR1 agonist kinetics. In fact, if the activation occurred in the gut lumen, the PD response would be expected to be linked to the intestinal transit kinetics. Secondly, application of an orally bioavailable GPBAR1 agonist produces identical secretion effects when applied either intravenously or per os. Thirdly, a GPBAR1 agonist with poor p.o. bioavailability was effective in promoting secretion from L-cells only when administered intravenously and not when given orally. Interestingly, the correlation of plasma GLP-1 and PYY levels indicates that in spite of the evident systemic activation, the PD response ultimately involves L-cell activation. We were further interested in addressing the translation of the mechanism found in rodent to non-human primates as a closer model to human. The treatment of mildly glucose-intolerant cynomolgus monkeys with a GPBAR1 agonist led to a clear and sustained increase in PYY plasma levels. In contrast to the mouse, the monkey PD response continued to develop after reaching the PK peak and elevated PYY levels were maintained even after compound washout. This pattern is characteristic of an indirect response, possibly driven by a stimulation of the peptide release and eventually the rate of peptide production.
Around 95% of BA are reabsorbed from the gut in the terminal ileum and are first-pass extracted by the liver. Although the uptake of BA in the liver is fairly efficient, a consistent fraction (10–30%) reaches the systemic circulation (Angelin et al., 1982). Therefore, BA levels spill over into the systemic circulation in the postprandial state, reaching levels of about 5–15 μmol·L−1 that are capable of activating GPBAR1. In line with our results, systemic increases of BA after a test meal were reported to correlate with GLP-1 and PYY levels (Roberts et al., 2011).
BA-mediated L-cell secretion may be relevant for the regulation of glucose homeostasis in humans. Postprandial BA responses are decreased in obese subjects, partly explaining the impaired GLP-1 and PYY responses (Glicksman et al., 2010) and they also correlate with increased insulin sensitivity in humans (Shaham et al., 2008). Furthermore, patients after bariatric surgery have higher systemic BA levels, which are positively correlated to peak GLP-1 levels (Patti et al., 2009).
The present data suggest that basolateral or indirect rather than luminal GPBAR1 activation is important for the secretagogue effect of BA. The kinetics of compound or BA absorption is in line with the in vivo setting in rats, where plasma levels of PYY peak despite the fact that nutrients and BA do not reach the distal ileum or colon (Fu-Cheng et al., 1997). Interestingly, similar GLP-1 kinetics were observed after a test meal in mice treated with an AER, that are used to sequester BA to L-cell-rich distal parts of the intestine. In this recent study, it was demonstrated by the use of GPBAR1 knockout mice that acute AER-induced GLP-1 secretion is mediated by GPBAR1 (Harach et al., 2012). This finding is in line with GLP-1-enhancing effects by AER in humans (Suzuki et al., 2007; Shang et al., 2010; Garg et al., 2011). It remains to be seen if, due to the impaired enterohepatic circulation, the postprandial systemic BA levels are increased upon AER treatment.
Several studies have demonstrated that nutrient-evoked cues in the duodenum are transmitted via the afferent vagus nerve to the CNS, in turn stimulating the vagal efferents to the distal gut (Rocca and Brubaker, 1999). In view of the expression of GPBAR1 in mesenteric neurons (Poole et al., 2010), BA from the circulation may activate a neuronal GPBAR1 and be part of the proximal-distal loop. In fact, GPBAR1 is colocalized with neurofilament M immunoreactivity (NFM-IR), which has been characterized as a marker of intrinsic primary afferent neurons in the mouse small intestine that innervate the mucosa and other neurons within the enteric circuitry (Furness et al., 2004).
In conclusion, our data provide the first evidence that systemic GPBAR1 agonists promote secretion by L-cells at the early phase by either direct stimulation of L-cells at the basolateral side, or indirectly which might involve neuronal expressed GPBAR1. However, systemic exposures by GPBAR1 agonists will inherently be linked to GPBAR1-mediated side effects such as gall bladder filling (Lavoie et al., 2010; Li et al., 2011; Duan et al., 2012) or eventually pancreatitis (Perides et al., 2010) and gastric adenocarcinomas (Cao et al., 2013). In fact, we can confirm the acute relaxing effect of the mouse gallbladder with RO5527239 (data not shown). A strategy to escape these side effects has been recently proposed by developing non-bioavailable GPBAR1 agonists (Duan et al., 2012). It remains to be shown if such chemical entities exhibit sufficient anti-diabetic effects given that our results show lack of acute secretion effects on L-cells. RO5527239 and tauro-RO5527239 emerged as highly efficacious GPBAR1 agonists for the study of GPBAR1 functions in rodents and non-human primates and their possible roles for the treatment of diabetes.
Acknowledgments
We are grateful to Doris Brugger, Christophe Flament, Vera Griesser, Tanja Minz, Anja Osterwald, Jean-Pierre Parys, Wolfgang Rapp, Angelina Wallier and Elisabeth Zirwes for their excellent technical assistance and Anish Konkar and Desmond Fitzgerald for critically reading the manuscript.
Glossary
- AER
anionic exchange resins
- BA
bile acids
- DCA
deoxycholic acid
- DPP4
dipeptidyl peptidase 4
- GLP-1
glucagon-like peptide 1
- LCA
lithocholic acid
- PMA
phorbol 12-myristate 13-acetate
- PYY
peptide tyrosine-tyrosine
- RT
room temperature
Conflicts of interest
All the authors are employees of F. Hoffmann-La Roche Ltd.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Targeting strategy of GPBAR1-KI mice.
Figure S2 GPBAR1 expression in GPBAR1-KI mice compared to wild-type littermates.
Table S1 Binding to receptors, ion channels, transporters, enzymes and cell based assays.
Table S2 Pharmacokinetic parameters for RO5527239 after single dose in C57BL/6J mice.
Table S3 Oligonucleotides used for qRT-PCR.
References
- Adrian TE, Ballantyne GH, Longo WE, Bilchik AJ, Graham S, Basson MD, et al. Deoxycholate is an important releaser of peptide YY and enteroglucagon from the human colon. Gut. 1993;34:1219–1224. doi: 10.1136/gut.34.9.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrian TE, Gariballa S, Parekh KA, Thomas SA, Saadi H, Al Kaabi J, et al. Rectal taurocholate increases L cell and insulin secretion, and decreases blood glucose and food intake in obese type 2 diabetic volunteers. Diabetologia. 2012;55:2343–2347. doi: 10.1007/s00125-012-2593-2. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelin B, Bjorkhem I, Einarsson K, Ewerth S. Hepatic uptake of bile acids in man. Fasting and postprandial concentrations of individual bile acids in portal venous and systemic blood serum. J Clin Invest. 1982;70:724–731. doi: 10.1172/JCI110668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature. 2002;418:650–654. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- Bock G, Dalla Man C, Micheletto F, Basu R, Giesler PD, Laugen J, et al. The effect of DPP-4 inhibition with sitagliptin on incretin secretion and on fasting and postprandial glucose turnover in subjects with impaired fasting glucose. Clin Endocrinol (Oxf) 2010;73:189–196. doi: 10.1111/j.1365-2265.2009.03764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Tian W, Hong J, Li D, Tavares R, Noble L, et al. Expression of bile acid receptor TGR5 in gastric adenocarcinoma. Am J Physiol Gastrointest Liver Physiol. 2013;304:G322–G327. doi: 10.1152/ajpgi.00263.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, McNulty J, Anderson D, Liu Y, Nystrom C, Bullard S, et al. Cholestyramine reverses hyperglycemia and enhances glucose-stimulated glucagon-like peptide 1 release in Zucker diabetic fatty rats. J Pharmacol Exp Ther. 2010;334:164–170. doi: 10.1124/jpet.110.166892. [DOI] [PubMed] [Google Scholar]
- Cipriani S, Mencarelli A, Bruno A, Renga B, Distrutti E, Santucci L, et al. GPBAR1 activation protects against gastrointestinal injury caused by non-steroidal anti-inflammatory drugs and ASA in mice. Br J Pharmacol. 2013;168:225–237. doi: 10.1111/j.1476-5381.2012.02128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon CF, Wamberg S, Bie P, Hughes TE, Holst JJ. Preservation of active incretin hormones by inhibition of dipeptidyl peptidase IV suppresses meal-induced incretin secretion in dogs. J Endocrinol. 2002;172:355–362. doi: 10.1677/joe.0.1720355. [DOI] [PubMed] [Google Scholar]
- Drucker DJ. Minireview: the glucagon-like peptides. Endocrinology. 2001;142:521–527. doi: 10.1210/endo.142.2.7983. [DOI] [PubMed] [Google Scholar]
- Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- Duan H, Ning M, Chen X, Zou Q, Zhang L, Feng Y, et al. Design, Synthesis, and antidiabetic activity of 4-phenoxynicotinamide and 4-phenoxypyrimidine-5-carboxamide derivatives as potent and orally efficacious TGR5 agonists. J Med Chem. 2012;55:10475–10489. doi: 10.1021/jm301071h. [DOI] [PubMed] [Google Scholar]
- Field BC, Wren AM, Peters V, Baynes KC, Martin NM, Patterson M, et al. PYY3-36 and oxyntomodulin can be additive in their effect on food intake in overweight and obese humans. Diabetes. 2010;59:1635–1639. doi: 10.2337/db09-1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu-Cheng X, Anini Y, Chariot J, Castex N, Galmiche JP, Roze C. Mechanisms of peptide YY release induced by an intraduodenal meal in rats: neural regulation by proximal gut. Pflugers Arch. 1997;433:571–579. doi: 10.1007/s004240050316. [DOI] [PubMed] [Google Scholar]
- Furness JB, Jones C, Nurgali K, Clerc N. Intrinsic primary afferent neurons and nerve circuits within the intestine. Prog Neurobiol. 2004;72:143–164. doi: 10.1016/j.pneurobio.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Garg SK, Ritchie PJ, Moser EG, Snell-Bergeon JK, Freson BJ, Hazenfield RM. Effects of colesevelam on LDL-C, A1c and GLP-1 levels in patients with type 1 diabetes: a pilot randomized double-blind trial. Diabetes Obes Metab. 2011;13:137–143. doi: 10.1111/j.1463-1326.2010.01320.x. [DOI] [PubMed] [Google Scholar]
- Glicksman C, Pournaras DJ, Wright M, Roberts R, Mahon D, Welbourn R, et al. Postprandial plasma bile acid responses in normal weight and obese subjects. Ann Clin Biochem. 2010;47(Pt 5):482–484. doi: 10.1258/acb.2010.010040. [DOI] [PubMed] [Google Scholar]
- Harach T, Pols TW, Nomura M, Maida A, Watanabe M, Auwerx J, et al. TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci Rep. 2012;2:430. doi: 10.1038/srep00430. doi: 10.1038/srep00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. 2005;329:386–390. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- Keitel V, Reinehr R, Gatsios P, Rupprecht C, Gorg B, Selbach O, et al. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45:695–704. doi: 10.1002/hep.21458. [DOI] [PubMed] [Google Scholar]
- Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. 2008;372:78–84. doi: 10.1016/j.bbrc.2008.04.171. [DOI] [PubMed] [Google Scholar]
- Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Haussinger D. The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology. 2009;50:861–870. doi: 10.1002/hep.23032. [DOI] [PubMed] [Google Scholar]
- Keitel V, Ullmer C, Haussinger D. The membrane-bound bile acid receptor TGR5 (Gpbar-1) is localized in the primary cilium of cholangiocytes. Biol Chem. 2010;391:785–789. doi: 10.1515/BC.2010.077. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski JR, Hubert J, Carrington PE, Chicchi GG, Mu J, Miller C, et al. The glucagon receptor is involved in mediating the body weight-lowering effects of oxyntomodulin. Obesity (Silver Spring) 2012;20:1566–1571. doi: 10.1038/oby.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie B, Balemba OB, Godfrey C, Watson CA, Vassileva G, Corvera CU, et al. Hydrophobic bile salts inhibit gallbladder smooth muscle function via stimulation of GPBAR1 receptors and activation of KATP channels. J Physiol. 2010;588(Pt 17):3295–3305. doi: 10.1113/jphysiol.2010.192146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Holmstrom SR, Kir S, Umetani M, Schmidt DR, Kliewer SA, et al. The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol Endocrinol. 2011;25:1066–1071. doi: 10.1210/me.2010-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubbers T, Bohringer M, Gobbi L, Hennig M, Hunziker D, Kuhn B, et al. 1,3-disubstituted 4-aminopiperidines as useful tools in the optimization of the 2-aminobenzo[a]quinolizine dipeptidyl peptidase IV inhibitors. Bioorg Med Chem Lett. 2007;17:2966–2970. doi: 10.1016/j.bmcl.2007.03.072. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, et al. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]
- Parker HE, Wallis K, le Roux CW, Wong KY, Reimann F, Gribble FM. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br J Pharmacol. 2012;165:414–423. doi: 10.1111/j.1476-5381.2011.01561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Houten SM, Bianco AC, Bernier R, Larsen PR, Holst JJ, et al. Serum bile acids are higher in humans with prior gastric bypass: potential contribution to improved glucose and lipid metabolism. Obesity (Silver Spring) 2009;17:1671–1677. doi: 10.1038/oby.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perides G, Laukkarinen JM, Vassileva G, Steer ML. Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology. 2010;138:715–725. doi: 10.1053/j.gastro.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisancie P, Dumoulin V, Chayvialle JA, Cuber JC. Luminal peptide YY-releasing factors in the isolated vascularly perfused rat colon. J Endocrinol. 1996;151:421–429. doi: 10.1677/joe.0.1510421. [DOI] [PubMed] [Google Scholar]
- Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14:747–757. doi: 10.1016/j.cmet.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole DP, Godfrey C, Cattaruzza F, Cottrell GS, Kirkland JG, Pelayo JC, et al. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil. 2010;22:814–825. doi: 10.1111/j.1365-2982.2010.01487.x. e227–e818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F, Habib AM, Tolhurst G, Parker HE, Rogers GJ, Gribble FM. Glucose sensing in L cells: a primary cell study. Cell Metab. 2008;8:532–539. doi: 10.1016/j.cmet.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RE, Glicksman C, Alaghband-Zadeh J, Sherwood RA, Akuji N, le Roux CW. The relationship between postprandial bile acid concentration, GLP-1, PYY and ghrelin. Clin Endocrinol (Oxf) 2011;74:67–72. doi: 10.1111/j.1365-2265.2010.03886.x. [DOI] [PubMed] [Google Scholar]
- Rocca AS, Brubaker PL. Role of the vagus nerve in mediating proximal nutrient-induced glucagon-like peptide-1 secretion. Endocrinology. 1999;140:1687–1694. doi: 10.1210/endo.140.4.6643. [DOI] [PubMed] [Google Scholar]
- Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A. 2008;105:16767–16772. doi: 10.1073/pnas.0808567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham O, Wei R, Wang TJ, Ricciardi C, Lewis GD, Vasan RS, et al. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol Syst Biol. 2008;4:214. doi: 10.1038/msb.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Q, Saumoy M, Holst JJ, Salen G, Xu G. Colesevelam improves insulin resistance in a diet-induced obesity (F-DIO) rat model by increasing the release of GLP-1. Am J Physiol Gastrointest Liver Physiol. 2010;298:G419–G424. doi: 10.1152/ajpgi.00362.2009. [DOI] [PubMed] [Google Scholar]
- Steinert RE, Beglinger C. Nutrient sensing in the gut: interactions between chemosensory cells, visceral afferents and the secretion of satiation peptides. Physiol Behav. 2011;105:62–70. doi: 10.1016/j.physbeh.2011.02.039. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Oba K, Igari Y, Matsumura N, Watanabe K, Futami-Suda S, et al. Colestimide lowers plasma glucose levels and increases plasma glucagon-like PEPTIDE-1 (7-36) levels in patients with type 2 diabetes mellitus complicated by hypercholesterolemia. J Nippon Med Sch. 2007;74:338–343. doi: 10.1272/jnms.74.338. [DOI] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis K, Walters JR, Gabe S. Short bowel syndrome: the role of GLP-2 on improving outcome. Curr Opin Clin Nutr Metab Care. 2009;12:526–532. doi: 10.1097/MCO.0b013e32832d23cd. [DOI] [PubMed] [Google Scholar]
- Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology. 2011;54:1421–1432. doi: 10.1002/hep.24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- Wynne K, Park AJ, Small CJ, Meeran K, Ghatei MA, Frost GS, et al. Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int J Obes (Lond) 2006;30:1729–1736. doi: 10.1038/sj.ijo.0803344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.