

Abstract
Background and Purpose
Ranolazine is an antianginal drug currently approved for treatment of angina pectoris in the United States. Recent studies have focused on its effects on neuronal channels and its possible therapeutic uses in the nervous system. We characterized how ranolazine affects the brain sodium channel, NaV1.2, and how its actions are modulated by low pH. In this way, we further explore ranolazine's potential as an anticonvulsant and its efficacy in conditions like those during an ischaemic stroke.
Experimental Approach
We performed whole-cell patch-clamp experiments on the voltage-gated sodium channel, NaV1.2. Experiments were performed with extracellular solution titrated to either pH 7.4 or pH 6.0 before and after ranolazine perfusion.
Key Results
Ranolazine accelerates onset and slows recovery of fast and slow inactivation. Ranolazine increases the maximum probability of use-dependent inactivation and reduces macroscopic and ramp sodium currents at pH 7.4. pH 6.0 reduced the slowing of fast inactivation recovery and inhibited use-dependent block by ranolazine. In the presence of ranolazine, the time constants of slow inactivation recovery and onset were significantly increased at pH 6.0 relative to pH 7.4 with 100 μM ranolazine.
Conclusions and Implications
Our work provides novel insights into the modulation of brain sodium channel, NaV1.2, by ranolazine. We demonstrate that ranolazine binds NaV1.2 in a state-dependent manner, and that the effects of ranolazine are slowed but not abolished by protons. Our results suggest that further research performed on channels with epilepsy-causing mutations may prove ranolazine to be an efficacious therapy.
Keywords: ranolazine, acidosis, sodium channel, NaV1.2, electrophysiology, brain, patch-clamp
Introduction
Ranolazine studies have focused primarily on its effects within the human heart. Electrophysiology studies determined that ranolazine displays anti-arrhythmic properties because it preferentially blocks late sodium currents (INaL) over peak fast sodium current (INa) (6 and 294 μM, respectively; Wu et al., 2004; Undrovinas et al., 2006). Ranolazine decreases calcium influx through the sodium/calcium exchanger (NCX) by blocking INaL, making it an effective treatment for chronic angina (Sossalla et al., 2008). Block of INaL reduces intracellular sodium concentrations and limits the driving force for the reverse mode of NCX (Sossalla et al., 2008).
Ranolazine also shortens repolarization in long-QT mutations (Moss et al., 2008), decreases the transmural dispersion of repolarization across the ventricular wall (Undrovinas et al., 2006) and reduces the risk factors for ventricular tachycardia and arrhythmia (Extramiana and Antzelevitch, 2004). Treatments including ranolazine significantly decreased the instance of cardiac arrhythmia (Scirica et al., 2007).
Ranolazine's actions outside the heart were first characterized on the skeletal and neuronal sodium channels, NaV1.4 and NaV1.7, respectively (Wang et al., 2008). Ranolazine blocks NaV1.4 (Wang et al., 2008) as well as the neuronal channels NaV1.1 (Kahlig et al., 2010) and NaV1.7 (Wang et al., 2008; Rajamani et al., 2008a). Ranolazine increases use-dependent inactivation (UDI), delays recovery from sodium channel inactivation and has a hyperpolarizing shift on steady-state inactivation curves in these channels (Wang et al., 2008; Estacion et al., 2010; Kahlig et al., 2010). Ranolazine also decreases the cell excitability of dorsal root ganglion neurons (Estacion et al., 2010). This work suggests that ranolazine may have application for the treatment of CNS disorders (Kahlig et al., 2010).
Therefore, the first goal of this paper is to further explore ranolazine's potential therapeutic application in neuronal disorders.
The second goal of this paper is to characterize extracellular proton modulation of ranolazine's effects. Extracellular protons block NaV1.2, hyperpolarize the steady-state inactivation curves and decrease the time constants of slow inactivation onset and recovery (Vilin et al., 2012). Previous studies show that ranolazine may be effective in treating cardiac ischaemia (Pepine and Wolff, 1999; Stone et al., 2010). During cardiac ischaemia or ischaemic stroke, extracellular pH may decrease to as low as pH 6.0 (Astrup et al., 1977; Siemkowicz and Hansen, 1981; Meyer, 1990; Maruki et al., 1993). One source of concern about this treatment is that decreased pH may alter drug action by protonating ranolazine (pKa = 7.2) (Rajamani et al., 2008b).
We characterized ranolazine's effects on NaV1.2 with extracellular solution titrated to both pH 7.4 and pH 6.0. Our results suggest that the effects of ranolazine on NaV1.2 are similar to those in other neuronal channels. We also show that the actions of ranolazine are inhibited but stylistically similar at pH 6.0.
Materials and methods
Chinese hamster ovary cells stably expressing the rat NaV1.2 channel (from Dr. W. A. Catterall) were grown at pH 7.4 in filter sterile DMEM (Life Technologies, NY, USA) with glutamine, supplemented with 2 g·L−1 NaCHO3, 100 units·mL−1 penicillin, 0.01 mg·mL−1 streptomycin, 50 mg·mL−1 G418, 5% FBS and maintained in a humidified environment at 37°C with 5% CO2. Twenty-four hours prior to electrophysiology experiments, cells were dissociated with 0.25% trypsin-EDTA (Life Technologies) and then plated on sterile cover slips.
Whole-cell recordings were performed in extracellular solution containing (in mM) 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2 and 10 HEPES (pH 7.4) or 10 MES (pH 6.0). Solutions were titrated with CsOH to pH 7.4 or pH 6.0. Pipettes were fabricated with a P-1000 puller using borosilicate glass (Sutter Instruments, CA, USA), dipped in dental wax to reduce capacitance, then thermally polished to a resistance of 1.0–1.5 MΩ. Low resistance electrodes were used to minimize series resistance between pipette and intracellular solution resulting in typical access resistances of 2.5 MΩ or less, thereby minimizing voltage measurement error. Pipettes were filled with intracellular solution, containing (in mM) 130 CsF, 10 NaCl, 10 HEPES and 10 EGTA titrated to pH 7.4.
All recordings were made using an EPC-9 patch-clamp amplifier (HEKA Elektronik, Lambrecht, Germany) digitized at 20 kHz using an ITC-16 interface (HEKA Elektronik, Lambrecht, Germany). Data were acquired and low-pass filtered (5 kHz) using PatchMaster/FitMaster software (HEKA Elektronik, Lambrecht, Germany) running on an Apple iMac (Apple Computer, Cupertino, CA, USA). Leak subtraction was performed online using a P/4 procedure. Leak subtraction was performed off-line for slow inactivation experiments, when necessary. Recordings were completed at room temperature. After whole-cell configuration was attained, currents were allowed to stabilize such that currents measured by successive trains of five 10 ms depolarizations at 1 Hz to 0 mV were similar. Run-down was assessed by comparing peak current amplitudes at the beginning and the end of recordings. Only cells showing less than 5% peak current reduction were included in analysis. The holding potential between protocols was −60 mV. Fitting and graphing were done using FitMaster software [HEKA Elektronik and Igor Pro (Wavemetrics, Lake Oswego, OR, USA)] with statistical information derived using SPSS statistical software (IBM Corporation, Armonk, NY, USA). Significance was determined at P < 0.05 using Student's ‘t’ test or one-way ANOVAs.
Conductance (G(V)) curves were calculated from the equation:
| (1) |
where G is conductance, Imax represents peak test pulse current, Vm is the test pulse voltage and ENa is the measured equilibrium potential.
The midpoint and apparent valence of activation were derived by plotting normalized conductance as a function of test potential. Data were then fitted with a Boltzmann equation:
| (2) |
where G/Gmax is normalized conductance, Vm is the test potential, z is the apparent valence, e0 is the elementary charge, V1/2 is the midpoint voltage, k is the Boltzmann constant, and T is temperature in °K.
Conductance block due to drug binding was analysed by normalizing conductance after drug perfusion to peak conductance before drug perfusion. The concentration dependence of conductance block was fit with a Hill equation:
| (3) |
where max is conductance without drug, X1/2 is the concentration of drug causing 50% block (IC50), X is drug concentration and rate is the Hill coefficient.
The time constants (τ) of open-state inactivation were derived using single exponential fits to the decay of current during each test pulse using the following equation:
| (4) |
where I is current amplitude, t is time, Offset is the asymptote of decay, and a is the amplitudes of the time constant τ.
Time constants of slow inactivation onset were fit with a single exponential (Equation 3). Time constants of fast and slow inactivation recovery and UDI data were fit with a double exponential using the following equation:
| (5) |
where I is current amplitude, t is time, Offset is the asymptote of decay, a1 and a2 are the relative amplitudes for the corresponding fast and slow time constants τ1 and τ2 respectively.
To assess steady-state FI and SI [steady-state fast inactivation (SSFI) and steady-state slow inactivation (SSSI), respectively] normalized current amplitude was plotted as a function of pre-pulse potential and fitted using the Boltzmann equation (equation 2). All data are presented as mean ± SEM.
Sodium channel nomenclature has previously been defined (Alexander et al., 2011).
Results
Activation
We measured the peak macroscopic currents from the neuronal voltage-gated sodium channel, NaV1.2, using 19 ms depolarizations from −130 mV to −90 mV through 70 mV in 10 mV intervals with 200 ms at holding potential between the test pulses. Sample traces at pH 7.4 and pH 6.0 are shown before and after perfusion of 100 μM ranolazine in Figure 1 A1, A2, B1 and B2. Perfusion of 100 μM ranolazine significantly reduced peak currents by 19.6 ± 1.0% at pH 7.4 [Figure 1 A1 (without drug) and A2 (ranolazine)] and 18.3 ± 1.3% at pH 6.0 [Figure 1 B1 (without drug) and B2 (ranolazine)]. Figure 1C shows the Boltzmann fits for NaV1.2 normalized to conductance at pH 7.4 and pH 6.0. G(V) curves recorded in control solutions and in the presence of 100 μM ranolazine are shown normalized to the peak conductance in control and drug respectively. Conductance in the presence of 100 μM ranolazine is normalized to the peak conductance in control solution in Figure 1D inset. The V1/2 of conductance was significantly depolarized at pH 6.0 (Table 1) compared to pH 7.4 (Table 1). Perfusion of 100 μM ranolazine caused a significant hyperpolarizing shift in the voltage dependence of conductance at normal pH 7.4 (Table 1) and at pH 6.0 (Table 1). The apparent valence of conductance was not measurably altered by pH or ranolazine (Table 1). The concentration-dependent block of peak conductance by ranolazine is shown in Figure 1D. Hill curves have IC50 values of 328 ± 29 μM at pH 7.4 and 274 ± 36 μM at pH 6.0.
Figure 1.
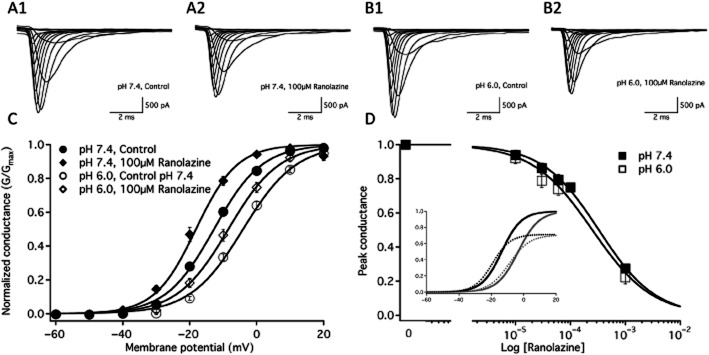
Macroscopic currents and conductance. Macroscopic current traces elicited between −90 and +70 mV before and after 100 μM ranolazine perfusion are shown at pH 7.4 in A1 and A2, respectively and at pH 6.0 in B1 and B2 respectively. C shows the conductance at pH 7.4 in control (closed circles, n = 33) and 100 μM ranolazine (closed diamonds, n = 12) as well as at pH 6.0 in control (open circles, n = 29) and 100 μM ranolazine (open diamonds, n = 14). Boltzmann curve parameters for conductance are listed in Table 1. D shows the concentration dependence of ranolazine block on the peak conductance of NaV1.2 at pH 7.4 (closed squares) and pH 6.0 (open squares). D inset shows conductance curves of the sodium channel at pH 7.4 (black) and pH 6.0 (grey) in the absence (solid lines) and presence of 100 μM ranolazine (dashed lines) as a function of membrane potential. In this case, the conductance in the presence of ranolazine is normalized to the peak conductance in the absence of ranolazine.
Table 1.
Conductance and steady-state fast inactivation
| G(V) V1/2 | G(V) z | SSFI V1/2 | SSFI z | |
|---|---|---|---|---|
| pH 7.4 | ||||
| NaV1.2 | −13.2 ± 0.8 mV | 3.8 ± 0.1 | −52.5 ± 1.5 mV | −4.0 ± 0.3 |
| NaV1.2 with 100 μM Ranolazine | −18.1 ± 1.2 mV*1 | 4.1 ± 0.1 | −58.7 ± 2.5 mV*1 | −3.5 ± 0.2 |
| pH 6.0 | ||||
| NaV1.2 | −4.4 ± 0.8 mV*1 | 3.6 ± 0.1 | −51.6 ± 2.7 mV | −3.4 ± 0.3 |
| NaV1.2 with 100 μM Ranolazine | −8.4 ± 1.1 mV*3*4 | 3.6 ± 0.1*3 | −57.0 ± 3.0 mV*4 | −3.3 ± 0.3 |
= P < 0.05 versus pH 7.4 without drug.
= P < 0.05 versus pH 7.4 with 100 μM Ranolazine.
= P < 0.05 versus pH 6.0 without drug.
Fast inactivation steady state
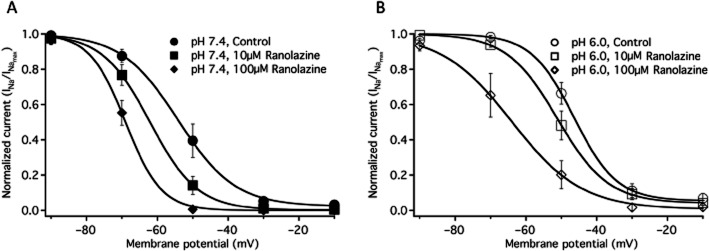
SSFI in NaV1.2 was measured with a −10 mV test pulse immediately following 500 ms conditioning pulses to −130 mV through 20 mV in 10 mV intervals. SSFI recorded in control solution and 100 μM ranolazine, at pH 7.4 and pH 6.0 are shown in Figure 2A and 2C respectively. Ranolazine perfusion caused significant, similar hyperpolarizing shifts in the V1/2 of fast inactivation at pH 7.4 and pH 6.0 (Figure 2, Table 1). There were no significant effects on the apparent valence of SSFI with changes in pH or ranolazine perfusion (Table 1).
Figure 2.

Fast inactivation. (A) Steady-state fast inactivation curves at pH 7.4 in control (closed circles, n = 7) and in 100 μM ranolazine (closed diamonds, n = 7). A inset shows the time constants of fast inactivation onset at pH 7.4 in control (closed circles n = 10) and in 100 μM ranolazine (closed diamonds, n = 10) at membrane potentials from −20 to +10 mV. B shows the recovery from fast inactivation at −130 mV in pH 7.4 control (closed circles, n = 14) and in 100 μM ranolazine (closed diamonds, n = 7). B inset shows the initial 32 ms of recovery. Time constants and amplitudes are listed in Table 1. Fast inactivation steady state curves at pH 6.0 in control (open circles, n = 6) and in 100 μM ranolazine (open diamonds, n = 6) are shown in C. C inset shows the time constants of fast inactivation onset at pH 6.0 in control (open circles n = 11) and in 100 μM ranolazine (open diamonds, n = 11) at membrane potentials from −20 to +10 mV. D shows the recovery from fast inactivation at −130 mV in pH 6.0 control (open circles, n = 9) and in 100 μM ranolazine (open diamonds, n = 6). D inset shows the initial 32 ms of recovery. Boltzmann curve parameters for steady-state fast inactivation are listed in Table 1. Time constants of open-state inactivation are listed in Table 2 and time constants and amplitudes of recovery are listed in Table 3.
Fast inactivation onset
Open-state fast inactivation onset was measured with single exponential fits of macroscopic current decay at voltages between −20 and +10 mV. Fast inactivation onset time constants, recorded without drug and in the presence of 100 μM ranolazine at pH 7.4 and pH 6.0, are shown in Figure 2A inset and 2C inset respectively (see Table 2 for specific values). At pH 7.4, ranolazine perfusion significantly accelerated the time constants of fast inactivation onset at −20 mV by 18% and at −10 mV by 10%. Extracellular pH 6.0 solution significantly slowed fast inactivation onset at −20 mV and −10 mV compared to control. Ranolazine perfusion accelerated fast inactivation onset as much as 28% between −20 mV and +10 mV at pH 6.0.
Table 2.
Open-state fast inactivation
| FI Onset τ −20 mV | FI Onset τ −10 mV | FIOnset τ 0 mV | FI Onset τ 10 mV | |
|---|---|---|---|---|
| pH 7.4 | ||||
| NaV1.2 | 1.63 ± 0.12 ms | 0.83 ± 0.04 ms | 0.56 ± 0.02 ms | 0.43 ± 0.02 ms |
| NaV1.2 with 100 μM Ranolazine | 1.34 ± 0.12 ms*1 | 0.75 ± 0.04 ms*1 | 0.52 ± 0.02 ms | 0.41 ± 0.02 ms |
| pH 6.0 | ||||
| NaV1.2 | 2.98 ± 0.28 ms*1 | 1.42 ± 0.12 ms*1 | 0.73 ± 0.06 ms | 0.49 ± 0.03 ms |
| NaV1.2 with 100 μM Ranolazine | 2.19 ± 0.18 ms*3*4 | 1.04 ± 0.08 ms*3*4 | 0.58 ± 0.04 ms*4 | 0.42 ± 0.03 ms*4 |
= P < 0.05 versus pH 7.4 without drug.
= P < 0.05 versus pH 7.4 with 100 μM Ranolazine.
= P < 0.05 versus pH 6.0 without drug.
Fast inactivation recovery
The rate of fast inactivation recovery was measured by depolarizing the membrane to −10 mV for 500 ms to fully fast-inactivate the channels. Voltage was the returned to a −130 ms interpulse potential. The current recovered during the interpulse was assessed during a test pulse to 0 mV, 0–1024 ms into the interpulse. Peak current measured from each test pulse was plotted as a function of interpulse duration and fitted with Equation 5. Figure 2B and D show fast inactivation recovery recorded at pH 7.4 and pH 6.0, respectively, with and without 100 μM ranolazine. The data from fast inactivation is listed in Table 3. At pH 7.4, ranolazine perfusion caused significant slowing of both the fast and slow time constants of recovery (Figure 2B and 2B inset, Table 3). Ranolazine also caused a significant increase in amplitude of the slow time constant of recovery at pH 7.4 (Table 3). At pH 6.0, ranolazine caused significant slowing of the fast and slow time constants of recovery as well (Figure 2D and 2D inset, Table 3), but had no effect on the amplitudes of either time constant. The slow time constant was smaller at pH 6.0 than at pH 7.4 before and after ranolazine perfusion.
Table 3.
Fast inactivation recovery
| FI Recovery τ 1 | FI Recovery A1 | FI Recovery τ 2 | FI Recovery A2 | |
|---|---|---|---|---|
| pH 7.4 | ||||
| NaV1.2 | 1.2 ± 0.1 ms | 0.79 ± 0.02 | 48.5 ± 7.9 ms | 0.20 ± 0.02 |
| NaV1.2 with 100 μM Ranolazine | 2.2 ± 0.4 ms*1 | 0.48 ± 0.03*1 | 325.1 ± 21.8 ms*1 | 0.51 ± 0.03*1 |
| pH 6.0 | ||||
| NaV1.2 | 1.1 ± 0.1 ms | 0.79 ± 0.02 | 24.5 ± 5.8 ms*1 | 0.14 ± 0.02 |
| NaV1.2 with 100 μM Ranolazine | 2.2 ± 0.3 ms*4 | 0.75 ± 0.04*3 | 53.7 ± 12.2 ms*3*4 | 0.24 ± 0.04*3 |
= P < 0.05 versus pH 7.4 without drug.
= P < 0.05 versus pH 7.4 with 100 μM Ranolazine.
= P < 0.05 versus pH 6.0 without drug.
UDI
UDI of NaV1.2 was measured using a train of 500, 10 ms depolarizations to 0 mV at a frequency of 10 Hz. UDI recorded without drug, and in the presence of 10 μM and 100 μM ranolazine at pH 7.4 and pH 6.0 is shown in Figure 3A and 3C respectively (see Table 4 for specific values). At pH 7.4, perfusion of 10 μM and 100 μM ranolazine increased UDI from 19% in control, to 38 and 74% respectively (Figure 3A, Table 4). At pH 6.0, ranolazine perfusion significantly increased UDI from 14% in the absence of ranolazine (Figure 3C, Table 4) to 21% and to 44% in the presence of 10 μM and 100 μM ranolazine respectively. The asymptote of the UDI curve was significantly increased at pH 6.0, relative to pH 7.4, in both control and ranolazine conditions. There was no significant change in the slow time constant of UDI development with ranolazine perfusion (Table 4); however, 100 μM ranolazine perfusion added a second, fast, component of inactivation (Figure 3A and 3C). The fast component of UDI was significantly accelerated at pH 7.4 with 100 μM ranolazine compared to pH 6.0 with 100 μM ranolazine (Table 4). The IC50 values of UDI due to ranolazine perfusion are 59.2 ± 13.7 μM at pH 7.4 and 182 ± 63.8 μM at pH 6.0 (Figure 3D inset).
Figure 3.

Use-dependent and ramp currents. A shows the average use-dependent inactivation of currents at pH 7.4 control (filled circles, n = 20), 10 μM ranolazine (filled squares, n = 6) and 100 μM ranolazine (filled diamonds, n = 7) over 500 traces at 10 Hz. Sample ramp currents elicited by a 500 ms depolarization from −130 to 20 mV are shown at pH 7.4 in control (Black) and after 100 μM ranolazine perfusion (Gray) in B. C shows the average use-dependent inactivation of currents at pH 6.0 control (open circles, n = 22), 10 μM ranolazine (open squares, n = 7) and 100 μM ranolazine (open diamonds, n = 8) over 500 traces at 10 Hz. C inset shows the concentration dependence of use-dependent block developed by the 500th pulse in a train of 10 Hz depolarizations to 0 mV at pH 7.4 (closed squares) and pH 6.0 (open squares). Sample ramp currents elicited by a 500 ms depolarization from −130 to 20 mV are shown at pH 6.0 in control (Black) and after 100 μM ranolazine perfusion (grey) in D. Time constants and asymptotes of use-dependent inactivation are in Table 4.
Table 4.
Use-dependent inactivation
| UDI Y0 | UDI τ 1 | UDI τ 2 | |
|---|---|---|---|
| pH 7.4 | |||
| NaV1.2 | 0.82 ± 0.02 | 25.8 ± 1.2 s | n/a |
| NaV1.2 with 10 μM Ranolazine | 0.62 ± 0.05*1 | 18.0 ± 0.5 s | n/a |
| NaV1.2 with 100 μM Ranolazine | 0.26 ± 0.06*1*2 | 30.5 ± 6.3 s | 1.0 ± 0.2 s |
| pH 6.0 | |||
| NaV1.2 | 0.86 ± 0.01*1 | 16.4 ± 1.2 s | n/a |
| NaV1.2 with 10 μM Ranolazine | 0.79 ± 0.01*2*4 | 23.3 ± 2.0 s | n/a |
| NaV1.2 with 100 μM Ranolazine | 0.56 ± 0.05*3*4*5 | 34.4 ± 6.3 s | 4.7 ± 0.6 s*3 |
= P < 0.05 versus pH 7.4 without drug.
= P < 0.05 versus pH 7.4 with 10 μM Ranolazine.
= P < 0.05 versus pH 7.4 with 100 μM Ranolazine.
= P < 0.05 versus pH 6.0 without drug.
= P < 0.05 versus pH 6.0 with 10 μM Ranolazine.
Window currents
Window currents were measured by a 0.3 mV·ms−1 ramp from −130 to 20 mV before and after ranolazine perfusion at pH 7.4 and pH 6.0. Figure 3B and 3D show sample ramp traces before and after 100 μM ranolazine perfusion at pH 7.4 and pH 6.0. At pH 7.4, perfusion of 100 μM ranolazine significantly decreased the peak window current by 20.8 ± 4.6% and the total charge moved by 24.5 ± 5.7% (Figure 3B). At pH 6.0, ranolazine perfusion did not significantly alter the peak current or the total charge moved (Figure 3D).
Slow inactivation steady state
SSSI was measured using a 30 s conditioning pulse at −130 mV through 10 mV in 10 mV increments followed by a 20 ms recovery pulse at −130 mV to recover fast-inactivated channels and then a 20 ms test pulse to −10 mV to measure channel availability. Figure 4A and 4B show averaged curves at pH 7.4 and pH 6.0 respectively. Data are shown recorded in without drug and in 10 μM ranolazine and 100 μM ranolazine solutions. At pH 7.4, there was a significant hyperpolarizing shift in the V1/2 of slow inactivation with perfusion of 10 μM ranolazine and 100 μM ranolazine (Figure 4A, Table 5). At pH 6.0, there was a significant hyperpolarizing shift in the V1/2 of slow inactivation only after 100 μM ranolazine perfusion (Figure 4B, Table 5). As the V1/2 of the slow inactivation curve was depolarized in pH 6.0 solution without drug, relative to pH 7.4, the hyperpolarizing shift due to ranolazine returned the steady-state curve to values closer to those at pH 7.4. There was no significant difference in the apparent valence of slow inactivation with changes in pH or ranolazine perfusion (Table 5).
Figure 4.
Slow inactivation steady state. Slow inactivation steady state curves are shown at pH 7.4 control (filled circles, n = 12), 10 μM ranolazine (filled squares, n = 9), and 100 μM ranolazine (filled diamonds, n = 5) in A. Slow inactivation steady state curves are shown at pH 6.0 control (open circles, n = 8), 10 μM ranolazine (open squares, n = 7) and 100 μM ranolazine (open diamonds, n = 7) in B. Boltzmann curve parameters are in Table 5.
Table 5.
Steady-state slow inactivation
| SSSI V1/2 | SSSI z | |
|---|---|---|
| pH 7.4 | ||
| NaV1.2 | −53.9 ± 2.5 mV | −4.8 ± 0.3 |
| NaV1.2 with 10 μM Ranolazine | −62.1 ± 2.1 mV*1 | −4.7 ± 0.3 |
| NaV1.2 with 100 μM Ranolazine | −66.7 ± 2.4 mV*1 | −4.9 ± 0.5 |
| pH 6.0 | ||
| NaV1.2 | −46.1 ± 1.7 mV*1 | −4.8 ± 0.3 |
| NaV1.2 with 10 μM Ranolazine | −50.7 ± 2.2 mV*2 | −4.1 ± 0.3 |
| NaV1.2 with 100 μM Ranolazine | −63.8 ± 4.5 mV*4*5 | −4.0 ± 0.4 |
= P < 0.05 versus pH 7.4 without drug.
= P < 0.05 versus pH 7.4 with 10 μM Ranolazine.
= P < 0.05 versus pH 6.0 without drug.
= P < 0.05 versus pH 6.0 with 10 μM Ranolazine.
Onset of slow inactivation and ranolazine block
The onset of slow inactivation was measured with conditioning pulses from 500 ms to 64 s at −10 mV followed by either a 500 ms (Figure 5A and 5C) or 2 s (Figure 5B and 5D) recovery period at −130 mV and a test pulse to −10 mV. Kinetic components of the single exponential fits to onset data under all conditions are shown in Table 6. After a 500 ms recovery pulse at pH 7.4, there was a significant decrease in the time constant of slow inactivation onset with 10 μM and 100 μM ranolazine perfusion, (Figure 5A). The asymptotes of slow inactivation were also significantly decreased, relative to pH 7.4 without drug, in 10 μM and 100 μM ranolazine. At pH 6.0, there was no significant change in the time constants of slow inactivation onset after a 500 ms recovery (Figure 5C). Similar to pH 7.4, there was a significant decrease in the asymptote of inactivation at pH 6.0 in 10 μM and 100 μM ranolazine, relative to pH 6.0 solution without drug. pH 6.0 did not affect the kinetics of onset relative to pH 7.4 in solutions without drug, but the time constants were significantly smaller at pH 7.4 than at pH 6.0 in the presence of ranolazine (see Table 6). After a 2 s recovery pulse, the level of inactivation was not large enough to accurately fit individual onsets from solutions without drug with exponential curves. After a 2 s recovery increasing the ranolazine concentration to 100 μM significantly decreased the time constants of slow inactivation at pH 7.4 (Figure 5B) and at pH 6.0 (Figure 5D), relative to the time constants in 10 μM ranolazine. The time constants of inactivation in drug solutions were not significantly different between pH 7.4 and pH 6.0 after a 2 s recovery.
Figure 5.

Onset of slow inactivation and ranolazine block. A shows the onset of slow inactivation and ranolazine block after a 500 ms recovery pulse at pH 7.4 control (filled circles, n = 8), 10 μM ranolazine (filled squares, n = 7) and 100 μM ranolazine (filled diamonds, n = 8). Onset of slow inactivation and ranolazine block after a 2 s recovery pulse at pH 7.4 control (filled circles, n = 8), 10 μM ranolazine (filled squares, n = 5) and 100 μM ranolazine (filled diamonds, n = 8) are shown in B. C shows the onset of slow inactivation and ranolazine block after a 500 ms recovery pulse at pH 6.0 control (open circles, n = 7), 10 μM ranolazine (open squares, n = 6) and 100 μM ranolazine (open diamonds, n = 7). Onset of slow inactivation and ranolazine block after a 2 s recovery pulse at pH 6.0 control (open circles, n = 7), 10 μM ranolazine (open squares, n = 6) and 100 μM ranolazine (open diamonds, n = 7) are shown in D. Time constants, amplitudes and asymptotes are in Table 6.
Table 6.
Slow inactivation onset
| SI Onset Y0 (500 ms) | SI Onset τ (500 ms) | SI Onset A (500 ms) | SI Onset Y0 (2 s) | SI Onset τ (2 s) | SI Onset A (2 s) | |
|---|---|---|---|---|---|---|
| pH 7.4 | ||||||
| NaV1.2 | 0.52 ± 0.05 | 19.4 ± 2.6 s | 0.42 ± 0.05 | n/a | n/a | n/a |
| NaV1.2 with 10 μM Ranolazine | 0.32 ± 0.04*1 | 9.4 ± 1.2 s*1 | 0.58 ± 0.03*1 | 0.65 ± 0.03 | 25.3 ± 3.8 s | 0.30 ± 0.04 |
| NaV1.2 with 100 μM Ranolazine | 0.19 ± 0.04*1 | 3.1 ± 0.3 s*1*2 | 0.44 ± 0.03*2 | 0.64 ± 0.04 | 11.5 ± 2.8 s*2 | 0.22 ± 0.02 |
| pH 6.0 | ||||||
| NaV1.2 | 0.58 ± 0.05 | 15.1 ± 3.1 s | 0.36 ± 0.04 | n/a | n/a | n/a |
| NaV1.2 with 10 μM Ranolazine | 0.31 ± 0.04*4 | 19.3 ± 2.6 s*2 | 0.60 ± 0.04*4 | 0.41 ± 0.07*2 | 34.3 ± 6.2 s | 0.51 ± 0.07*2 |
| NaV1.2 with 100 μM Ranolazine | 0.07 ± 0.02*3*4*5 | 10.2 ± 1.6 s*3 | 0.77 ± 0.04*3*4*5 | 0.25 ± 0.06*3 | 11.5 ± 2.2 s*5 | 0.61 ± 0.05*3 |
= P < 0.05 versus pH 7.4 without drug.
= P < 0.05 versus pH 7.4 with 10 μM Ranolazine.
= P < 0.05 versus pH 7.4 with 100 μM Ranolazine.
= P < 0.05 versus pH 6.0 without drug.
= P < 0.05 versus pH 6.0 with 10 μM Ranolazine.
Recovery from slow inactivation and ranolazine block
Recovery from slow inactivation was measured after a 4 s (Figure 6A and 6C with insets) or 32 s (Figure 6B and 6D with insets) depolarization to −10 mV followed by a 20 ms to 60 s recovery at −130 mV and a test pulse at −10 mV. Parameters of double exponential fits to data under all conditions are shown in Table 7. After a 4 s onset at pH 7.4, 100 μM ranolazine increased the fast and slow time constants of recovery, while increasing the amplitude of the fast component (Figure 6A and 6A inset, Table 7). In contrast, at pH 6.0, there was a significant slowing of the slow time constant of recovery and increase in the slow time constant amplitude with 100 μM ranolazine perfusion (Figure 6C and 6C inset, Table 7), but the fast time constant of recovery was unchanged. At pH 6.0, the amplitude of the slow time constant was significantly larger in 100 μM ranolazine than in pH 7.4 with 100 μM ranolazine. Perfusion of 10 μM ranolazine had no significant effect on the fast or slow time constants of recovery at either pH. At pH 7.4 after a 32 s onset, similar to after a 4 s onset, there was significant slowing of the fast time constant of slow inactivation recovery, but after both 10 and 100 μM ranolazine perfusion (Figure 6B and 6B inset). At pH 6.0 after a 32 s onset, ranolazine perfusion significantly increased the slow time constant of slow inactivation recovery (Figure 6D and 6D inset) similar to after a 4 s onset. Unlike after a 4 s onset, the fast time constant at pH 6.0 was significantly slowed in 100 μM ranolazine when compared to solutions without drug. In 100 μM ranolazine after a 32 s onset, as after a 4 s onset, the slow component of recovery was a significantly larger proportion of the total recovery at pH 6.0 compared to pH 7.4.
Figure 6.

Recovery from slow inactivation and ranolazine block. A shows the recovery from slow inactivation and ranolazine block after a 4 s onset at pH 7.4 control (filled circles, n = 6), 10 μM ranolazine (filled squares, n = 7) and 100 μM ranolazine (filled diamonds, n = 6). Recovery from slow inactivation and ranolazine block after a 32 second onset at pH 7.4 control (filled circles, n = 8), 10 μM ranolazine (filled squares, n = 7) and 100 μM ranolazine (filled diamonds, n = 6) are shown in B. C shows the recovery from slow inactivation and ranolazine block after a 4 s onset at pH 6.0 control (open circles, n = 5), 10 μM ranolazine (open squares, n = 6) and 100 μM ranolazine (open diamonds, n = 7). Recovery from slow inactivation and ranolazine block after a 32 s onset at pH 6.0 control (open circles, n = 6), 10 μM ranolazine (open squares, n = 6) and 100 μM ranolazine (open diamonds, n = 7) are shown in D. Time constants, amplitudes and asymptotes are in Table 7. All insets show the initial 5 s of recovery in their respective condition.
Table 7.
Slow inactivation recovery
| SI Recovery τ 1 (4 s) | SI Recovery A 1 (4 s) | SI Recovery τ 2 (4 s) | SI Recovery A 2 (4 s) | SI Recovery τ 1 (32 s) | SI Recovery A 1 (32 s) | SI Recovery τ 2 (32 s) | SI Recovery A 2 (32 s) | |
|---|---|---|---|---|---|---|---|---|
| pH 7.4 | ||||||||
| NaV1.2 | 116 ± 8 ms | 0.55 ± 0.04 | 4.2 ± 1.3 s | 0.14 ± 0.03 | 266 ± 63 ms | 0.71 ± 0.04 | 6.3 ± 1.1 s | 0.27 ± 0.04 |
| NaV1.2 with 10 μM Ranolazine | 324 ± 89 ms | 0.67 ± 0.06 | 5.77 ± 2.87 s | 0.17 ± 0.05 | 933 ± 190 ms*1 | 0.78 ± 0.07 | 9.0 ± 3.3 s | 0.22 ± 0.06 |
| NaV1.2 with 100 μM Ranolazine | 1080 ± 89 ms*1*2 | 0.94 ± 0.02*1*2 | 37.2 ± 26 s*1*2 | 0.07 ± 0.02*2 | 1450 ± 150 ms*1*2 | 0.73 ± 0.03 | 18.4 ± 4.3 s*1*2 | 0.24 ± 0.03 |
| pH 6.0 | ||||||||
| NaV1.2 | 104 ± 26 ms | 0.44 ± 0.05 | 2.7 ± 0.8 s | 0.24 ± 0.05 | 213 ± 60 ms | 0.60 ± 0.03*1 | 5.7 ± 1.2 s | 0.29 ± 0.03 |
| NaV1.2 with 10 μM Ranolazine | 108 ± 34 ms | 0.49 ± 0.07 | 6.1 ± 2.8 s | 0.23 ± 0.01 | 340 ± 89 ms*2 | 0.54 ± 0.03 | 13.6 ± 1.6 s*2*4 | 0.44 ± 0.03*4 |
| NaV1.2 with 100 μM Ranolazine | 134 ± 27 ms*3 | 0.45 ± 0.04*3 | 19.7 ± 2.3 s*4*5 | 0.39 ± 0.03*3*4*5 | 606 ± 128 ms*3*4 | 0.18 ± 0.03*3*4*5 | 16.3 ± 1.3 s*4 | 0.83 ± 0.03*3*4*5 |
= P < 0.05 versus pH 7.4 without drug.
= P < 0.05 versus pH 7.4 with 10 μM Ranolazine.
= P < 0.05 versus pH 7.4 with 100 μM Ranolazine.
= P < 0.05 versus pH 6.0 without drug.
= P < 0.05 versus pH 6.0 with 10 μM Ranolazine.
Discussion and conclusion
Ranolazine is used clinically as an antianginal drug as it reduces diastolic tension (Sossalla et al., 2008). Ranolazine is also thought to be an anti-arrhythmic because it decreases transmural dispersion across the heart wall (Undrovinas et al., 2006). Our understanding of ranolazine now extends to its effects on neuronal channels, NaV1.7 and NaV1.1, and the skeletal muscle channel, NaV1.4 (Wang et al., 2008; Kahlig et al., 2010). Ranolazine's effects have not been characterized in NaV1.2, a CNS sodium channel implicated in generalized epilepsy with febrile seizures plus (GEFS+) (Sugawara et al., 2001). Also, despite evidence that it may be useful in the treatment of ischaemic conditions (Pepine and Wolff, 1999; Stone et al., 2010), the effects of ranolazine on channel electrophysiology at low pH have not been tested. This study examined the actions of ranolazine on NaV1.2, and how its effects are modulated by extracellular protons.
Sodium channel mutations in NaV1.2 are associated with several types of epilepsy including GEFS+ and benign familial infantile seizures (Sugawara et al., 2001; Berkovic et al., 2004; George, 2005; Scalmani et al., 2006). These mutations are characterized by increased activation or impaired inactivation of the channel leading to increased sodium currents and neuronal hyperexcitability. Therefore, the pharmacological intervention in epilepsy is often targeted block of neuronal sodium channels. Antiepileptics, such as carbamazepine and diphenylhydantoin, block sodium channels in a use-dependent manner inhibiting repetitive action potential firing (Willow et al., 1985). Besides sodium channel mutations, seizures may also occur after ischaemic strokes. Early and late seizures have been recorded along with onset of epilepsy following a stroke (Camilo and Goldstein, 2004). Understanding effects of antiepileptic drugs at low pH is therefore important.
Ranolazine block of the neuronal sodium channels NaV1.1 and NaV1.7 has previously been characterized at concentrations between 1 and 100 μM (Wang et al., 2008; Rajamani et al., 2008a; Estacion et al., 2010; Kahlig et al., 2010). Here, we characterize ranolazine effects at concentrations of 10 and 100 μM. In vivo predicted plasma concentrations of ranolazine range from 2 to 10 μM with predicted brain concentration of one-third the plasma concentration (Kahlig et al., 2010). The minimal concentration used is, therefore, within possible plasma concentrations, but above the concentrations predicted in the CNS.
We first characterized how ranolazine and low pH affect the fast kinetics of the sodium channel NaV1.2. We showed that ranolazine introduces a hyperpolarizing shift on conductance (Figure 1B and D) and fast inactivation steady state (Figure 2A and C) curves at pH 7.4 and pH 6.0. Hyperpolarizing shifts have previously been shown to occur in the fast inactivation curves of NaV1.7 and NaV1.8 (Rajamani et al., 2008a. At pH 6.0, the G(V) curve is depolarized relative to pH 7.4. Ranolazine caused a leftward shift in the V1/2 of activation at pH 6.0, returning it closer to the V1/2 recorded at pH 7.4. We also saw significant hyperpolarizing shifts in the V1/2 of activation in the presence of 10, 30 and 60 μM ranolazine (data not shown).
As reported in previous studies, ranolazine accelerated the onset of fast inactivation (Figure 2A inset and 2C inset) (Rajamani et al., 2008a) at both pH 7.4 and pH 6.0. Because onset was slowed at pH 6.0, ranolazine returned the time course of inactivation closer to values recorded at pH 7.4. We found that ranolazine slowed the recovery from fast inactivation (Figure 2 B and D) as shown previously for NaV1.1 (Kahlig et al., 2010). Interestingly, our fast inactivation recovery followed a double exponential curve even in control conditions. We attribute the slow component of fast inactivation recovery to intermediate inactivation induced by the long conditioning pulse. In conditions without drug, this component was faster at pH 6.0 than at pH 7.4. Ranolazine increased the time constant and amplitude of this component at pH 7.4, but not at pH 6.0. These results suggest ranolazine stabilizes the inactivated state of the channel, but this stabilization is impeded by extracellular protons.
Our next experiments focused on channel block induced by ranolazine. Ranolazine blocks late currents and use-dependent currents in both cardiac and neuronal sodium channels (Sossalla et al., 2008; Wang et al., 2008; Rajamani et al., 2008a; Estacion et al., 2010; Kahlig et al., 2010). Consistent with previous studies, (Rajamani et al., 2008a; Estacion et al., 2010) we observed tonic block of NaV1.2 by ranolazine. Tonic block was not measurably different between pH values, with 100 μM ranolazine causing approximately 20% block. We showed that ranolazine concentrations between 10 and 100 μM increased the level of UDI at both pH 7.4 and 6.0 (Figure 3A and 3C). The level of block at pH 6.0, however, was less than at pH 7.4. This also demonstrates that the actions of ranolazine can be impeded by extracellular protons. This was further confirmed by the absence of measurable ranolazine effects on window currents at pH 6.0, despite ranolazine's reduction of peak window current and the total charge moved at pH 7.4 (Figure 3B and 3D). It should be noted that peak INa in NaV1.2 is reduced by ∼32% at pH 6.0 (Vilin et al., 2012). Window currents at pH 6.0 thus had smaller amplitudes making accurate measurements of ranolazine block more difficult. Previous studies have used mutated channels with larger window currents to measure the effects of ranolazine (Kahlig et al., 2010) or have not shown significant effects of ranolazine on window currents (Estacion et al., 2010). It may also be possible, given the slowed binding of ranolazine at pH 6.0, that a 500 ms ramp was unable to elicit the level of drug binding observed at pH 7.4. Finally, UDI experiments also showed that, at pH 6.0, the effect of ranolazine occurred over a longer time period than at pH 7.4. This suggested the need to study ranolazine using longer protocols.
Our final experiments examined slow inactivation and used longer pre-pulse lengths, up to 1 min. Previous research suggested that ranolazine stabilizes a slow inactivation-like state in NaV1.7 (Rajamani et al., 2008a). Our study on NaV1.2 found that 10 and 100 μM ranolazine shifted the V1/2 of slow inactivation to more negative potentials at pH 7.4 and 100 μM ranolazine had a significant effect at pH 6.0 (Figure 4A and 4B). We also showed that ranolazine accelerates onset of slow inactivation and delays recovery (Figures 5 and 6). Interestingly, at pH 7.4 ranolazine increased the amplitude of the fast component of recovery while at pH 6.0, ranolazine increased the amplitude of the slow time constant. Slower kinetics at pH 6.0 may explain why ranolazine had less noticeable effects on short protocols, such as those that measured fast inactivation recovery and UDI. It should be noted that at pH 7.4 after a 4 s onset, the slow component of recovery was very small, making it difficult to accurately fit.
Our slow inactivation onset and recovery experiments show that at pH 7.4 the actions of the drug occur over shorter time scales then at pH 6.0. This suggests that drug binding kinetics are slowed by higher concentrations of extracellular protons. One of ranolazine's two pKa values is 7.2; a shift from pH 7.4 to pH 6.0 would be expected to increase the proportion of protonated ranolazine molecules by approximately 61%. The extra charge on the molecule is predicted to make it more hydrophilic and, therefore, may impede access to its binding site. This mechanism could account for the observed pH dependent differences in use-dependent block and lack of difference in tonic block of conductance. The slowed kinetics of ranolazine binding, as suggested by slow-inactivation protocols, suggest ranolazine binding will occur slower in trains of successive depolarizations at pH 6.0. It is also possible that protonation of the channel is interfering with the drug binding. At pH 6.0, there are depolarizing shifts in steady-state activation and slow inactivation. Ranolazine affinity is higher for channels in the open and states, therefore decreased occupancy at these states at lower pH could impair drug binding (Estacion et al., 2010).
Our results provide insight into the interaction between protons and ranolazine, and validates previous research into its possible use as an antiepileptic (Kahlig et al., 2010). The effects of ranolazine on channel fast inactivation and activation were limited even at high concentrations as was previously shown in the NaV1.1 isoform (Kahlig et al., 2010). Like both phenytoin and lamotrigine, ranolazine also has little effect on the resting channel currents; however, ranolazine is able to block the channel in a use-dependent manner at lower concentrations much like these two anti-convulsants (Mantegazza et al., 2010; Thompson et al., 2011). Ranolazine is also similar to other antiepileptics in its ability to stabilize the slow-inactivated states (Lang et al., 1993; Xie et al., 1995). The stabilization of the slow-inactivated state and the increases in UDI are expected to decrease sodium currents over longer periods of repetitive channel opening, thereby limiting cell excitability in the manner of other antiepileptics (Willow et al., 1985).
Lastly, our data suggest directions for future experiments on ranolazine. One set of experiments would focus on ranolazine's ability to modulate channels with mutations underlying inheritable epilepsy. The second would focus on the binding mechanism of ranolazine and why extracellular protons slow this process, thus providing more information on ranolazine's effectiveness in treating ischaemia and low pH conditions. One experiment to test the hypothesis that ranolazine protonation impairs the ability of the drug to cross the plasma membrane is to introduce the drug intracellularly.
Acknowledgments
The authors thank Dr. Yury Y. Vilin, Mohammed Hassan-Ali and Csilla Egri for input on experimental technique, David K Jones for his insightful advice and remarks on data collection and interpretation.
This work was supported by a research grant from Gilead Sciences, Inc., a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (PCR), and an NSERC Undergraduate Research Scholarship Award (CHS).
Glossary
- ENa
Reversal potential for sodium
- G(V)
Conductance
- GEFS+
Generalized epilepsy with febrile seizures plus
- INa
fast sodium current
- INaL
late sodium current
- NaV1.1
Voltage-gated sodium channel encoded by the SCN1A gene
- NaV1.2
Voltage-gated sodium channel encoded by the SCN2A gene
- NaV1.4
Voltage-gated sodium channel encoded by the SCN4A gene
- NaV1.7
Voltage-gated sodium channel encoded by the SCN9A gene
- NaV1.8
Voltage-gated sodium channel encoded by the SCN10A gene
- NCX
sodium calcium exchanger
- &tau
Time Constant
- SSFI
steady-state fast inactivation
- SSSI
steady-state slow inactivation
- UDI
Use-dependent inactivation
- V1/2
midpoint
- Vm
membrane potential
- z,
apparent valence
Conflict of interest
S. Rajamani is an employee of Gilead Sciences, Inc. manufacturer and distributor of ranolazine. This work was supported in part by a research grant from Gilead Sciences, Inc.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition (2011) Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrup J, Symon L, Branston N, Lassen N. Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke. 1977;8:51–57. doi: 10.1161/01.str.8.1.51. [DOI] [PubMed] [Google Scholar]
- Berkovic S, Heron S, Giordano L, Marini C, Guerrini R, Kaplan R, et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol. 2004;55:550–557. doi: 10.1002/ana.20029. [DOI] [PubMed] [Google Scholar]
- Camilo O, Goldstein L. Seizures and epilepsy after ischemic stroke. Stroke. 2004;35:1769–1775. doi: 10.1161/01.STR.0000130989.17100.96. [DOI] [PubMed] [Google Scholar]
- Estacion M, Waxman SG, Dib-Hajj SD. Effects of ranolazine on wild-type and mutant hNaV 1.7 channels and on DRG neuron excitability. Mol Pain. 2010;6:1–13. doi: 10.1186/1744-8069-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Extramiana F, Antzelevitch C. Amplified Transmural Dispersion of Repolarization as the Basis for Arrhythmogenesis in a Canine Ventricular-Wedge Model of Short-QT Syndrome. Circulation. 2004;110:3661–3666. doi: 10.1161/01.CIR.0000143078.48699.0C. [DOI] [PubMed] [Google Scholar]
- George A. Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlig KM, Lepist I, Leung K, Rajamani S, George AL. Ranolazine selectively blocks persistent current evoked by epilepsy – associated NaV1.1 mutations. Br J Pharmacol. 2010;161:1414–1426. doi: 10.1111/j.1476-5381.2010.00976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang DG, Wang CM, Cooper BR. Lamotrigine, Phenytoin and Carbamazepine Interactions on the Sodium Current Present in N4TG1 Mouse Neuroblastoma Cells. J Pharmacol Exp Ther. 1993;266:829–835. [PubMed] [Google Scholar]
- Mantegazza M, Curia G, Biagini G, Ragsdale D, Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol. 2010;9:413–424. doi: 10.1016/S1474-4422(10)70059-4. [DOI] [PubMed] [Google Scholar]
- Maruki Y, Koehler RC, Eleff SM, Traystman RJ. Intracellular pH during reperfusion influences evoked potential recovery after complete cerebral ischemia. Stroke. 1993;24:697–703. doi: 10.1161/01.str.24.5.697. [DOI] [PubMed] [Google Scholar]
- Meyer FB. Intracellular brain pH and ischemic vasoconstriction in the white New Zealand rabbit. Stroke. 1990;21:IV117–IV119. [PubMed] [Google Scholar]
- Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol. 2008;19:1289–1293. doi: 10.1111/j.1540-8167.2008.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepine CJ, Wolff AA. A controlled trial with a novel anti-ischemic agent, ranolazine, in chronic stable angina pectoris that is responsive to conventional antianginal agents. Am J Cardiol. 1999;84:46–50. doi: 10.1016/s0002-9149(99)00190-3. [DOI] [PubMed] [Google Scholar]
- Rajamani S, Shryock JC, Belardinelli L. Block of tetrodotoxin-sensitive, NaV1.7 and tetrodotoxin-resistant, NaV1.8, Na Channels by ranolazine. Channels. 2008a;2:449–460. doi: 10.4161/chan.2.6.7362. [DOI] [PubMed] [Google Scholar]
- Rajamani S, Shryock JC, Belardinelli L. Rapid kinetic interactions of ranolazine with HERG K channels. J Cardiovasc Pharmacol. 2008b;51:581–589. doi: 10.1097/FJC.0b013e3181799690. [DOI] [PubMed] [Google Scholar]
- Scalmani P, Rusconi R, Armatura E, Zara F, Avanzini G, Franceschetti S, et al. Effects in neocortical neurons of mutations of the NaV1.2 Na+ Channel causing benign familial neonatal-infantile seizures. J Neurosci. 2006;26:10100–10109. doi: 10.1523/JNEUROSCI.2476-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM, et al. Effects of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non – ST-segment – elevation acute coronary syndrome: results from the metabolic efficiency with ranolazine for less ischemia in non – ST – elevation acute coronary syndrome – thrombolysis in myocardial infarction 36 (MERLIN – TIMI 36) randomized controlled trial. Circulation. 2007;116:1647–1652. doi: 10.1161/CIRCULATIONAHA.107.724880. [DOI] [PubMed] [Google Scholar]
- Siemkowicz E, Hansen A. Brain extracellular ion composition and EEG activity following 10 minutes ischemia in normo- and hyperglycemic rats. Stroke. 1981;12:236–240. doi: 10.1161/01.str.12.2.236. [DOI] [PubMed] [Google Scholar]
- Sossalla S, Wagner S, Rasenack EC, Ruff H, Weber SL, Schondube FA, et al. Ranolazine Improves diastolic dysfunction in isolated myocardium from failing human hearts – role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;45:32–43. doi: 10.1016/j.yjmcc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Stone PH, Chaitman BR, Stocke K, Sano J, DeVault A, Koch GG. The Anti-Ischemic Mechanism of Action of Ranolazine in Stable Ischemic Heart Disease. J Am Coll Cardiol. 2010;56:934–942. doi: 10.1016/j.jacc.2010.04.042. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Tsurubuchi Y, Agarwala K, Ito M, Fukuma G, Mazaki-Miyazaki E, et al. A missense mutation of the Na+ channel αII subunit gene Nav1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A. 2001;98:6384–6389. doi: 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CH, Kahlig KM, George AL. SCN1A splice variants exhibit divergent sensitivity to commonly used antiepileptic drugs. Epilepsia. 2011;52:1000–1009. doi: 10.1111/j.1528-1167.2011.03040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J Cardiovasc Electrophysiol. 2006;17:S169–S177. doi: 10.1111/j.1540-8167.2006.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilin YY, Peters CH, Ruben PC. Acidosis differentially modulates inactivation in NaV1.2, NaV1.4, and NaV1.5 channels. Front Pharmacol. 2012;3:109. doi: 10.3389/fphar.2012.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Calderon J, Wang S-Y. State- and use-dependent block of muscle NaV1.4 and neuronal NaV1.7 voltage-gated Na channel isoforms by ranolazine. Mol Pharmacol. 2008;73:940–948. doi: 10.1124/mol.107.041541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willow M, Gonoi T, Catterall WA. Voltage clamp analysis of the inhibitory actions of diphenylhydantoin and carbamazepine on voltage-sensitive sodium channels in neuroblastoma cells. Mol Pharmacol. 1985;27:549–558. [PubMed] [Google Scholar]
- Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L. Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther. 2004;310:599–605. doi: 10.1124/jpet.104.066100. [DOI] [PubMed] [Google Scholar]
- Xie X, Lancaster B, Peakman T. Interaction of the antiepileptic drug lamotrigine with recombinant rat brain type IIA Na channels and with native Na channels in rat hippocampal neurones. Pflugers Arch. 1995;430:437–446. doi: 10.1007/BF00373920. [DOI] [PubMed] [Google Scholar]