

Abstract
In innate immune system cells, such as macrophages and dendritic cells, deployment of inducible gene expression programmes in response to microbes and danger signals requires highly precise regulatory mechanisms. The inflammatory response has to be tailored based on both the triggering stimulus and its dose, and it has to be unfolded in a kinetically complex manner that suits the different phases of the inflammatory process. Genomic characterization of regulatory elements in this context indicated that transcriptional regulators involved in macrophage specification act as pioneer transcription factors (TFs) that generate regions of open chromatin that enable the recruitment of TFs activated in response to external inputs. Therefore, competence for responses to a specific stimulus is programmed at an early stage of differentiation by factors involved in lineage commitment and maintenance of cell identity, which are responsible for the organization of a cell-type-specific cis-regulatory repertoire. The basic functional and organizational principles that regulate inflammatory gene expression in professional cells of the innate immune system provide general paradigms on the interplay between differentiation and environmental responses.
Keywords: chromatin, epigenome, inflammation, enhancers, Pu.1, macrophages
1. Introduction
Transcription of eukaryotic genes is a highly coordinated process that is regulated in time and space. At the heart of transcriptional control is the vast amount of DNA-encoded regulatory information contained in mammalian genomes and located both right upstream of transcription start sites and at distal regulatory elements, particularly enhancers.
Cell-type-specific usage of the vast genomic repertoire of cis-regulatory sequences is accomplished mainly by the combinatorial activity of sequence-specific transcription factors (TFs) involved in lineage determination and maintenance of cell identity [1]. Genomic regions that are active as enhancers in a given cell type typically reside in regions with altered chromatin structure, as revealed by hypersensitivity to DNAse enzymes and nucleosomal depletion [2–5]. In addition, genome-wide studies indicate that enhancers exhibit a characteristic chromatin ‘signature’, consisting of monomethylation of lysine 4 in histone H3 (H3K4me1) in the absence of significant trimethylation (H3K4me3) [6–9]. Additional marks associated with active enhancers include binding of histone acetyltransferases such as p300 and CBP, and histone acetylation (most notably, but not at all exclusively H3K27) [10–13]. High-resolution maps of these histone modifications indicated that the regions of the genome bearing a chromatin signature of enhancers are generated in a cell-type-specific manner [12,14–17]. According to current models, the cell-type specificity of these regulatory regions is due to the presence of binding sites for cell-type-specific TFs that are directly implicated in their functionalization. Evidence accumulated during the past few years suggests that lineage-specific TFs organize the enhancer landscape required for differential recruitment of TFs involved in gene expression programmes during development or in response to external stimuli [18–22]. Although this model implies that the enhancer repertoire in a given cell type is predetermined, the emerging picture indicates that chromatin modifications at these genomic regulatory elements can be highly dynamic in order to allow cell-type-specific epigenomes to acquire their characteristic properties or to respond to signals received from the microenvironment. In this context, the epigenome of cells whose main biological function is to sense and to react to environmental inputs, such as cells of the innate immune system, has to rapidly change according to the received signals. This review will focus on the epigenomic landscape of inflammatory cells as a paradigmatic example of how the genome can be organized to enable highly specialized responses to a broad array of external stimuli.
2. The epigenome of inflammatory cells
The innate immune system provides initial protection against infection, injury and tissue stress. Genes encoding proteins with antimicrobial and proinflammatory activities must be rapidly and highly induced in the presence of an invading microbe as well as upon release of the intracellular content of damaged tissues. At the same time, inflammatory genes must be maintained in a transcriptionally repressed state under normal homeostatic conditions.
(a). General features of the inflammatory gene expression programme
The complexity of the inflammatory response requires several hundreds genes to be activated in a kinetically complex fashion, with some genes rapidly activated immediately after the stimulus (‘primary’ response genes) and others induced with slower kinetics (‘secondary’ response genes and some slowly activated primary response genes). Regardless of their activation kinetics, primary response genes are formally defined as those genes that can be induced without de novo protein synthesis, while secondary genes require new protein synthesis [23]. Moreover, some genes, such as the one encoding TNFα, are activated with similar patterns in almost every type of inflammatory responses while others (that encode proteins with more specialized functions) are tightly regulated in a stimulus-specific and cell-type-restricted manner [24]. During an inflammatory response, a large number of genes are coordinately induced by a limited set of TFs that are constitutively expressed by many cell types, such as NF-κB, IFN-regulatory factors (IRFs) and AP-1 family members. In spite of the involvement of these general, broadly expressed regulators, inflammatory gene expression is both cell-type- and stimulus-specific, an observation that until recently did not have an adequate mechanistic explanation. Clearly, the transcriptional selectivity has to be established by other regulatory layers.
First of all, analysis of the genomic features of the promoters of primary response genes revealed a striking enrichment for CpG islands, a sequence feature that directly or indirectly (via recruitment of the general transcriptional machinery) interferes with the assembly of stable nucleosomes [25–27]. Conversely, promoters with a lower CpG content are in general associated with secondary response genes. Consistently, nucleosome remodelling and displacement by the SWI/SNF complex is necessary for the induction of secondary response genes, while most early primary response genes are SWI/SNF-independent. These observations suggest that the sole presence of a CpG island is sufficient to create a nucleosomal state permissive for rapid gene activation in response to a stimulus. Interestingly, whereas the promoters of secondary response genes undergo stimulus-dependent H3K4me3 and H3 acetylation, the promoters of SWI–SNF-independent primary response are almost invariantly associated already in the basal state with high levels of chromatin modifications characteristic of active chromatin [26]. In addition, primary response genes associated with CpG islands are constitutively associated with RNA polymerase II and transcribed at low levels [26]. Overall, it is clear that a different sequence-programmed chromatin organization at individual inflammatory genes lays the grounds for different expression kinetics.
(b). Organization and function of cis-regulatory regions in inflammatory cells
Recent studies on macrophages, cells that play a central role in the inflammatory response, linked changes in chromatin structure to molecular events occurring during lineage commitment and development that provide competence for subsequent transcriptional induction. ChIP-Seq studies revealed that almost the entire macrophage enhancer repertoire is constitutively bound by the ETS TF Pu.1/Spi1 [10]. The ETS family includes almost 30 members that can be assigned to four classes based on their binding specificity [28]. Pu.1 and its paralogues, Spib and Spic, recognize a highly specific sequence that differs at a few critical positions (mainly at the 5′ of the binding site) from the binding sites of all other ETS proteins, thus laying the foundations for their specificity [28]. Pu.1 is the essential macrophage-determining TF: it is constantly expressed at high levels in macrophages and is required to induce and to maintain macrophage differentiation and viability [29]. Analysis of the genomic distribution of Pu.1 in macrophages showed that it is constitutively associated with nearly all genomic enhancers marked by H3K4me1 [10]. In B cells, where Pu.1 concentration is about 10-fold lower than in macrophages, Pu.1 distribution (as well as the enhancer repertoire) is completely different, which might reflect a higher dependence on cooperative interactions provided by B-cell-specific partner TFs [30].
Taken together, these studies suggest that Pu.1, presumably in combination with critical regulators of macrophage development, binds enhancer elements during macrophage development to promote the creation of a macrophage-specific enhancer landscape. Importantly, Pu.1 binding is sufficient to promote the deposition of H3K4me1, enabling changes in chromatin structure. Specifically, the creation of a nucleosome-free region by Pu.1 is essential for the recruitment of non-cell-type-specific TFs responsive to external stimuli, such as NF-κB, AP-1 or IRF family members or for the binding of transcriptional repressors, such as Bcl-6 [31]. Moreover, recent results showed that Pu.1 also contributes to the basal activation state and H3K4 trimethylation of a subset of inducible promoters [32].
These results clearly suggest a model where the cell-type specificity of genes induced in response to a stimulus is determined during development when cis-regulatory regions for the inducible genes become associated with, and functionalized by, lineage-determining TFs.
3. Pioneer transcription factors and the control of cis-regulatory regions
Recent technological advances have started to elucidate the timing and mechanisms by which specific enhancers acquire unique chromatin signatures. Evidence accumulated during the last few years indicates that during development some TFs start marking tissue-specific regulatory elements. Pioneer factors are functionally defined as sequence-specific DNA-binding proteins able to bind to their target sites when embedded in a nucleosomal context that is not permissive for binding of other TFs. Subsequent recruitment of chromatin remodellers by pioneer factors results in stable local opening of the chromatin, thus making it competent for other factors to bind (figure 1). Once bound, pioneer factors act in some cases as placeholders that will be replaced by other TFs at later stages of development [33]. In this context, a relay model, involving the replacement of pluripotent factors at some primed regions by related lineage-specific TFs has been proposed, such as the replacement of Sox2 by Sox4 on primed regions during B-cell differentiation [34].
Figure 1.
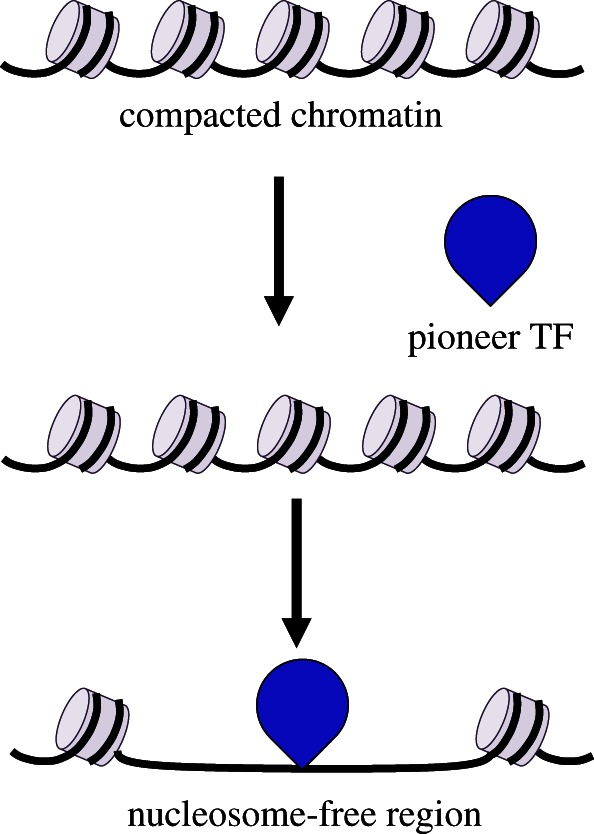
Pioneer TFs bind and activate cis-regulatory regions. During differentiation, pioneer TFs have nucleosome-binding properties that allow them to actively open condensed chromatin and to recruit other TFs, chromatin modifiers and nucleosome remodellers. Pioneer TFs binding is sufficient to enable change in chromatin structure to create a nucleosome-free region.
Also in completely differentiated cells, the emerging picture is that pioneer TFs can access their binding sites even when wrapped in nucleosomes. Consistent with this model, these lineage-specific TFs are actively involved in determining the baseline accessible chromatin landscape in order to facilitate the recruitment of other TFs unable to invade nucleosomes, including many of those responsive to environmental stimuli.
(a). Role of PU.1 as a global genome organizer in inflammatory cells
In macrophages, the observations reported above suggest a model wherein the global landscape determined by Pu.1 provides the context in which the dynamic TF–chromatin interactions in response to specific environmental inputs occur. Pu.1 expression in non-myeloid cells or in Pu.1-negative myeloid progenitors is sufficient to induce nucleosome-free DNA stretches limited on both sides by H3K4me1-marked nucleosomes at the same genomic regions identified as enhancers in macrophages [10]. Therefore, these data clearly indicate that Pu.1 is not a simple marker of enhancers but indeed controls formation and accessibility of the entire genomic regulatory repertoire of macrophages. Such activity implies that Pu.1 must be able to bind nucleosomal sites, attracting chromatin-remodelling factors to stabilize nucleosome displacement. Whether Pu.1 has the ability to directly recruit chromatin-remodelling complexes remains to be established. Importantly, the role of Pu.1 as master regulator implies that it is able to activate and maintain as enhancers the genomic regions it binds to: the continuous presence of Pu.1 is absolutely required for the maintenance of the macrophage epigenome and to enable the subsequent activation of the inflammatory gene expression programme [1].
Recently, a study on GATA1 binding during erythroid differentiation revealed that the catalytic subunit BRG1 of the Swi/Snf chromatin-remodelling complex mediates nucleosome repositioning at GATA1 enhancers. GATA1-mediated recruitment of BRG1 and subsequent nucleosome shifting facilitate binding of TAL1, a key TF for haematopoiesis [2]. An alternative possibility is mere competition between nucleosomes and TF for the same site. Studies on binding dynamics at genomic scale recently suggested a model wherein nucleosomes and TFs compete for binding to the same regulatory regions. TF binding to these regions occurs in short pulses, not sufficient for efficient transcription. When the nucleosome is removed or its affinity for DNA is decreased, the TF can quickly achieve stable binding to its target sequence [35].
This mechanism of regulation of enhancers controlling environmental responses is obviously not restricted to innate immunity cells but applies to other systems and implies a prerequisite for chromatin priming by one TF for the secondary recruitment of other regulatory factors (summarized in table 1). For example, AP-1 can facilitate the selective access of the glucocorticoid receptor (GR) to specific sites in the genome by maintaining an accessible chromatin environment in a murine epithelial cell line [41]. In breast cancer cells, the Forkhead protein FOXA1/HNF3α was shown to be required for ER binding, thus probably functioning as a pioneer factor [38]. FOXA1 influences genome-wide chromatin accessibility and almost all ER–chromatin interactions and gene expression changes are dependent on the presence of FOXA1. Consistent with this idea, FoxA1 can facilitate the access of regulated TFs, as exemplified by the androgen receptor (AR), acting on distinct classes of enhancers in prostate cancer cells [37].
Table 1.
Summary of studies on master regulators that generate regions of open chromatin enabling the recruitment of stimulus-activated TFs in different cell types.
| cell type | master regulator | stimulus | stimulus-activated TFs | references |
|---|---|---|---|---|
| dendritic cells | Pu.1 | LPS | NF-κB, STATs, AP-1, IRFs | [36] |
| LNCaP (prostate epithelium) | FoxA1 | 5α-dihydrotestosterone | AR | [37] |
| MCF-7 (breast epithelium) | FoxA1 | tamoxifen, oestradiol | ERα | [38] |
| MCF-7 (breast epithelium) | FoxA1, AP2γ | oestradiol | ERα | [39] |
| U2OS (osteoblast-like cells) | GATA4 | oestradiol | ERα | [40] |
| 3134 (breast epithelium) | AP-1 | dexamethasone | GR | [41] |
| ESCs | OCT4 | TGFβ | Smad2/3 | [42] |
| myotubes | Myo-D | TGFβ | Smad2/3 | [42] |
| Pro-B cells | Pu.1 | TGFβ | Smad2/3 | [42] |
| macrophages | Pu.1 | LPS | Bcl-6 | [31] |
| macrophages | Pu.1 | LPS | NF-κB | [10] |
In conclusion, lineage-specific factors coupled with other general transcriptional factors generate a cell-type-specific enhancer network, allowing other regulated factors to access regulatory DNA embedded in an originally not accessible context.
(b). Dynamic changes of cis-regulatory regions in response to stimuli
So far, several studies have reported that dynamic changes in chromatin marks correlate with transcriptional activity. H3K4me1 represents a general mark of distal regulatory regions and additional modifications can distinguish between enhancers that are active and those that are poised and can subsequently be activated during developmental transitions or in response to external stimuli. Recent findings pointed out that only a fraction of distal H3K4me1 positive regions involved in modulating transcription in a given cell type are also marked by H3K27 acetylation [43–45]. These enhancers are defined as active, as opposed to inactive and poised enhancers containing H3K4me1 only [43,44]. Further computational analyses defined additional sub-categories of active (H3K4me1/H3K27Ac positive), poised (H3K4me1/H3K27me3 positive), or intermediate (H3K4me1 positive/H3K27Ac negative) enhancers using H3K27Ac in combination with other chromatin features [46].
The enhancer repertoire marked by H3K4me1 evolves during development in response to associated changes in the repertoire of TFs expressed during (and controlling) different stages of differentiation. For example, during the developmental progression from the multipotent haematopoietic progenitor to a committed lymphoid or myeloid cell stage, lineage-restricted enhancer elements were found to be already primed by H3K4me1 prior to differentiation [47]. Upon commitment to the B or myeloid lineages, an increase in H3K4me1 was observed across genomic regions associated with a B-cell or myeloid lineage gene expression programme, respectively. Thus, developmental transitions are associated with dynamic changes in the H3K4me1 repertoire.
More importantly, the mechanism by which the same stimulus is able to activate a distinct enhancers repertoire in different cells has been described. From a conceptual point of view, the general principle is that master regulators are responsible for determining the cell-type-specific effects of the same signalling pathway. In a paradigmatic case, the cell-type-specific effects of TGFβ signalling were found to be determined by the interaction of the TGFβ-activated TFs Smad2/3 with TFs that specify and maintain cell identity [42]. The genomic distribution of Smad3 was found to overlap Oct4 in embryonic stem cells (ESCs), Myod1 in myotubes and Pu.1 in pro-B cells. These cell-type-specific master regulators allow Smad3 binding by establishing open chromatin accessible regions first and by tethering Smad3 to them via protein–protein interactions when the signal is delivered [42].
In the innate immune system, external inputs such as proinflammatory stimuli are expected to have a broad impact on chromatin organization and marking at cis-regulatory elements. In macrophages, a recent genome-wide analysis confirmed that thousands of cis-regulatory regions gain H4 acetylation upon the proinflamatory stimulus lipopolysaccharide (LPS) as expected [48]. These studies also suggested a specific and non-redundant role of the histone deacetylase HDAC3 in controlling acetylation levels at a subset of genomic regions. In this context, it would be of interest to study how distinct stimuli can reorganize the enhancer repertoire of professional innate immune cells to clearly define general principles regulating the dynamic use of the available cis-regulatory information. It is possible that the impact of a broad array of stimuli on such highly specialized cells goes far beyond simple changes in acetylation and results in a reorganization of a subset of enhancers.
(c). Combinatorial control of transcription factors on cis-regulatory regions
From a mechanistic point of view, pioneer TFs acting as global genomic organizers in a lineage-restricted manner, need additional restricted or non-restricted TFs to bind and activate specific subsets of enhancers. Specific rules controlling functional cooperation in the presence or the absence of cooperative binding remain however to be defined. As discussed above, in macrophages Pu.1 controls the establishment of the enhancer repertoire. However, how it cooperates at a genomic level with other TFs to define or activate specific subsets of enhancers is poorly understood.
In this context, a major breakthrough was recently provided by Amit and co-workers [36] in a study describing genomic occupancy of a large number of TFs in mouse dendritic cells. A high-throughput chromatin immunoprecipitation method (HT-ChIP) was applied to build genome-wide dynamic maps of 25 TFs and four chromatin marks at different time-points following LPS stimulation [36]. Analysis of these dynamics showed that TF binding falls into at least three broad classes. The first class identified TFs with a very pervasive association with almost all regulatory elements in the genome. This group includes the pioneer TF Pu.1 and a very few additional TFs, such as Cebpb. The broad distribution of these TFs is compatible with their role as chromatin openers that facilitate access of a second group of TFs called ‘primers’, such as Junb, Irf4 and Atf3, that are able to prime for activation regions that are associated with stimulus-dependent gene induction. A third set of TFs (that includes NF-κB and Stat family members) bind dynamically specific set of genes in a stimulus-dependent manner and control gene expression induction (figure 2). Most importantly, the new idea that emerges from this study is that the type of transcriptional response to stimulation is established prior to stimulus delivery. In other words, many TF–DNA interactions are established predominantly at early stages before the treatment with the inflammatory stimulus. Therefore, the potential for proinflammatory genes to be transcriptionally induced is established before stimulation by binding of TF subset to their genomic cis-regulatory regions.
Figure 2.

Master Regulator Pu.1 organizes the enhancer landscape required for stimulus-induced transcription in innate immune system cells. The lineage-determining TF Pu.1 pervasively binds to regulatory elements in the genome of macrophages and dendritic cells. Pu.1 binding is sufficient to promote the deposition of H3K4me1 in a nucleosome-free region. Pu.1 binding is essential for the access of a second group of TFs called ‘primers’, such as Junb, Irf4 and Atf3 for dendritic cells, able to prime for activation regions that are associated with stimulus-dependent gene induction. Environmental stimuli (e.g. response to LPS) trigger the recruitment of stimulus-dependent TFs, such as NF-κB, AP-1, IRFs and STATs family members, to a specific set of proinflammatory genes.
4. Concluding remarks and perspectives
Recent advances in high-throughput sequencing technologies have provided an increasingly sophisticated picture of the mechanisms that regulate inflammatory gene expression programmes in cells of the innate immune system. The data reviewed here indicate that the molecular events occurring during lineage commitment, such as TF binding and changes in chromatin structure, provide competence for subsequent transcriptional induction. However, it is important to conclude by emphasizing that this general framework cannot fully describe the complexity of the inflammatory response and more in general of inducible responses activated by external stimuli. A higher level of data integration, such as that achieved by the ENCODE (Encyclopedia of DNA Elements) consortium project will allow in the future to obtain a more detailed view of coordinated changes in gene expression in response to stimulation [49–52]. In particular, DNAse I footprints coupled with ChIP-seq occupancy maps might be useful to better characterize highly cell-selective and dynamic occupancy patterns and the combinatorial regulation of tissue-specific TF that underlies specific functions.
Additional aspects will have to be included in these models, such as the role of TFs in controlling the three-dimensional network of interactions between distal regulatory elements and target genes, as well as the role of non-coding RNAs, including the enhancer-templated ones [37,53–56]. In this context, topological studies performed in different model system, such as the α- and β-globin gene loci, the immunoglobulin and other antigen receptor gene loci, the imprinted H19–Igf2 locus and the Hox gene clusters, have provided evidence that regulatory DNA sequences can control transcription over a long distance by physically contacting target genes via chromatin looping [57–60]. It will be of interest to define general principles describing how chromatin contacts dynamically change in response to a changing environment, such as the delivery of an inflammatory stimulus.
It is also important to realize that individual genome-wide binding experiments provide a static picture of a dynamic process, where the information of a single TF binding is not enough to predict the regulatory outcome on the target gene. In addition, the current ChIP-seq technologies are not sufficient to determine the true nature of transcription in live cells or at single cell levels. Technical improvements in this technology will allow identifying at greater resolution all TFs binding events in a specific regulatory region and ultimately to understand how they impact on gene expression.
Acknowledgements
Studies of transcriptional regulation in cells of the innate immune system in GN laboratory are funded by ERC (to G.N.) and Italian Ministry of Health (to S.G.).
References
- 1.Natoli G. 2010. Maintaining cell identity through global control of genomic organization. Immunity 33, 12–24 10.1016/j.immuni.2010.07.006 (doi:10.1016/j.immuni.2010.07.006) [DOI] [PubMed] [Google Scholar]
- 2.Hu G, et al. 2011. Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res. 21, 1650–1658 10.1101/gr.121145.111 (doi:10.1101/gr.121145.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He HH, et al. 2010. Nucleosome dynamics define transcriptional enhancers. Nat. Genet. 42, 343–347 10.1038/ng.545 (doi:10.1038/ng.545) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. 2008. Dynamic regulation of nucleosome positioning in the human genome. Cell 132, 887–898 10.1016/j.cell.2008.02.022 (doi:10.1016/j.cell.2008.02.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song L, et al. 2011. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 21, 1757–1767 10.1101/gr.121541.111 (doi:10.1101/gr.121541.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heintzman ND, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 10.1038/ng1966 (doi:10.1038/ng1966) [DOI] [PubMed] [Google Scholar]
- 7.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 10.1016/j.cell.2007.05.009 (doi:10.1016/j.cell.2007.05.009) [DOI] [PubMed] [Google Scholar]
- 8.Visel A, Rubin EM, Pennacchio LA. 2009. Genomic views of distant-acting enhancers. Nature 461, 199–205 10.1038/nature08451 (doi:10.1038/nature08451) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou VW, Goren A, Bernstein BE. 2011. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 12, 7–18 10.1038/nrg2905 (doi:10.1038/nrg2905) [DOI] [PubMed] [Google Scholar]
- 10.Ghisletti S, et al. 2010. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity 32, 317–328 10.1016/j.immuni.2010.02.008 (doi:10.1016/j.immuni.2010.02.008) [DOI] [PubMed] [Google Scholar]
- 11.Visel A, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858 10.1038/nature07730 (doi:10.1038/nature07730) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heintzman ND, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 10.1038/nature07829 (doi:10.1038/nature07829) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z, et al. 2008. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903 10.1038/ng.154 (doi:10.1038/ng.154) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ernst J, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 10.1038/nature09906 (doi:10.1038/nature09906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.May D, et al. 2011. Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 44, 89–93 10.1038/ng.1006 (doi:10.1038/ng.1006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pennacchio LA, Loots GG, Nobrega MA, Ovcharenko I. 2007. Predicting tissue-specific enhancers in the human genome. Genome Res. 17, 201–211 10.1101/gr.5972507 (doi:10.1101/gr.5972507) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blow MJ, et al. 2010. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 42, 806–810 10.1038/ng.650 (doi:10.1038/ng.650) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ram O, et al. 2011. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell 147, 1628–1639 10.1016/j.cell.2011.09.057 (doi:10.1016/j.cell.2011.09.057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He A, Kong SW, Ma Q, Pu WT. 2011. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl Acad. Sci. USA 108, 5632–5637 10.1073/pnas.1016959108 (doi:10.1073/pnas.1016959108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tijssen MR, et al. 2011. Genome-wide analysis of simultaneous GATA1/2, RUNX1, FLI1, and SCL binding in megakaryocytes identifies hematopoietic regulators. Dev. Cell 20, 597–609 10.1016/j.devcel.2011.04.008 (doi:10.1016/j.devcel.2011.04.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Junion G, Spivakov M, Girardot C, Braun M, Gustafson EH, Birney E, Furlong EE. 2012. A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell 148, 473–486 10.1016/j.cell.2012.01.030 (doi:10.1016/j.cell.2012.01.030) [DOI] [PubMed] [Google Scholar]
- 22.Bulger M, Groudine M. 2011. Functional and mechanistic diversity of distal transcription enhancers. Cell 144, 327–339 10.1016/j.cell.2011.01.024 (doi:10.1016/j.cell.2011.01.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herschman HR. 1991. Primary response genes induced by growth factors and tumor promoters. Annu. Rev. Biochem. 60, 281–319 10.1146/annurev.bi.60.070191.001433 (doi:10.1146/annurev.bi.60.070191.001433) [DOI] [PubMed] [Google Scholar]
- 24.Nau GJ, Richmond JF, Schlesinger A, Jennings EG, Lander ES, Young RA. 2002. Human macrophage activation programs induced by bacterial pathogens. Proc. Natl Acad. Sci. USA 99, 1503–1508 10.1073/pnas.022649799 (doi:10.1073/pnas.022649799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, Sidow A. 2011. Determinants of nucleosome organization in primary human cells. Nature 474, 516–520 10.1038/nature10002 (doi:10.1038/nature10002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramirez-Carrozzi VR, et al. 2009. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138, 114–128 10.1016/j.cell.2009.04.020 (doi:10.1016/j.cell.2009.04.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hargreaves DC, Horng T, Medzhitov R. 2009. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145 10.1016/j.cell.2009.05.047 (doi:10.1016/j.cell.2009.05.047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei GH, et al. 2010. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 29, 2147–2160 10.1038/emboj.2010.106 (doi:10.1038/emboj.2010.106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nerlov C, Graf T. 1998. PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev. 12, 2403–2412 10.1101/gad.12.15.2403 (doi:10.1101/gad.12.15.2403) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinz S, et al. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 10.1016/j.molcel.2010.05.004 (doi:10.1016/j.molcel.2010.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, Dent AL, Tangirala RK, Evans RM. 2010. Bcl-6 and NF-κB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 24, 2760–2765 10.1101/gad.1998010 (doi:10.1101/gad.1998010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Escoubet-Lozach L, et al. 2011. Mechanisms establishing TLR4-responsive activation states of inflammatory response genes. PLoS Genet. 7, e1002401. 10.1371/journal.pgen.1002401 (doi:10.1371/journal.pgen.1002401) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaret KS, Carroll JS. 2011. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 25, 2227–2241 10.1101/gad.176826.111 (doi:10.1101/gad.176826.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liber D, Domaschenz R, Holmqvist PH, Mazzarella L, Georgiou A, Leleu M, Fisher AG, Labosky PA, Dillon N. 2010. Epigenetic priming of a pre-B cell-specific enhancer through binding of Sox2 and Foxd3 at the ESC stage. Cell Stem Cell 7, 114–126 10.1016/j.stem.2010.05.020 (doi:10.1016/j.stem.2010.05.020) [DOI] [PubMed] [Google Scholar]
- 35.Lickwar CR, Mueller F, Hanlon SE, McNally JG, Lieb JD. 2012. Genome-wide protein-DNA binding dynamics suggest a molecular clutch for transcription factor function. Nature 484, 251–255 10.1038/nature10985 (doi:10.1038/nature10985) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garber M, et al. 2012. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol. Cell 47, 810–822 10.1016/j.molcel.2012.07.030 (doi:10.1016/j.molcel.2012.07.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang D, et al. 2011. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474, 390–394 10.1038/nature10006 (doi:10.1038/nature10006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. 2011. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 43, 27–33 10.1038/ng.730 (doi:10.1038/ng.730) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan SK, Lin ZH, Chang CW, Varang V, Chng KR, Pan YF, Yong EL, Sung WK, Cheung E. 2011. AP-2γ regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. Embo J. 30, 2569–2581 10.1038/emboj.2011.151 (doi:10.1038/emboj.2011.151) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miranda-Carboni GA, Guemes M, Bailey S, Anaya E, Corselli M, Peault B, Krum SA. 2011. GATA4 regulates estrogen receptor-alpha-mediated osteoblast transcription. Mol. Endocrinol. 25, 1126–1136 10.1210/me.2010-0463 (doi:10.1210/me.2010-0463) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biddie SC, et al. 2011. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell 43, 145–155 10.1016/j.molcel.2011.06.016 (doi:10.1016/j.molcel.2011.06.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mullen AC, et al. 2011. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 147, 565–576 10.1016/j.cell.2011.08.050 (doi:10.1016/j.cell.2011.08.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Creyghton MP, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA 107, 21 931–21 936 10.1073/pnas.1016071107 (doi:10.1073/pnas.1016071107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 10.1038/nature09692 (doi:10.1038/nature09692) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, Zhao K. 2009. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 4, 80–93 10.1016/j.stem.2008.11.011 (doi:10.1016/j.stem.2008.11.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zentner GE, Tesar PJ, Scacheri PC. 2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 21, 1273–1283 10.1101/gr.122382.111 (doi:10.1101/gr.122382.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mercer EM, et al. 2011. Multilineage priming of enhancer repertoires precedes commitment to the B and myeloid cell lineages in hematopoietic progenitors. Immunity 35, 413–425 10.1016/j.immuni.2011.06.013 (doi:10.1016/j.immuni.2011.06.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen X, et al. 2012. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl Acad. Sci. USA 109, E2865–E2874 10.1073/pnas.1121131109 (doi:10.1073/pnas.1121131109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Project Consortium et al. 2012. An integrated encyclopedia of DNA elements in the human genome. ENCODE Nature 489, 57–74 10.1038/nature11247 (doi:10.1038/nature11247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thurman RE, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489, 75–82 10.1038/nature11232 (doi:10.1038/nature11232) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neph S, et al. 2012. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489, 83–90 10.1038/nature11212 (doi:10.1038/nature11212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerstein MB, et al. 2012. Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100 10.1038/nature11245 (doi:10.1038/nature11245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Santa F, et al. 2010. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 8, e1000384. 10.1371/journal.pbio.1000384 (doi:10.1371/journal.pbio.1000384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rinn JL, Chang HY. 2012. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166 10.1146/annurev-biochem-051410-092902 (doi:10.1146/annurev-biochem-051410-092902) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Natoli G, Andrau JC. 2012. Noncoding transcription at enhancers: general principles and functional models. Annu. Rev. Genet. 46, 1–19 10.1146/annurev-genet-110711-155459 (doi:10.1146/annurev-genet-110711-155459) [DOI] [PubMed] [Google Scholar]
- 56.Kim TK, et al. 2010. Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187 10.1038/nature09033 (doi:10.1038/nature09033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA. 2012. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 149, 1233–1244 10.1016/j.cell.2012.03.051 (doi:10.1016/j.cell.2012.03.051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo C, Gerasimova T, Hao H, Ivanova I, Chakraborty T, Selimyan R, Oltz EM, Sen R. 2011. Two forms of loops generate the chromatin conformation of the immunoglobulin heavy-chain gene locus. Cell 147, 332–343 10.1016/j.cell.2011.08.049 (doi:10.1016/j.cell.2011.08.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murrell A, Heeson S, Reik W. 2004. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat. Genet. 36, 889–893 10.1038/ng1402 (doi:10.1038/ng1402) [DOI] [PubMed] [Google Scholar]
- 60.Montavon T, Soshnikova N, Mascrez B, Joye E, Thevenet L, Splinter E, de Laat W, Spitz F, Duboule D. 2011. A regulatory archipelago controls Hox genes transcription in digits. Cell 147, 1132–1145 10.1016/j.cell.2011.10.023 (doi:10.1016/j.cell.2011.10.023) [DOI] [PubMed] [Google Scholar]