

Abstract
Atmospheric organic nitrogen (ON) appears to be a ubiquitous but poorly understood component of the atmospheric nitrogen deposition flux. Here, we focus on the ON components that dominate deposition and do not consider reactive atmospheric gases containing ON such as peroxyacyl nitrates that are important in atmospheric nitrogen transport, but are probably not particularly important in deposition. We first review the approaches to the analysis and characterization of atmospheric ON. We then briefly summarize the available data on the concentrations of ON in both aerosols and rainwater from around the world, and the limited information available on its chemical characterization. This evidence clearly shows that atmospheric aerosol and rainwater ON is a complex mixture of material from multiple sources. This synthesis of available information is then used to try and identify some of the important sources of this material, in particular, if it is of predominantly natural or anthropogenic origin. Finally, we suggest that the flux of ON is about 25 per cent of the total nitrogen deposition flux.
Keywords: nitrogen, organic nitrogen, atmosphere
1. Introduction
The role of organic matter in the atmosphere attracts interest and much current research, both as a vector of global biogeochemical cycles and also because of the potential role of aerosol organic matter within the Earth's radiation budget and in cloud processes [1–4]. The focus in this contribution is on a sub-component of the atmospheric organic matter, organic nitrogen (ON) and its role within the global nitrogen cycle, a cycle which is known to be considerably disturbed by human activity [5]. We concentrate here on ON in rain and aerosol. There are a few gaseous ON compounds, including peroxyacyl nitrates, HCN, HNCO and CH3CN and low molecular weight alkyl nitrates that are important components of the global nitrogen transport system. However, these compounds are not efficiently deposited, although they can become efficiently deposited after chemical reaction [6], and hence probably not of major importance in terms of overall nitrogen deposition and are not specifically considered here. Seitzinger & Sanders [7] evaluated the biological availability of atmospheric ON and concluded that a large part of it was rapidly used by micro-organisms, and hence its deposition is likely to have ecological impacts. There have been recent reviews of aspects of atmospheric ON [8,9], and so here we do not attempt a comprehensive review but build on those other reviews and focus on new insights into sources of ON and the importance of this material in the global nitrogen cycle.
2. The quantification of atmospheric organic nitrogen in aerosols and precipitation
ON in rain and aerosol samples represents the total nitrogen (TN) content minus the amounts of nitrate and ammonium (the dominant inorganic nitrogen components), i.e.
| 2.1 |
Nitrate and ammonium concentrations are measured individually. The TN is measured after strong oxidation of samples, which is assumed to convert all organic-bound nitrogen species present in the sample to a readily measured inorganic form [8]. While there has been some discussion in the scientific literature about the best oxidizing agent, the three commonly used approaches of photochemical, chemical and high-temperature oxidation all appear to be able to efficiently oxidize the ON to inorganic forms [8]. These chemical analyses are usually done on filtered rain samples or aqueous extracts of aerosol samples collected on filters, and hence the ON measured represents dissolved ON (DON) in rain or water-soluble ON (WSON) in aerosols. The amount of WSON and DON measured will reflect the filter size used to create the dissolved/particulate separation, and Chen et al. [10] have size-fractionated the WSON to further characterize speciation. In one study, an additional insoluble aerosol ON component has been identified and shown to be three times larger than the soluble WSON [11]. In another study, where total ON rather than WSON was measured, the rather high proportion of ON to TN compared with other studies is consistent with a substantial insoluble ON component [12]. This insoluble ON component deserves further attention in terms of its abundance and bioavailabilty.
Sampling methods need careful evaluation to ensure the samples adequately represent the distribution of ON between the gas, liquid and aerosol phases at the time of collection, and appropriate storage and analysis of atmospheric ON is necessary in order to avoid contamination, loss of material by biological action and gains or losses of material by exchange with the gas phase [8]. Recently, Gonzaléz-Benítez et al. [13] and Zamora et al. [14] reported a significant amount of relatively volatile WSON in aerosols collected in Scotland and Miami and Barbados, respectively.
The approach embodied in equation (2.1) follows that used for measurements of DON in freshwater and marine systems. It has the advantage of approximating to the total amount of soluble nitrogen in the sample and, therefore, sets an upper limit on that potential biologically available nitrogen delivered to the receiving biological system. However, the approach does not say anything about the form of the soluble ON present, and hence its source or bioavailability. The approach embodied in equation (2.1) also means that the analytical uncertainty in the estimation of the ON concentration is associated with the propagation of errors from three individual analyses, and this often leads to large analytical uncertainties in the measurements of the concentration and fluxes of ON [8]. This problem is particularly serious for the measurement of ON in aerosol and rainwater, because the proportions of the ON are generally similar to the concentrations of both nitrate and ammonium, and so all three errors contribute significantly. This contrasts with, for example, the measurement of DON in surface ocean waters where the nitrate and ammonium concentrations are very low compared with DON, so the analytical uncertainties in the DON concentrations are relatively small [15].
3. The characterization of atmospheric organic nitrogen
An alternative and complementary analytical approach to total ON measurement for improving our understanding of the sources and significance of ON in the atmosphere is to measure directly the concentration of individual ON compounds or groups of compounds in rainwater or aerosol extracts. This approach has been used by many research groups because of a particular interest in the compounds in question, or to evaluate the significance of that compound or group of compounds in terms of the total amount of ON present (see [8] for a detailed review). Compounds analysed in individual studies include amines, amino acids, urea, nitrophenols, alkyl amides, N-heterocyclic alkaloids and organic nitrates. In some cases, some of these compounds have been found to be important components of the ON at particular sites, and some of these cases are discussed below. However, it is clear that none of these compounds dominate the ON composition on a global basis. Recent studies using ultra-high resolution Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR) with electrospray ionisation [16,17] identified several thousand ON compounds in rain samples collected in New Jersey and Bermuda. These studies found significant differences between relatively pristine marine and more polluted continental rain samples, but at both sites more than two thirds of the organic N compounds measured contained reduced nitrogen, and most of these compounds contained a single nitrogen atom as well as oxygen atoms. Altieri et al. [16] suggested that oligomerization reactions involving amino acids would be consistent with the ON composition seen in continental rains. In the Bermuda rains [17], less influenced by continental sources, a more diverse ON composition was evident suggesting additional sources, possibly including breakdown of more complex nitrogen-containing organic molecules such as proteins. Carbamylation reactions involving isocyanates may also yield compounds consistent with these compositional patterns [18]. The extent to which the results from these detailed characterization studies of a few rain samples from two sites can be extrapolated more widely is unknown.
The analyses of individual compounds have shown that in certain places and at certain times, some particular groups of compounds make an important contribution to atmospheric ON. Cornell et al. [19] and Mace et al. [20] showed that urea could be important (greater than 20%) in some locations, but other studies [21,22] find that this is a minor or very minor component of the organic N at other locations such as the Mediterranean. Recently, Zhang et al. [23] suggested that urea represents more than 40 per cent of DON in rain in some areas of China where urea is widely used as a fertilizer. Srinivas et al. [24] suggested urea use contributed to high WSON in air flowing out of northeastern India. Globally about 40 per cent nitrogen fertilizer use is as urea, much of it in Asia ([25], and consistent with more recent manufacturer data; http://www.fertilizer.org/ifa/HomePage/STATISTICS/Production-and-trade (accessed 25 September 2012)). Additional sources of urea may arise from animal waste [20]. Hence, it is likely that urea is an important component of the atmospheric ON in some regions.
Amino acids represent a significant component of the ON in some sites (e.g. about 50% in Tasmania [20]) but not others (e.g. less than 2 per cent Amazonia and Mediterranean [21,22,26]). Mace et al. [20] suggest that the sources of the amines may be proteins transported and degraded in the atmosphere, and that some of the site-to-site differences may reflect degradation of both proteins and amines in collected samples. Facchini et al. [27] report dimethyl and diethyl amine salts contributing 35 per cent to the total ON concentration in aerosols at Mace Head in western Ireland with other primary and tertiary amines not detected. Much lower concentrations of these compounds were reported at Cape Verde [28] and on Crete [26] with a resultant much lower contribution to the observed ON. These differences may reflect local or regional differences in marine primary production, because a marine source is suggested for the amines in these studies. The atmospheric sources and cycling of amines has been reviewed [29].
Aerosol mass spectrometry is now allowing the analysis of individual aerosol particles. Interpretation of the resulting dataset can be challenging, but can provide a different perspective on the characterization of atmospheric ON. Sun et al. [30] recently reported on the characterization of aerosols in New York City using this approach. This technique predominantly samples the fine-mode aerosol. Sun et al. were able to statistically cluster the data into five organic aerosol components, one of which was identified as nitrogen rich, and indeed appears to be the only one of the five components to contain nitrogen. The authors suggest that this nitrogen-rich fine-mode organic aerosol is formed by gas-to-particle reactions and adsorption to pre-existing aerosol, and its’ composition is dominated by amino groups.
Reviewing this literature leads to the conclusion that the ON is still largely uncharacterized and probably composed of a diverse, variable and likely complex mixture of organic compounds, and some new technologies are beginning to provide some insights into this complexity. This compositional complexity is similar to the situation for marine and terrestrial organic matter [31,32]. The measurement of individual compounds or groups of compounds is valuable to help elucidate the cycle of those compounds and their biogeochemical role, to investigate the sources and cycling of potentially harmful anthropogenic compounds and as tracers of particular sources of nitrogen. However, such analyses are unlikely to identify a single dominant ON source in the atmosphere. Thus, the analysis of the bulk ON as defined in equation (2.1) remains a useful pragmatic approach that allows us to at least put bounds on the biogeochemical role of atmospheric ON in the global nitrogen cycle.
4. Sources of atmospheric organic nitrogen
The size distribution of aerosol atmospheric ON can offer clues to its sources and cycling. Primary sources of atmospheric particles (such as soil dust and sea spray) are usually associated with coarse-mode aerosol particles (diameter more than 1 µm), whereas aerosols formed in the atmosphere from gas-phase precursors are usually in the smaller aerosol accumulation mode (diameter less than 1 µm; [33]), although the adsorption of gas-phase components to pre-existing aerosol can complicate this picture. Thus, for example, aerosol ammonium sulfate formed from the reaction of ammonia and sulfuric acid is found predominantly in the fine mode, whereas elements associated with soil dust and sea spray components such as aluminium and sodium are found predominantly in the coarse mode. Nitrate is also found mainly in the coarse mode in the marine atmosphere because of a specific reaction of gaseous nitric acid with sea salt [33]. The partial oxidation of organic matter in the atmosphere generally adds polar oxygenated or nitrogenous functional groups (e.g. ketones or alcohol groups), which tend to make the products of oxidation less volatile and more water soluble, and hence more likely to transfer from the gas to aerosol phase [3].
The available measurements of the size distribution of aerosol ON again indicate complex and multiple sources. In remote marine air masses such as Hawaii and Tasmania [19,20], WSON is reported to be associated with the fine mode, whereas in samples more influenced by terrestrial and anthropogenic sources more of the WSON is associated with coarse-mode material [10]. Lesworth et al. [34] found that on average over the North Atlantic Ocean, WSON concentrations within the fine and coarse mode were approximately equal, but the percentage fine mode did increase somewhat in more remote regions. Zamora et al. [14] and Srinivas et al. [24], both report significant amounts of coarse- and fine-mode WSON, with the fine-mode dominant. Violaki & Mihalopoulis [26] found the WSON to be predominantly associated with the fine-mode aerosol (68%) with anthropogenic and biomass burning sources particularly important, while dust sources were an important source of coarse-mode WSON. Chen et al. [10] separated the WSON into a high (greater than 1000 Da) and low molecular-weight fraction for aerosols collected in Taiwan. They found 57 per cent of the WSON in the high molecular-weight fraction and observed that the two fractions did not covary, and they therefore argued that the sources of the two fractions were somewhat different.
Another approach to investigating ON sources is to consider the relationship between ON concentration and various tracers of atmospheric processes and emission sources. Similarly, comparisons of results from sites under the influence of very different source regimes and/or where the source regime varies strongly with season or air flow can be particularly instructive in illustrating the relative importance of potential sources. Comparison with specific tracers also demonstrates complex and multiple sources, with particular sources dominant in certain regions. Four particular sources have been identified as being important, based on the use of air-parcel back trajectories and chemical tracers.
(i) Soil dust particularly evident where air that had recently passed over the Sahara desert was sampled [22,26,34]. Note this source may be associated with soil organic matter itself or with the adsorption of ON onto these dust particles.
(iii) Marine emission both direct and via emissions of gaseous precursors [20,35].
(iv) Anthropogenic and agricultural sources [10,19,20,24,26,36–38].
One feature that is clear from sampling at sites impacted by transport from both terrestrial and marine regions [19,20,39] is that, while a marine source for some atmospheric ON can be clearly identified, concentrations are higher when the air flow is from land rather than from the oceans. This implies that terrestrial sources, whether anthropogenic or natural, are stronger than marine sources. However, in remote regions there is clear evidence for the emission of WSON from ocean sources [12,27] and that compounds from these marine emissions may be an important component of the aerosol [40,41].
In a recent review, Cornell [9] summarized available data on rainwater DON and showed a clear geographical pattern with highest concentrations in continental areas and particularly over Asia (mean concentrations more than 100 µmol l−1) with much lower concentrations in remote marine locations (approx. 5 µmol l−1). A similar trend is evident in the compilation of Zhang et al. [23].
Rainwater concentrations are inevitably strongly dependent on rainfall amounts, so in some ways aerosol concentrations offer an easier way to assess large-scale geographical patterns and concentration ranges (table 1). However, aerosol concentrations are still influenced by rainfall amounts. Mace et al. [21], for instance, showed that average Amazonian aerosol WSON is 3.5 nmol m−3 in the wet season and 61 nmol m−3 in the dry season, although part of this difference reflects the biomass burning which takes place in the dry season. The data in table 1 again suggest concentrations are higher in areas more influenced by continental sources compared with remote ocean regions, with a total concentration range of about a factor of 50. Thus, there are clearly important sources on land, although marine emissions may be important in remote areas.
Table 1.
Aerosol water-soluble ON (WSON) from selected sites that are remote from local influences to illustrate large-scale patterns of concentrations.
| site | WSON (nmol m−3) | references |
|---|---|---|
| Taiwan | 76 | [10] |
| NE India/Bay of Bengal | 35, 46 | [24] |
| Northern California | 16 | [42] |
| Crete | 12 | [26] |
| Delaware | 7 | [11] |
| North Atlantic Saharan outflow | 14.5 | [34] |
| North Atlantic remote | 5.3 | [34] |
| South Atlantic | 6 | [34] |
| Miami | 3 | [14] |
| Southern Ocean | 1–3 | [20,34] |
| North Pacific remote | 1.2, 3.3 | [19,35] |
| Barbados | 1.5 | [14] |
Isotopic analysis of both carbon and nitrogen in the aerosol has been undertaken to try to characterize sources. Carbon 12/13 isotopic analysis can clearly distinguish between marine and terrestrial organic matter, and the addition of carbon 14 analysis allows contributions of modern and fossil carbon to be estimated, with the latter predominantly anthropogenic. The results of such analyses suggest that at terrestrial sites in Europe and North America, both modern and fossil carbon contribute significantly to the aerosol organic matter [3]. At Mace Head, a coastal site in NW Europe, the aerosol organic carbon is on average 80 per cent of marine origin in air masses arriving from over the North Atlantic, whereas in air masses arriving from the east impacted by European emissions, the proportions of fossil, marine and continental non-fossil carbon are approximately equal [41]. Miyazaki et al. [35] also found marine organic carbon emissions to be the main source of organic carbon in aerosols collected over the western North Pacific, although earlier studies at very remote island sites suggested that the terrestrial organic carbon source was more important [43]. Attempts to use nitrogen isotopes to characterize ON sources have yielded less clear-cut results which are, therefore, consistent with a wide range of nitrogen sources as discussed by Cape et al. [8].
Kanakidou et al. [44] have adapted a global organic carbon model to create the first global model of atmospheric ON. The authors use C : N ratios specific to certain sources alongside their organic carbon flux estimates to estimate ON fluxes, together with a specific emission source for amines from the ocean. The model is then used to estimate inputs to the ocean. The model yields organic carbon and inorganic fluxes consistent with other estimate and credible ON flux distributions. Comparison of the model with available data suggests the model underestimates fluxes of ON, which the authors suggest reflects errors in the sources and model parameterization. In this model, the authors estimate the total emission of ON (including that formed in the atmosphere) to be 27.4 Tg N yr−1 with 45 per cent from combustion sources, 32 per cent from primary biogenic particles, 1 per cent from soil dust, 20 per cent from ocean particulate emissions and 3 per cent from ocean amine emissions. They estimate 46 per cent of the ON emissions are anthropogenic, which includes biomass-burning emissions in this calculation.
5. Contribution of organic nitrogen to the overall nitrogen cycle
The relative contribution of DON and WSON to the total measured N has been calculated in many studies with estimates ranging from a few per cent to more than 40 per cent [9], with clear patterns in individual studies often difficult to determine. However, taking an overview of several datasets, some useful patterns begin to emerge. It should be noted that in all cases these refer to atmospheric DON and WSON. There is less information on such relationships for the insoluble forms of ON in the atmosphere, which may be present in relatively large amounts [11,12] as noted earlier, although Duan et al. [45] did report similar relations for total organic N to those discussed below for soluble organic N.
As noted by Zhang et al. [23], concentrations of DON and soluble TN are particularly high in samples from China, which may be related to the use of urea fertilizers as discussed earlier. Figure 1 shows the relationship between average DON and soluble TN (DON + nitrate + ammonium) in rainwater from many sites around the world. The figure illustrates several important points. The scatter in figure 1 shows that the concentrations and the relative proportions of DON to TN are variable, presumably reflecting variations in source strengths and atmospheric processing with time and place. Although the relationship is very noisy, there is a clear statistically significant correlation between soluble TN and DON. As the nitrate and ammonium concentrations are heavily impacted by anthropogenic emissions, the relationship in figure 1 implies a significant anthropogenic source for DON.
Figure 1.
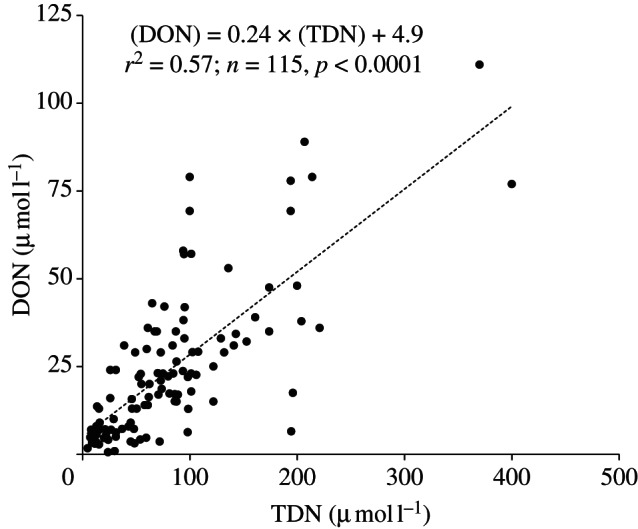
Plot of published long-term average rainwater DON and total dissolved nitrogen (TDN) concentrations from sites around the world, based on the data compilations of references [6,9,23] including a best fit correlation.
The slope of the relationship in figure 1 (0.24) implies that DON represents on average 24 per cent of soluble TN on a global basis. In an independent study of aerosols rather than rainwater, Lesworth et al. [34] analysed aerosol samples along a sampling transect crossing over the central Atlantic Ocean from north to south which also reveals a similar (and even stronger) correlation and an identical slope for this independent dataset. Chen et al. [10] and Zhang et al. [42] similarly report an average percentage WSON in TN in Taiwanese aerosols of 26 per cent and 20 per cent in Californian aerosols, respectively, but lower WSON percentages have been reported in other studies: 13 per cent in Crete [26], 16 per cent in the outflow from northeast India over the Bay of Bengal [24], 7 per cent in Miami and 10 per cent in Barbados [14], and 6 per cent in Delaware, USA [11]. Note, however, that Zamora et al. [14] report a semi-volatile organic N aerosol that is of similar magnitude to the involatile fraction, and inclusion of this fraction would significantly increase the reported percentage ON fractions at the Miami and Barbados sites. Kanakidou et al. [44] estimated ON to be 26 per cent of total N deposition on a global basis using their modelling approach.
Despite the complexity of ON sources and their variability in space and time, there is a coherent, if statistically noisy, relationship with other nitrogen species (figure 1). This suggests a relationship between the emission, transport, transformation and deposition of ON and other nitrogen species. As noted above, this does imply that a substantial component of the atmospheric ON is anthropogenic, and this conclusion is important in understanding the role of ON in the global nitrogen cycle. The relationship also offers a pragmatic method of estimating ON fluxes based on the better understood nitrate and ammonium fluxes, albeit without a fundamental mechanistic understanding of the ON sources and cycling. This approach was adopted, for instance, by Duce et al. [46] who assumed ON to be 30 per cent of TN.
Zhang et al. [23] suggest that on a global basis the correlation between WSON and ammonium in rainwater is stronger than with nitrate. Cape et al. [36] also report a strong correlation between ammonium and WSON in UK rainwater, and Srinivas et al. [24] report a very strong correlation of WSON and ammonium in aerosol collected over the Bay of Bengal. The global cycling of oxidized and reduced inorganic nitrogen (i.e.  versus
versus 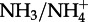 ) is different, reflecting the different reactions and lifetimes of the gaseous precursors and the different aerosol size distributions, at least in marine environments [33]. Hence, the stronger correlation of ON with reduced rather than oxidized nitrogen may reflect the atmospheric processing of ON being similar to that of reduced nitrogen, or that their sources are similar, or indeed both. As noted above, Altieri et al. [16,17] suggested that the rainwater WSON at their sampling site mostly contained reduced forms of nitrogen, and Sun et al. [30] argue for a dominant role for amine compounds in New York City fine-mode aerosol. Taken together, these lines of evidence suggest that an important route for the formation of atmospheric ON, at least in the fine mode, may be via interactions between organic matter and reduced nitrogen, presumably ammonia and amine compounds or HNCO and related gases. There is some laboratory experimental evidence for these kinds of reactions [47].
) is different, reflecting the different reactions and lifetimes of the gaseous precursors and the different aerosol size distributions, at least in marine environments [33]. Hence, the stronger correlation of ON with reduced rather than oxidized nitrogen may reflect the atmospheric processing of ON being similar to that of reduced nitrogen, or that their sources are similar, or indeed both. As noted above, Altieri et al. [16,17] suggested that the rainwater WSON at their sampling site mostly contained reduced forms of nitrogen, and Sun et al. [30] argue for a dominant role for amine compounds in New York City fine-mode aerosol. Taken together, these lines of evidence suggest that an important route for the formation of atmospheric ON, at least in the fine mode, may be via interactions between organic matter and reduced nitrogen, presumably ammonia and amine compounds or HNCO and related gases. There is some laboratory experimental evidence for these kinds of reactions [47].
6. Conclusions
Atmospheric ON is evidently a significant component of the atmospheric nitrogen cycle. Most studies to date have focused on ON that is soluble in water either as DON in rain or after aqueous extraction from aerosols. This component represents on average about 25 per cent of total atmospheric soluble fixed nitrogen (soluble ON plus nitrate plus ammonium), although this proportion is highly variable in space and time. This soluble ON appears to be bioavailable and composed of a wide range of different ON compounds. This DON and WSON have diverse sources including both direct emissions, adsorption of gases to pre-existing aerosol particles and the formation of new particles within the atmosphere, as illustrated schematically in figure 2. In addition, there is evidence for an additional substantial component of water-insoluble ON in the atmosphere. This may include soil organic matter and biological debris which will ultimately also be bioavailable. There is some emerging evidence that suggests that the soluble ON may be dominated by reduced nitrogen, and hence that a major route for the formation of this material may involve reactions of organic matter with ammonia/ammonium or low molecular-weight gaseous reduced nitrogen compounds. This may be particularly important in the fine-mode aerosol, whereas biological and soil particle sources may be important in the coarse mode.
Figure 2.

A cartoon illustrating the diversity of atmospheric ON precursors including primary sources and formation by secondary processes (VOC, volatile organic carbon; N-PAH, nitrogen containing polycyclic aromatic hydrocarbons).
Further study of atmospheric ON is needed to understand its sources and cycling and to allow its incorporation into atmospheric nitrogen models, and assess its impact on the global nitrogen cycle and on atmospheric chemistry and physics.
Acknowledgements
A review of this kind is only possible because of the efforts of many researchers around the world who contribute to the scientific understanding of atmospheric ON. We thank them all for their efforts. We also thank two anonymous reviewers for their helpful and constructive comments on an earlier version of this manuscript. The work on this paper was supported in part by NERC UK grant no. NE/F017359/1.
References
- 1.Seinfeld JH, Pankow JF. 2003. Organic atmospheric particulate matter. Annu. Rev. Phys. Chem. 54, 121–140 10.1146/annurev.physchem.54.011002.103756 (doi:10.1146/annurev.physchem.54.011002.103756) [DOI] [PubMed] [Google Scholar]
- 2.Kanakidou M, et al. 2005. Organic aerosol and global climate modelling: a review. Atmos. Chem. Phys. 5, 1053–1123 10.5194/acp-5-1053-2005 (doi:10.5194/acp-5-1053-2005) [DOI] [Google Scholar]
- 3.Hallquist M, et al. 2009. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 9, 5155–5236 10.5194/acp-9-5155-2009 (doi:10.5194/acp-9-5155-2009) [DOI] [Google Scholar]
- 4.Gouw JD, Jiminez JL. 2009. Organic aerosols in the Earth's atmosphere. Environ. Sci. Technol. 43, 7614–7618 10.1021/es9006004 (doi:10.1021/es9006004) [DOI] [PubMed] [Google Scholar]
- 5.Galloway JN, Townsend AR, Erisman JW, Bekunda M, Cai Z, Freney JR, Martinelli LA, Seitzinger SP, Sutton MA. 2008. Transformations of the nitrogen cycle: recent trends, questions and potential solutions. Science 320, 889–892 10.1126/science.1136674 (doi:10.1126/science.1136674) [DOI] [PubMed] [Google Scholar]
- 6.Cornell SE, Jickells TD, Cape JN, Rowland AP, Duce RA. 2003. Organic nitrogen deposition on land and coastal environments: a review of methods and data. Atmos. Environ. 37, 2173–2191 10.1016/S1352-2310(03)00133-X (doi:10.1016/S1352-2310(03)00133-X) [DOI] [Google Scholar]
- 7.Seitzinger SP, Sanders RW. 1999. Atmospheric inputs of dissolved organic nitrogen stimulate estuarine bacteria and phytoplankton. Limnol. Oceanogr. 44, 721–730 10.4319/lo.1999.44.3.0721 (doi:10.4319/lo.1999.44.3.0721) [DOI] [Google Scholar]
- 8.Cape JN, Cornell SE, Jickells TD, Nemitz E. 2011. Organic nitrogen in the atmosphere—where does it come from? A review of sources and methods. Atmos. Res. 102, 30–48 10.1016/j.atmosres.2011.07.009 (doi:10.1016/j.atmosres.2011.07.009) [DOI] [Google Scholar]
- 9.Cornell SE. 2011. Atmospheric nitrogen deposition: revising the question of the importance of the organic component. Environ. Pollut. 159, 2214–2222 10.1016/j.envpol.2010.11.014 (doi:10.1016/j.envpol.2010.11.014) [DOI] [PubMed] [Google Scholar]
- 10.Chen H-Y, Chen L-D, Chiang Z-Y, Hung C-C, Lin F-J, Chou W-C, Gong G-C, Wen L-S. 2010. Size fractionation and molecular composition of water-soluble inorganic and organic nitrogen in aerosols of a coastal environment. J. Geophys. Res. 115, D22307. 10.1029/2010JD014157 (doi:10.1029/2010JD014157) [DOI] [Google Scholar]
- 11.Russell KM, Keene WC, Maben JR, Galloway JN, Moody JL. 2003. Phase partitioning and dry deposition of atmospheric nitrogen at the mid-Atlantic US coast. J. Geophys. Res. 108, 4656. 10.1029/2003JD003736 (doi:10.1029/2003JD003736) [DOI] [Google Scholar]
- 12.Miyazaki Y, Kawamura K, Sawano M. 2010. Size distribution of organic nitrogen and carbon in remote marine aerosols: evidence of marine biological origin based on their isotopic ratios. Geophys. Res. Lett. 37, L06803. 10.1029/2010GL042483 (doi:10.1029/2010GL042483) [DOI] [Google Scholar]
- 13.Gonzaléz-Benítez JM, Cape JN, Heal MR. 2010. Gaseous and particulate water-soluble organic and inorganic nitrogen in rural air in southern Scotland. Atmos. Environ. 44, 1506–1514 10.1016/j.atmosenv.2010.01.016 (doi:10.1016/j.atmosenv.2010.01.016) [DOI] [Google Scholar]
- 14.Zamora LM, Prospero JM, Hansell DA. 2011. Organic nitrogen in aerosols and precipitation at Barbados and Miami: implications regarding sources, transport and deposition to the western subtropical Atlantic. J. Geophys. Res. 116, D20309. 10.1029/2011JD015660 (doi:10.1029/2011JD015660) [DOI] [Google Scholar]
- 15.Bronk DA. 2002. Dynamics of DON. In Biogeochemistry of marine dissolved organic matter (eds Hansell DA, Carlson CA.), pp. 153–247 San Diego, CA: Academic Press [Google Scholar]
- 16.Altieri K, Turpin BJ, Seitzinger SP. 2009. Composition of dissolved organic nitrogen in continental precipitation investigated by ultra-high resolution FT-ICR Mass Spectrometry. Environ. Sci. Technol. 43, 6950–6955 10.1021/es9007849 (doi:10.1021/es9007849) [DOI] [PubMed] [Google Scholar]
- 17.Altieri KE, Hastings MG, Peters AJ, Sigman DM. 2012. Molecular characterization of water soluble organic nitrogen in marine rainwater by ultra-high resolution electrospray ionization mass spectrometry. Atmos. Chem. Phys. 12, 3557–3571 10.5194/acp-12-3557-2012 (doi:10.5194/acp-12-3557-2012) [DOI] [Google Scholar]
- 18.Roberts JM, et al. 2011. Isocyanic acid in the atmosphere and its possible link to smoke-related health effects. Proc. Natl Acad. Sci. USA 108, 8966–8971 10.1073/pnas.1103352108 (doi:10.1073/pnas.1103352108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornell S, Mace K, Coeppicus S, Duce R, Huebert B, Jickells T, Zhuang L-Z. 2001. Organic nitrogen in Hawaiian rain and aerosol. J. Geophys. Res. 106, 7973–7983 10.1029/2000JD900655 (doi:10.1029/2000JD900655) [DOI] [Google Scholar]
- 20.Mace KA, Duce RA, Tindale NW. 2003. Organic nitrogen in rain and aerosol at Cape Grimm, Tasmania, Australia. J. Geophys. Res. 108, ACH 3-1–3-14 10.1029/2002JD003051 (doi:10.1029/2002JD003051) [DOI] [Google Scholar]
- 21.Mace KA, Artaxo P, Duce RA. 2003. Water-soluble organic nitrogen in Amazon Basin aerosols during the dry (biomass burning) and wet seasons. J. Geophys. Res. 108, ACH 14-1–14-10 10.1029/2003JD003557 (doi:10.1029/2003JD003557) [DOI] [Google Scholar]
- 22.Mace KA, Kubilay N, Duce RA. 2003. Organic nitrogen in rain and aerosol in the eastern Mediterranean atmosphere: an association with atmospheric dust. J. Geophys. Res. 108, ACH 5-1–5-11. 10.1029/2002JD002997 (doi:10.1029/2002JD002997) [DOI] [Google Scholar]
- 23.Zhang Y, Song L, Liu XJ, Li WQ, Lü SH, Zheng LX, Bai ZC, Cai GY, Zhang FS. 2012. Atmospheric organic nitrogen deposition in China. Atmos. Environ. 46, 195–204 10.1016/j.atmosenv.2011.09.080 (doi:10.1016/j.atmosenv.2011.09.080) [DOI] [Google Scholar]
- 24.Srinivas B, Sarin NM, Sarma VVSS. 2011. Atmospheric dry deposition of inorganic and organic nitrogen to the Bay of Bengal: impact of continental outflow. Mar. Chem. 127, 170–179 10.1016/j.marchem.2011.09.002 (doi:10.1016/j.marchem.2011.09.002) [DOI] [Google Scholar]
- 25.Matthews E. 1994. Nitrogenous fertilizers: global distribution of consumption and associated emissions of nitrous oxide and ammonia. Glob. Biogeochem. Cycles 8, 411–439 10.1029/94GB01906 (doi:10.1029/94GB01906) [DOI] [Google Scholar]
- 26.Violaki K, Mihalopoulos N. 2010. Water-soluble organic nitrogen (WSON) in size-segregated atmospheric particles over the Eastern Mediterranean. Atmos. Environ. 44, 4339–4345 10.1016/j.atmosenv.2010.07.056 (doi:10.1016/j.atmosenv.2010.07.056) [DOI] [Google Scholar]
- 27.Facchini MA, et al. 2008. Important source of marine secondary organic aerosol from biogenic amines. Environ. Sci. Technol. 42, 9116–9121 10.1021/es8018385 (doi:10.1021/es8018385) [DOI] [PubMed] [Google Scholar]
- 28.Müller C, Iinuma Y, Karstensen J, van Pinteren D, Lehmann S, Gnauk T, Hermann H. 2009. Seasonal variation of aliphatic amines in marine sub-micrometer particles at the Cape Verde islands. Atmos. Chem. Phys. 9, 9587–9597 10.5194/acp-9-9587-2009 (doi:10.5194/acp-9-9587-2009) [DOI] [Google Scholar]
- 29.Ge X, Wexler AS, Clegg SL. 2011. Atmospheric amines. I. A review. Atmos. Environ. 45, 524–546 10.1016/j.atmosenv.2010.10.012 (doi:10.1016/j.atmosenv.2010.10.012) [DOI] [Google Scholar]
- 30.Sun Y-L, et al. 2011. Characterisation of the sources and processes of organic and inorganic aerosols in New York City with a high-resolution time-of-flight aerosol mass spectrometer. Atmos. Chem. Phys. 11, 1581–1602 10.5194/acp-11-1581-2011 (doi:10.5194/acp-11-1581-2011) [DOI] [Google Scholar]
- 31.Benner R. 2002. Chemical composition and reactivity. In Biogeochemistry of marine dissolved organic matter (eds Hansell DA, Carlson CA.), pp. 35–90 San Diego, CA: Academic Press [Google Scholar]
- 32.Schmidt MWI, et al. 2011. Persistence of soil organic matter as an ecosystem property. Nature 478, 49–56 10.1038/nature10386 (doi:10.1038/nature10386) [DOI] [PubMed] [Google Scholar]
- 33.Raes F, van Dingenen R, Vignati E, Wilson J, Putaud J-H, Seinfeld JH, Adams P. 2000. Formation and cycling of aerosols in the global troposphere. Atmos. Environ. 34, 4215–4240 10.1016/S1352-2310(00)00239-9 (doi:10.1016/S1352-2310(00)00239-9) [DOI] [Google Scholar]
- 34.Lesworth T, Baker AR, Jickells T. 2010. Aerosol organic nitrogen over the remote Atlantic Ocean. Atmos. Environ. 44, 1887–1893 10.1016/j.atmosenv.2010.02.021 (doi:10.1016/j.atmosenv.2010.02.021) [DOI] [Google Scholar]
- 35.Miyazaki Y, Kawamura K, Jung J, Furutani H, Uematsu M. 2011. Latitudinal distributions of organic nitrogen and organic carbon in marine aerosols over the western North Pacific. Atmos. Chem. Phys. 11, 3037–3049 10.5194/acp-11-3037-2011 (doi:10.5194/acp-11-3037-2011) [DOI] [Google Scholar]
- 36.Cape JN, Anerson M, Rowland AP, Wilson D. 2004. Organic nitrogen in precipitation across the United Kingdom. Water Air Soil Pollut. 4, 25–35 10.1007/s11267-005-3010-3 (doi:10.1007/s11267-005-3010-3) [DOI] [Google Scholar]
- 37.Zhang Y, Zheng IX, Liu XJ, Jickells TD, Cape JN, Goulding K, Fangmeier A, Zhang FS. 2008. Evidence for organic N deposition and its anthropogenic sources in China. Atmos. Environ. 42, 1035–1041 10.1016/j.atmosenv.2007.12.015 (doi:10.1016/j.atmosenv.2007.12.015) [DOI] [Google Scholar]
- 38.Bencs L, Krata A, Horemans B, Buczyńska AJ, Dirtu AC, Godoi AFL, Godoi RHM, Potgieter-Vermaak S, Van Grieken R. 2009. Atmospheric nitrogen fluxes at the Belgian coast: 2004–2006. Atmos. Environ. 43, 3786–3798 10.1016/j.atmosenv.2009.04.002 (doi:10.1016/j.atmosenv.2009.04.002) [DOI] [Google Scholar]
- 39.Spokes LJ, Yeatman SG, Cornell SE, Jickells TD. 2000. Nitrogen deposition to the eastern Atlantic Ocean. The importance of south-easterly flow. Tellus 52B, 37–49 10.1034/j.1600-0889.2000.00062.x (doi:10.1034/j.1600-0889.2000.00062.x) [DOI] [Google Scholar]
- 40.O'Dowd CD, Facchini MC, Cavalli F, Ceburnis D, Mircea M, Decesari S, Fuzzi S, Yoon YJ, Putaud J-P. 2004. Biogenically driven organic contribution to marine aerosol. Nature 431, 676–680 10.1038/nature02959 (doi:10.1038/nature02959) [DOI] [PubMed] [Google Scholar]
- 41.Ceburnis D, et al. 2011. Quantification of the carnoaceous matter origin in submicron marine aerosol by 13C and 14C isotope analysis. Atmos. Chem. Phys. 11, 8593–8606 10.5194/acp-11-8593-2011 (doi:10.5194/acp-11-8593-2011) [DOI] [Google Scholar]
- 42.Zhang Q, Anatasio C, Jimenez-Cruz M. 2002. Water-soluble organic nitrogen in atmospheric fine particles (PM2.5) from northern California. J. Geophys. Res. 107, AAC 3-1–AAC 3-9 10.1029/2001JD000870 (doi:10.1029/2001JD000870) [DOI] [Google Scholar]
- 43.Buat-Ménard P, Cachier H, Chesselet R. 1989. Sources of particulate carbon in the marine atmosphere. In Chemical oceanography, vol. 10 (eds Duce RA, Riley JP, Chester R.), pp. 251–279 London, UK: Academic Press [Google Scholar]
- 44.Kanakidou M, et al. 2012. Atmospheric fluxes of organic N and P to the global ocean. Glob. Biogeochem. Cycles 26, GB2022. 10.1007/s00128-008-9560-0 (doi:10.1007/s00128-008-9560-0) [DOI] [Google Scholar]
- 45.Duan F, Liu X, He K, Dong S. 2009. Measurements and characteristics of nitrogen-containing compounds in atmospheric particulate matter in Beijing, China. Bull. Environ. Contam. Toxicol. 82, 332–337 10.1126/science.1150369 (doi:10.1126/science.1150369) [DOI] [PubMed] [Google Scholar]
- 46.Duce RA, et al. 2008. Impacts of atmospheric anthropogenic nitrogen on the open ocean. Science 320, 893–897 10.1126/science.1150369 (doi:10.1126/science.1150369) [DOI] [PubMed] [Google Scholar]
- 47.Chang SG, Novakov T. 1967. Formation of pollution particulate nitrogen compounds by NO–soot and NH3–soot gas-particle reactions. Atmos. Environ. 9, 495–504 10.1016/0004-6981(75)90109-2 (doi:10.1016/0004-6981(75)90109-2) [DOI] [Google Scholar]