

Abstract
The recent discovery of a small-molecule benzosuberene-based phenol that demonstrates remarkable picomolar cytotoxicity against selected human cancer cell lines and strongly inhibits tubulin polymerization (1–2 µM) inspired the design and synthesis of a variety of new, structurally diverse benzosuberene derivatives. An efficient synthetic route to functionalized benzosuberenes was developed. This methodology utilized a Wittig reaction, followed by a selective alkene reduction and ring-closing cyclization to form the core benzosuberone structure. This synthetic route facilitated the preparation of a 6-nitro-1-(3′,4′,5′-trimethoxyphenyl) benzosuberene derivative and its corresponding 6-amino analogue in good yield. The 6-amino analogue was a strong inhibitor of tubulin polymerization (1.2 µM), demonstrated enhanced cytotoxicity against the human cancer cell lines examined (GI50 = 33 pM against SK-OV-3 ovarian cancer, for example), and exhibited a concentration dependent disruption of a pre-established capillary-like network of tubules formed from human umbilical vein endothelial cells.
Introduction
The discovery of small-molecule anticancer agents that demonstrate nanomolar to subnanomolar cytotoxicity against human cancer cell lines is noteworthy, and such compounds have the potential to become drug candidates through an appropriately focused development program. The discovery of compounds that demonstrate picomolar cytotoxicity is even more exciting. In previous studies,1–3 we identified a functionalized benzosuberene-based phenol 1 as one such compound (Fig. 1). Its structure is reminiscent of both colchicine4,5 and combretastatin A-4 (CA4),6 which are natural products that are potent inhibitors of tubulin assembly.7 Both compounds interact with tubulin at a small-molecule binding site named the colchicine site (Fig. 1). Compound 1 also bears structural similarity to a dihydronaphthalene analogue 2 (discovered previously in our laboratory) that is strongly cytotoxic and inhibitory against tubulin assembly.1,8 In addition to its antiproliferative mechanism of action, both CA4 and compound 2 damage tumor vasculature.8,9 Preliminary studies with the benzosuberene phenol 1, in appropriate prodrug form, also suggest vascular damage as one component of its overall mechanism of action as an antitumor agent.2 This makes it likely that the benzosuberene amino derivative 3 would function through a similar mechanistic pathway.
Figure 1.

Representative Small-Molecule Inhibitors of Tubulin Assembly.
Small-molecule anticancer agents that target solid-tumor vasculature represent an important emerging area of research significance.9,10 Solid tumors require nutrients and oxygen provided by a network of vasculature, which has distinct morphological differences compared with a corresponding vascular network feeding normal healthy tissue.11 Tumor vasculature is highly disorganized with abnormal bulges, blind ends, and shunts.12 It is also characterized as leaky and discontinuous. It is these physiological dissimilarities that collectively offer a therapeutic advantage for the selective targeting and disruption of tumor vasculature through small-molecule drug intervention.
Tumor vasculature can be efficiently targeted through two distinct mechanistic pathways.13 Angiogenic inhibiting agents (AIAs) prevent new blood vessel growth.14 Vascular disrupting agents (VDAs), on the other hand, damage existing tumor vasculature through a series of cell-signaling pathways.15,16 One important class of small-molecule VDAs bind to tubulin heterodimers at the colchicine site and thereby potently inhibit tubulin assembly. The colchicine site is located on the β-subunit of the αβ-tubulin heterodimer, near the interface between the two subunits.17
In the late 1970’s, the combretastatins were discovered by Pettit and co-workers in the South African bush willow tree, Combretum caffrum. Combretastatin A-1 (CA1)18–20 and combretastatin A-4 (CA4)6 are two of the most potent compounds isolated from C. caffrum, and both have pronounced biological activity as VDAs and as inhibitors of tubulin polymerization. In prodrug forms, both of these compounds, as well as the synthetic combretastatin amino-analogue 7, are currently in human clinical trials.21–25
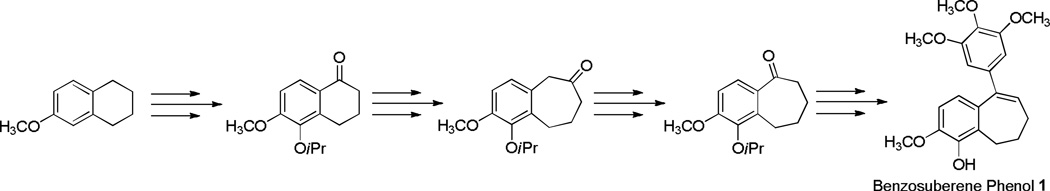
Our original synthetic methodology towards the benzosuberene molecular scaffold utilized a cyanogen azide ring expansion reaction,1,26–28 and, while reliable, the yield for this overall process was relatively low (Scheme 1). Accordingly, we developed a new, efficient synthetic strategy towards the functionalized six-seven fused ring system. This new method facilitated the preparation of the nitro-benzosuberene analogue 4 and its corresponding amino derivative 3. These two new benzosuberene analogues were evaluated for their ability to inhibit tubulin assembly and for their cytotoxicity against three human cancer cell lines.
Scheme 1.
Results and Discussion
Our improved synthetic approach to the target benzosuberene analogues relied on a sequential Wittig reaction, selective reduction, cyclization strategy to assemble the requisite 6,7-ring fusion. Key features of this overall synthetic route include both improved yields and fewer reaction steps. While other synthetic approaches to this type of 6,7-fused ring system are known1,29,30 (including Friedel-Crafts cyclization31), the strategy that we employed proved to be highly efficient. A Wittig reaction using 3-(carboxypropyl)triphenylphosphonium bromide, potassium tert-butoxide, and 3-methoxy-2-nitrobenzaldehyde resulted in compound 8 in good yield (Scheme 2, Route 1). A selective reduction of the alkene moiety to afford compound 9 was achieved using a 10% Pd/C catalyst with 1,4-cyclohexadiene in excess. Reduced analogue 9 underwent cyclization upon treatment with Eaton’s reagent (7.7 wt% P2O5 in methanesulfonic acid)32 to afford nitrobenzosuberone 1033 in good yield. Alternatively, nitrobenzosuberone 10 was prepared by nitration of benzosuberone 15. The regioisomeric mixture obtained (10, 37% and 12, 49%) was separated chromatographically (Scheme 2, Route 2).33 Dropwise addition of nitrobenzosuberone 10 to 3,4,5-trimethoxyphenyllithium (prepared in situ from the corresponding aryl bromide) gave tertiary alcohol 11, which underwent elimination upon exposure to acetic acid to generate nitrobenzosuberene 4. The nitro group in compound 4 was reduced to its corresponding amine 3 upon treatment with zinc dust in acetic acid. Eaton's reagent has been successfully employed for cyclization to form tetralone (6,6-fused) and benzosuberone (6,7-fused) ring systems,32,37,38 and we were encouraged by the efficient extension of this methodology to these functionalized benzosuberene core structures.
Scheme 2.

Improved Ring-Cyclization Methodologies for the Construction of Benzosuberene-based Compounds.
Inhibition of Tubulin Assembly
The two new benzosuberene analogues 3 and 4 were found to be potent inhibitors of tubulin assembly (IC50 = 1.2 µM and 2.5 µM, respectively (Table 1)), comparable to CA1, CA4, and the benzosuberene phenol 1. A binding assay utilizing tritium-labeled colchicine indicated that amino-benzosuberene 3, at a concentration of 5 µM, inhibited colchicine binding by 91% (Table 1). At the lower concentration of 1 µM, amine 3 inhibited colchicine binding by 65%. This is comparable with the activity of CA4 (100% inhibition at 5 µM and 88% inhibition at 1 µM). The nitro analogue 4 was less active as an inhibitor of colchicine binding.
Table 1.
Inhibition of tubulin polymerization and colchicine binding.
Cell Line Studies
In addition, the compounds were evaluated for their cytotoxicity against human non-small cell lung (NCI-H460), prostate (DU-145), and ovarian (SK-OV-3) cancer cell lines. The amino benzosuberene analogue demonstrated remarkable cytotoxicity against ovarian cancer with a GI50 value of 32.9 pM. In addition, the compound was strongly cytotoxic against the non-small cell lung and prostate cell lines. While somewhat less active than the parent benzosuberene phenol 1, the amino derivative 3 was more active against each cell line than the natural products CA4 and CA1. The nitrobenzosuberene analogue 4 was significantly less cytotoxic than any of the comparison compounds (Table 2).
Table 2.
Cytotoxicity studies against human cancer cell lines NCI-H460, DU-145, SK-OV-3.
Endothelial Tube Disruption
HUVECs, plated in medium supplemented with growth factors onto Matrigel™ as a model for the extracellular matrix, form a network of capillary-like tubules (Fig. 2, Control). The disruption of these tubules by individual compounds provides an in vitro assessment for vascular disrupting agents (VDAs). After 16 h, the endothelial tubule network was treated for 2 h with varying concentrations of 3, 4, or CA4 (for comparison as a positive control), or vehicle (medium containing 1% DMSO, negative control). Representative photographs from a minimum of three independent experiments are shown in Fig. 2. Tubule disruption and cell rounding was observed for 3 at 0.10 µM, and this effect was greatly increased at a concentration of 1 µM (Fig. 2A). Compound 4 was much less active and demonstrated no significant effects at concentrations of 0.01 or 0.10 µM. Only minor disruption of tubules was observed at 1.0 µM compound 4, although a substantial number of cells were beginning to round (Fig. 2B). In comparison, tubule disruption activity was detected at 0.01 µM for CA4, and this activity was more pronounced with increasing concentrations (Fig. 2C).
Figure 2.

HUVEC tube disruption. Effects of a 2 h treatment with (A) Cmpd. 3, (B) Cmpd. 4, and (C) CA4 on a pre-established network of capillary-like tubules formed from HUVECS seeded onto Matrigel™ in a growth factor rich medium over a period of 16 h. CA4 was used as a positive control. All experiments including the vehicle control contained a final concentration of 1% DMSO. These results are representative of the photographic record that was obtained with a minimum of three independent experiments for each compound concentration and observing 9 fields/well (40× objective).
Hydrochloride Salt Formation
To facilitate planned in vivo experiments in mice, a hydrochloride salt 16 was prepared synthetically from amine 3 in an effort to obtain a more water-soluble derivative (Scheme 3). Although the salt derivative 16 demonstrated limited solubility in water, it was possible to obtain complete dissolution (NMR study used approximately 3 mg of compound 16 in approximately 1.0 mL of D2O). Obtaining a clear (to the naked eye) solution took several days. A more robust solubilization strategy involved adding DMSO (with vigorous vortexing and/or ultrasound to achieve a clear solution) followed by the addition of water (to achieve a favorable formulation for in vivo experiments in mice). The amine salt 16 demonstrates cytotoxicity (in vitro) in the DU-145 prostate cancer cell line comparable to its parent aminobenzosuberene analogue 3 (Table 1).
Scheme 3.

Synthesis of the HCl salt 16 of Amine Benzosuberene 3.
Conclusions
In summary, we have developed a sequential Wittig reaction, selective reduction, Eaton’s reagent-mediated cyclization strategy as an efficient synthetic route for the preparation of functionalized benzosuberene derivatives. An aminobenzosuberene analogue 3, prepared by this method, demonstrated pronounced cytotoxicity against human cancer cell lines. In addition, this compound was a potent inhibitor of tubulin polymerization and effectively disrupted pre-established endothelial tubes. In a collective sense, these data position compound 3 as an excellent candidate for further evaluation (through additional biochemical/biological studies and in vivo tumor imaging) as an anticancer, vascular disrupting agent.
Supplementary Material
Acknowledgments
The authors are grateful to the Cancer Prevention and Research Institute of Texas (CPRIT (RP100406), to K.G.P. and M.L.T.), Oxigene Inc. (to K.G.P. and M.L.T), the Welch Foundation (grant no. AA-1278 to K.G.P.), and the National Cancer Institute Award Number 5R01CA140674 (to K.G.P. and M.L.T) for their financial support of this project, and to the NSF for funding the Varian 500 MHz NMR spectrometer (grant no. CHE-0420802). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The authors also thank Dr. Alejandro Ramirez (Mass Spectrometry Core Facility, Baylor University) for mass spectroscopic analysis, Dr. Craig Moehnke for assistance with NMR studies, and Dr. James Karban and Dr. Michelle Nemec (Director) for use of the shared Molecular Biosciences Center at Baylor University.
Footnotes
Electronic Supplementary Information (ESI) available: [Synthesis and characterization of compounds 3, 4, 8–16; as well as the methodology used in the tubulin assembly, colchicine binding, SRB assays, and endothelial tube disruption assay. This material is available online.]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Sriram M, Hall JJ, Grohmann NC, Strecker TE, Wootton T, Franken A, Trawick ML, Pinney KG. Bioorg. Med. Chem. 2008;16:8161–8171. doi: 10.1016/j.bmc.2008.07.050. [DOI] [PubMed] [Google Scholar]
- 2.Pinney KG, Trawick ML, Tanpure RP, George CS, Sriram M, Strecker TE, Charlton-Sevcik A, Liu L, Mason R, Siims BG, Chaplin DJ. The discovery and development of highly potent benzosuberene anti-cancer agents; CPRIT Innovations in Cancer Prevention and Research Conference; Austin, TX. 2010. Nov 17, [Google Scholar]
- 3.Sriram M. Ph.D. Dissertation. Waco, TX: Baylor University; 2007. [Google Scholar]
- 4.Boyland E, Boyland ME. Biochem. J. 1937;31:454–460. doi: 10.1042/bj0310454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGown AT, Fox BW. Anticancer Drug Des. 1989;3:249–254. [PubMed] [Google Scholar]
- 6.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Experientia. 1989;45:209–211. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 7.Lin CM, Ho HH, Pettit GR, Hamel E. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 8.Pinney KG. Molecular Recognition of the Colchicine Binding Site as a Design Paradigm for the Discovery and Development of Vascular Disrupting Agents. In: Siemann DW, editor. Vascular-Targeted Therapies in Oncology. John Wiley & Sons, Ltd; 2006. pp. 95–121. [Google Scholar]
- 9.Vincent L, Kermani P, Young LM, Cheng J, Zhang F, Shido K, Lam G, Bompais-Vincent H, Zhu Z, Hicklin DJ, Bohlen P, Chaplin DJ, May C, Rafii S. J. Clin. Invest. 2005;115:2992–3006. doi: 10.1172/JCI24586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mason RP, Zhao D, Liu L, Trawick ML, Pinney KG. Integr. Biol. 2011;3:375–387. doi: 10.1039/c0ib00135j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baluk P, Hashizume H, McDonald DM. Curr. Opin. Genet. Dev. 2005;15:102–111. doi: 10.1016/j.gde.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Siemann DW. Cancer Treat. Rev. 2011;37:63–74. doi: 10.1016/j.ctrv.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siemann DW, Bibby MC, Dark GG, Dicker AP, Eskens FA, Horsman MR, Marme D, Lorusso PM. Clin. Cancer Res. 2005;11:416–420. [PubMed] [Google Scholar]
- 14.Ferrara N, Kerbel RS. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 15.Tozer GM, Kanthou C, Baguley BC. Nat. Rev. Cancer. 2005;5:423–435. doi: 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]
- 16.Kanthou C, Tozer GM. Blood. 2002;99:2060–2069. doi: 10.1182/blood.v99.6.2060. [DOI] [PubMed] [Google Scholar]
- 17.Nogales E, Wolf SG, Downing KH. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 18.Pettit GR, Thornhill AJ, Moser BR, Hogan F. J. Nat. Prod. 2008;71:1561–1563. doi: 10.1021/np800179g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pettit GR, Singh SB, Niven ML, Hamel E, Schmidt JM. J. Nat. Prod. 1987;50:119–131. doi: 10.1021/np50049a016. [DOI] [PubMed] [Google Scholar]
- 20.Tanpure RP, Strecker TE, Chaplin DJ, Siim BG, Trawick ML, Pinney KG. J. Nat. Prod. 2010;73:1093–1101. doi: 10.1021/np100108e. [DOI] [PubMed] [Google Scholar]
- 21.Mooney CJ, Nagaiah G, Fu P, Wasman JK, Cooney MM, Savvides PS, Bokar JA, Dowlati A, Wang D, Agarwala SS, Flick SM, Hartman PH, Ortiz JD, Lavertu PN, Remick SC. Thyroid. 2009;19:233–240. doi: 10.1089/thy.2008.0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demers B, Vrignaud P, Bissery M. J. Clin. Oncol. (Meet. Abstr.) 2006;24:13074. [Google Scholar]
- 23.Siemann DW, Chaplin DJ, Walicke PA. Expert Opin. Investig. Drugs. 2009;18:189–197. doi: 10.1517/13543780802691068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garon EB, Kabbinavar FF, Neidhart JA, Neidhart JD, Gabrail NY, Oliveira MR, Lu S, Balkissoon J. J. Clin. Oncol. (Meet. Abstr.) 2011;29 (suppl; abstr 7559). [Google Scholar]
- 25.Mainwaring PN, Lickliter J, Brown MP, Millward M, Chaplin DJ, Goldberg Z. J. Clin. Oncol. (Meet. Abstr.) 2010;28:15s. (suppl; abstr TPS164). [Google Scholar]
- 26.Wohl R. J. Org. Chem. 1973;38:3862–3864. [Google Scholar]
- 27.McMurry JE, Coppolino AP. J. Org. Chem. 1973;38:2821–2827. [Google Scholar]
- 28.Miller RB, Gutierrez CG. J. Org. Chem. 1978;43:1569–1573. [Google Scholar]
- 29.Fillion E, Fishlock D, Wilsily A, Goll JM. J. Org. Chem. 2005;70:1316–1327. doi: 10.1021/jo0483724. [DOI] [PubMed] [Google Scholar]
- 30.Patti RK, Waynant KV, Herndon JW. Org. Lett. 2011;13:2848–2851. doi: 10.1021/ol200825j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allinger NL, Jones ES. J. Org. Chem. 1962;27:70–76. [Google Scholar]
- 32.Tanis VM, Moya C, Jacobs RS, Little RD. Tetrahedron. 2008;64:10649–10663. [Google Scholar]
- 33.Carpenter PD, Peesapati V, Proctor GR. J. Chem. Soc., Perkin Trans. 1. 1979:103–107. [Google Scholar]
- 34.Monk KA, Siles R, Hadimani MB, Mugabe BF, Ackley JF, Studerus SW, Edvardsen K, Trawick ML, Garner CM, Rhodes MR, Pettit GR, Pinney KG. Bioorg. Med. Chem. 2006;14:3231–3244. doi: 10.1016/j.bmc.2005.12.033. [DOI] [PubMed] [Google Scholar]
- 35.Pinney KG, Mejia MP, Villalobos VM, Rosenquist BE, Pettit GR, Verdier-Pinard P, Hamel E. Bioorg. Med. Chem. 2000;8:2417–2425. doi: 10.1016/s0968-0896(00)00176-0. [DOI] [PubMed] [Google Scholar]
- 36.Pettit GR, Grealish MP, Herald DL, Boyd MR, Hamel E, Pettit RK. J. Med. Chem. 2000;43:2731–2737. doi: 10.1021/jm000045a. [DOI] [PubMed] [Google Scholar]
- 37.Negoro N, Sasaki S, Mikami S, Ito M, Suzuki M, Tsujihata Y, Ito R, Harada A, Takeuchi K, Suzuki N, Miyazaki J, Santou T, Odani T, Kanzaki N, Funami M, Tanaka T, Kogame A, Matsunaga S, Yasuma T, Momose Y. ACS Med. Chem. Lett. 2010;1:290–294. doi: 10.1021/ml1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benzosuberene 1 (originally prepared according to ref. 1) has now been prepared by a separate synthetic strategy: Chen Z, O’Donnell CJ, Maderna A. Tetrahedron Lett. 2012;53:64–66.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.