

Abstract
Targeted therapy for cancer, which is specifically directed toward the cancer without any potential for effects outside of controlling the tumor, is a gold standard for treatment. Ewing’s sarcoma contains the potential target EWS-FLI1, as a result of a pathognomonic chromosomal translocation. The EWS-FLI1 fusion protein includes the EWS domain, a potent transcriptional activator alongside the highly conserved FLI1 ets DNA-binding domain. Because of the combination of these domains, the EWS-FLI1 fusion protein acts as an aberrant transcription factor whose expression results in cellular transformation. EWS-FLI1 functions by binding to normal cellular protein partners in transcription and splicing, similar to how a virus would corrupt normal cellular machinery for virion production. Therefore, understanding the protein-protein interactions of EWS-FLI1 and the pathways that are regulated by these partnerships will inform both oncogenesis and therapeutics. This review describes the known protein partners and transcriptional targets of EWS-FLI1, while proposing strategies for exploiting these partnerships with targeted therapy.
Background
The Ewing’s sarcoma family of tumors (ESFT) is composed of highly malignant tumors of bone and soft tissue occurring in children, adolescents, and young adults, requiring novel, targeted therapies to exploit tumor vulnerability based on ESFT ontogeny, oncogenesis, and tumor-maintenance pathways (reviewed in ref. 1). ESFT are defined by the characteristic chromosomal translocation t (11:22) and its fusion protein product EWS-FLI1. The translocation, or a related variant, occurs in 95% of tumors (2), between the central exons of the EWSR1 gene (for “Ewing Sarcoma Breakpoint region 1”; chromosome 22), to the central exons of an ets family gene; either FLI1 (“Friend Leukemia integration 1”; chromosome 11) or ERG (“v-ets erythroblastosis virus E26 oncogene homolog”; chromosome 21), t(11;22) and t(21;22), respectively.
The ontogeny of ESFT has been debated for more than 50 years, in the quest for the tumors’ cell of origin. To identify the cell of origin, multiple models have been created that express EWS-FLI1 in a variety of cell types, which delete EWS-FLI1 from ESFT cell lines, and which directly compare endogenous ESFT cell lines with primordial cell lines. Although early evidence favored a neuroectodermal origin, many recent studies have identified a primitive mesenchymal cell as the potential origin of ESFT (for reviews see refs. 3, 4). In fact, one unanswered question is whether the pathognomonic translocation is the initial event that leads to ESFT development, or if a predisposing genetic lesion leads to the translocation.
Whether initial event, or second hit, the EWS-FLI1 fusion protein is not only critical for tumor maintenance, but its elimination may be a therapeutic Achilles’ “heal” of ESFT. This pathognomonic fusion protein participates in the interconnected pathways of transcription and splicing. Dissection of the key protein partners will likely lead to strategies for disruption that will potentially benefit patients with ESFT by creating novel therapies; in addition, the analysis may be informative about the mysteries of mesenchymal solid tumor oncogenesis.
This review focuses upon the aspects of EWS-FLI1 related to its protein-protein interactions. In fact, linking protein-protein interactions to specific pathway regulation is a new frontier of ESFT research. Exploration and resolution of this frontier may not only shed light on the process of oncogenesis, but may also lead to potentially novel targets for therapeutic development.
EWS-FLI1 a central regulator of ESFT
The cloning of EWSR1-FLI1 and recognition of the expressed fusion protein product in ESFT began an era of molecular analysis of its ontogeny, maintenance, and therapy (5). EWS-FLI1 was quickly identified as a transcriptional activator, on the basis of its FLI1 ets binding domain and the EWS domain’s potently activated transcription (6, 7). Each tumor has a single translocation that combines truncated EWS at either exons 7, 8, 9, or 10 with exons 5, 6, 7, or 8 of FLI1, leading to a series of protein variants. As cDNA technology evolved, seminal studies showed that expression patterns of cDNA linked tumors with EWS-FLI1 expression, but did not distinguish between these fusion protein variants (8, 9). Many targets of EWS-FLI1, both direct and indirect, have been implicated in ESFT tumor maintenance (10). Protein partners of EWS-FLI1 were identified as part of the spliceosome, and subsequently, EWS-FLI1 was implicated as a modulator of splicing (11). Biochemical purification and characterization of EWS-FLI1 revealed it to be a disordered protein (12), and the intrinsic disorder of EWS-FLI1 is critical for its transcriptional activity (13).
EWS-FLI1 protein contains intrinsically disordered regions
A basic definition of disordered proteins is the lack of a stable structure when the disordered protein is isolated. The inherent flexibility and movement in disordered proteins allow rapid, yet specific, complex formation and dissociation that are critical for higher level transcriptional regulation in eukaryotes. Thus, the characterization and composition of protein complexes containing EWS-FLI1 are central to revealing its functional pathways. The disordered nature of EWS-FLI1 allows for its participation in multiprotein nuclear complexes. No direct enzymatic activity has been proscribed to EWS-FLI1, nor are any putative enzymatic domains recognized. Therefore, we rely on EWS-FLI1’s protein-protein interaction to identify its activity and dissect the functional pathways. EWS-FLI1 protein interactions lead to regulation of transcriptional targets (14, 15), through processes that modulate mRNA synthesis or splicing. EWS-FLI1 interacts with other proteins on the basis of its intrinsic disorder, rather than currently characterized protein interaction domains (such as SH2, PDZ, etc.). Thus, the intrinsically disordered regions of EWS-FLI1 facilitate protein-protein complexes that lead to oncogenesis.
Disordered proteins have evolved with the higher cellular functions and compartmentalization that correspond to prokaryotes evolving into eukaryotes (16-18). The amino acid composition and/or sequence of proteins dictates intrinsic disorder. The abundance of prolines, charged and polar residues, combined with depletion of cysteins, and hydrophobic and aromatic amino acids are hallmarks of intrinsically disordered proteins. This combination of amino acids prevents such proteins from forming singular, fixed structures. These “structureless” peptides and/or proteins do have specific features when analyzed by nuclear magnetic resonance (19). They exist as dynamic ensembles within which atom positions and backbone Ramachandran angles exhibit extreme temporal and spatial fluctuations without specific equilibrium values. It is important to recognize that disordered proteins are not incorrectly folded rejects of the endoplasmic reticulum directed toward proteosomal degradation, but are proteins whose functions rely upon intrinsic movement to participate in multiprotein complexes like transcription. The characterization and role of disordered proteins in human disease represents a novel intersection of biochemistry and pathology (reviewed in ref. 20).
The purification of EWS-FLI1 was challenged by many of the biochemical properties of disordered proteins, including low and/or poor solubility and aggregation. By calculating the overall hydrophobicity in relation to the charge of EWS-FLI1, the molecule was described as intrinsically disordered (12). Importantly, EWS-FLI1 transcriptional activation was dependent on the intrinsic disorder (13). Three different algorithms (VSL2, VLXT, VL3) show the almost 100% prediction of disorder from the amino-terminal of EWS up to the DNA-binding domain of FLI1 (data are not shown). The DNA-binding domain composed of helix-turn-helix is relatively ordered, whereas the far-carboxy region is also intrinsically disordered. Understanding that EWS-FLI1 is a disordered protein is critical to probing and characterizing its protein-protein interactions.
EWS-FLI1 partners with proteins in transcriptional apparatus
Disordered protein interactions have relatively high specificity and low affinity (21). Some have postulated that the very properties of EWS-FLI1 intrinsic disorder allow for the precise regulation of transcription (12). Although a key event in transcriptional regulation occurs when transcription factors bind to regulatory elements in the DNA, it is the recruitment of cofactors that either initiate or repress transcription. Although EWS-FLI1 shares the ets binding core sequence with other ets family members, their divergent regulated targets may be explained by the differences in protein-protein interactions (Fig. 1).
Fig. 1.
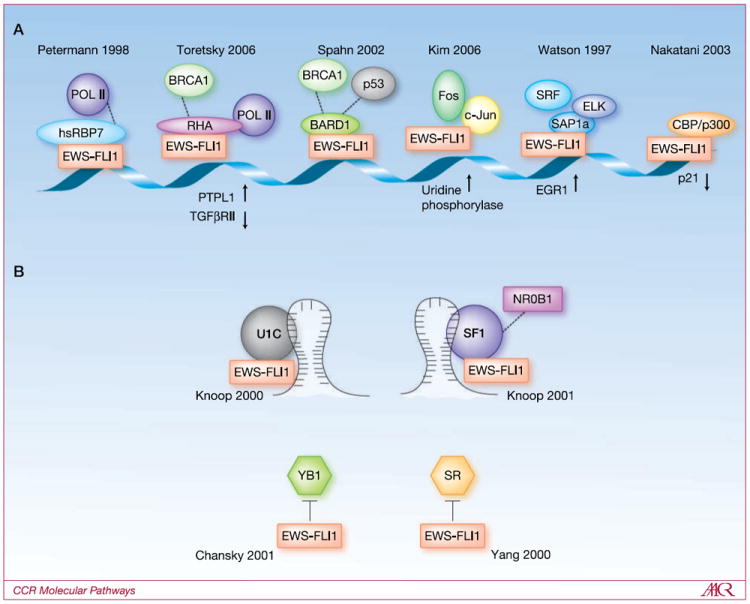
EWS-FLI1 partners with multiple proteins that regulate mRNA transcription and splicing. A, EWS-FLI1 protein-protein interaction partners in transcription. EWS-FLI1 interacts with hsRBP7, RHA, BARD1, C-JUN, SAP1a, CBP/p300. The name of the gene product is listed below the interaction when a given complex is directly linked to that promoter. All references were reviewed that contained evidence of partner proteins for EWS-FLI1 and included if they contained at least two independent validation experiments. The description of each protein interaction is described in the text. Dotted lines indicate well-documented interactions between proteins described in models that did not specifically include EWS-FLI1. B, EWS-FLI1 protein-protein interaction partners in splicing. U1C and SF1 proteins interact with EWS-FLI1 to modulate splicing. Interactions with YB1 and SR inhibit or alter splicing. Dotted lines indicate well-documented interactions between proteins described in models that did not specifically include EWS-FLI1.
Early investigations identified that EWS-FLI1 binds to DNA in ternary protein complexes, with binding partners dependent upon adjacent DNA sequences (22). The EWS-FLI1 interaction with FOS-JUN dimers, which bind AP1 sequences, regulates a number of EWS-FLI1–modulated transcripts, including uridine phosphorlyase (23). These investigations confirmed a critical role for the far carboxy-terminus of EWS-FLI1 in protein-protein interactions, which was predicted from earlier functional studies (24). In fact, the far-carboxy region of EWS-FLI1 is predicted to be intrinsically disordered, which would support its critical interaction with other transcriptional regulators. Thus, the far-carboxy region of EWS-FLI1 likely determines the adjacent protein binding to DNA regulatory elements. As a transcriptional regulator, EWS-FLI1 not only binds to DNA, but also directly interacts with the transcriptional apparatus.
RNA polymerase holozyme II (Pol II) is a 12-subunit, heteromeric complex that is the synthetic apparatus of mRNA. The wild-type EWS protein binds to the pretran-scription complex of TFIID, as well as the synthetic hRPB3 subunit of Pol II (25). Using an immunoprecipitation approach and confirmatory sedimentation gradient experiments, EWS-FLI1 was not found in the same transcriptional complexes as EWS (25). The authors were careful to point out that EWS-FLI1 binding to transcriptional proteins may have been missed because of transient interactions with lower affinity. The hRBP7 subunit of Pol II was found to bind EWS-FLI1 and was confirmed to bind to the EWS- portion (26). An additional report suggests that Pol II phosphorylation differentially determines EWS-FLI1’s direct binding (27). The differential binding of EWS compared with EWS-FLI1 supports different roles for the proteins. There is evidence both in support of and against the hypothesis that EWS-FLI1 acts as a dominant negative to EWS in oncogenesis.
Additional components of the transcriptional apparatus described as partners of EWS-FLI1 include Creb-binding protein (CBP; Fig. 1A; refs. 28, 29). CBP is also a functional acetylase, yet the acetylation of EWS-FLI1 has not been described. BARD1 was identified as a protein partner using a yeast two-hybrid approach, but the functional role has not been pursued (30).
RNA helicase A (RHA) was identified as a protein partner of EWS-FLI1 using phage display (31). RHA is a multifunctional RNA helicase, which is part of both the basal transcriptional apparatus (32), as well as the posttranscriptional RNA metabolism (33). RHA was found to be present at promoters occupied by EWS-FLI1 and was required for optimal transformation of murine fibroblasts (31). Reduced EWS-FLI1 activity and tumorigenesis was observed after using mutation, peptide, and small molecule approaches to disrupt RHA from binding to EWS-FLI1 (34).
NR0B1 (DAX1) was first identified as a transcriptional target of EWS-FLI1 using a cDNA array that compared profiles of EWS-FLI1 to FLI1-expressing cells (35, 36). NR0B1 putatively regulates cell cycle in ESFT cell lines (37). When the EWS-FLI1 level was reduced with RNA interference, NR0B1 transcription levels were reduced, suggesting EWS-FLI1 regulates NR0B1 transcription. NR0B1 was also necessary, but not sufficient, for the transformation of fibroblasts by EWS-FLI1 (36). EWS-FLI1 not only activates transcription of NR0B1, but the two proteins also seem to partner at sites of DNA binding (38).
EWS-FLI1 protein interactions include the spliceosome
Posttranscriptional mRNA splicing is a critical regulatory aspect of ultimate protein production. Using an unbiased yeast two-hybrid approach, the splicing component snRNP U1C was identified as a protein partner of EWS-FLI1 (Fig. 1B; ref. 11). EWS-FLI1 functionally altered splice site selection of the E1A transcript (39). Additional proteins involved in the EWS-FLI1 spliceosome, but not identified as directly binding to EWS-FLI1, are the TASR proteins (27) and YB-1 (40). More recently, EWS-FLI1 showed altered splice site selection of cyclin D leading to a more stable cell cycle agonist (41). The small molecule YK-4-279 that blocks RHA binding to EWS-FLI1 showed decreased cyclin D levels in ESFT cells (34). Recently, a proteomic approach identified multiple spliceosome components binding to EWS, but EWS-FLI1 was not evaluated (42).
Posttranslational modifications of EWS-FLI1 influence protein interactions
The posttranslational modifications of EWS-FLI1 have been relatively elusive, and to date, this area of investigation suffers a paucity of published findings. If ESFT cells are stressed with a potent toxin that induces double-strand DNA breakage, EWS-FLI1 is phosphorylated on specific threonine residues (43). In a more general approach, ESFT cells treated with okadaic acid, as a general poison of serine-threonine phosphatases, showed enhanced phosphorylation; however, the residues were not identified (44). O-GlcNAcylation of EWS-FLI1 enhances transcription and seems to be reciprocally related to phosphorylation (44). The effects of these posttranslational modifications on EWS-FLI1 protein-protein interactions and on overall ESFT cellular growth remain to be investigated.
Transcriptome of ESFT regulated by EWS-FLI1
Although EWS-FLI1 partnering is relatively novel, EWS-FLI1 transcriptional targets have long been recognized as important mediators of oncogenesis. The pathways that are modulated by these transcriptional targets are thought to summarize the Ewing’s sarcoma phenotype. Although a recent study describes a complete functional map of ESFT, it does so at the level of an integrative analysis of gene expression profiling experiments (45). Specific downstream targets of EWS-FLI1 have been investigated on the basis of expression array analysis and, in many cases, follow-up functional investigation.
Transcriptional targets of EWS-FLI1 include transcriptional modulators, signaling molecules, cell cycle regulators, metabolism, and angiogenesis genes (Table 1). Direct targets of EWS-FLI1 are those that have been confirmed with techniques such as chromatin immunoprecipitation, whereas indirect targets have not yet been shown to have this direct association of EWS-FLI1 with the gene promoter. Although many of the initially discovered targets have been reviewed (10), new targets that are more directly associated with cell fate have been identified. Recently, Enhancer of Zeste 2 (EZH2) was identified as a direct target of EWS-FLI1 (46, 47). EZH2 directly resulted in upregulation of genes having roles in neuroectodermal and endothelial differential in ESFT cells. Other recently identified direct targets include caveolin (48), Aurora kinase A (49), hTERT (50), uridine phosphorylase (51), GSTM4 (52), PLD2 (53), and GLI1 (54, 55).
Table 1.
EWS-FLI1’s transcriptional targets
| Transcriptional regulators | Signaling pathway regulation | Cell cycle | Metabolism | Angiogenesis-extracellular matrix |
|---|---|---|---|---|
| EZH2 | IGFBP3 | Cyclin E | Uridine phosphorylase | VEGF-A |
| NR0B1 | PTPL1 | p21 | GSTM4 | MMP-1 |
| GLI1 | Caveolin | P57KIP* | — | PLD2 |
| Id2 | hTERT | CYCLIN D1* | — | Tenascin C |
| EGR1 | TGFβRII | — | — | — |
| NKX2.2* | AURKA | — | — | — |
| — | PDGFC* | — | — | — |
Indirect targets of EWS-FLI1.
Clinical-Translational Advances
Targeting the oncoprotein EWS-FLI1
EWS-FLI1 has only been identified in tumor cells, thus providing a unique therapeutic target. EWS-FLI1, as a single oncogene, is able to transform fibroblasts, although the cellular context and augmenting proteins are necessary for oncogenesis (56). Reduction of EWS-FLI1 by antisense oligodeoxynucleotides (57-59), antisense RNA expressed from a vector (59-61), and small interfering RNA (siRNA) delivered via nanoparticles (62), all inhibit the proliferation of ESFT cell lines and xenografted tumors. The lack of clinical translation of these macromolecule-based strategies lies in the challenge of pharmacologic delivery (63).
Targeted inhibition represents a significant challenge because EWS-FLI1 lacks intrinsic enzymatic function, as described above. One approach to identifying EWS-FLI1 inhibitors is based upon high-throughput transcript activation (64). An alternative was sought to identify clinically approved agents with a similar cDNA signature as compared with decreased EWS-FLI1 levels. Neither of these methods has led to validated, specific EWS-FLI1 inhibitors. Disruption of key EWS-FLI1 protein-protein interactions is a strategy suggested to regulate other ets-family proteins (65), thus potentially useful for EWS-FLI1.
Fortunately, the intrinsic flexibility and movement of disordered proteins not only allows for protein-protein interaction, leading to formation of transcriptional complexes, but also provides an opportunity for small molecule targeting. Disruption of protein-protein interactions is thought to be a very challenging, albeit surmountable, target for small molecule therapeutics (66-68). Investigators are actively exploring the methodology of identifying small molecule protein-protein interaction inhibitors (69). In order to effectively target EWS-FLI1 and develop assays for small molecule optimization, we sought valid protein partners of EWS-FLI1. An elegant correlation showed that the region of RHA that bound to EWS-FLI1 contained the degenerate peptide sequence (E9R) from phage display (31). E9R has been an important tool for validating the interaction of EWS-FLI1/RHA as a therapeutic target (34).
The recombinant EWS-FLI1 was a critical reagent for screening a library of small molecules to identify compounds with direct binding. Molecules identified from a National Cancer Institute Developmental Therapeutics Program library were secondarily screened as having “drug-like” qualities by collaborators with medicinal chemistry expertise. A lead scaffold was selected, followed by combinatorial expansion, on the basis of decorating one of the phenol rings. Among the novel structures, YK-4-279 was selected for further study on the basis of assays showing dissociation of recombinant RHA from EWS-FLI1 (34).
In addition to EWS-FLI1 protein-protein interaction targeting, some investigators are inhibiting key transcriptional targets. An example is MLN8237, a small molecule inhibitor of Aurora kinase A (AURKA). Recently, AURKA was shown as a transcriptional target of ESFT, and in a screen of pediatric tumors using the AURKA inhibitor, ESFT xenografts were particularly sensitive (70). Insulin-like growth factor receptor I (IGF-IR) inhibitors, small molecules, and antibodies are in widespread clinical trials for patients with ESFT. These trials are based upon data that IGF-IR is critical for EWS-FLI1 transformation, as well as an abundance of preclinical studies showing xenograft efficacy (56, 71, 72). Recently, a first phase I study of an IGF-IR antibody showed clinical regression in patients with ESFT (73).
Integration of EWS-FLI1 directed therapy into clinical use
At this time, no direct, targeted therapeutics against EWS-FLI1 are in clinical use. However, the evolution of proteomics with enhanced biochemical understanding of disordered proteins will likely lead to novel therapeutics. Given the high mortality of patients with metastatic or recurrent disease, who ultimately become refractory to non-targeted therapy, novel and effective therapeutics are needed. The small molecule YK-4-279 is the first molecule to directly target EWS-FLI1 (34), and, if developed clinically, would potentially add to the clinical arsenal against ESFT. Additional therapies that target the “targets” will be interesting to explore, as the targeted pathways of EWS-FLI1 may enhance the vulnerability of ESFT. The development of EWS-FLI1–specific therapies will likely benefit patients with ESFT and will inform the mechanism of cancers that have the Achilles’ heel of a chromosomal translocation-generated oncoprotein.
Acknowledgments
The authors would like to dedicate this review in honor of Mrs. Shirley Howard and the Children’s Cancer Foundation of Baltimore, Maryland. Their support was critical in maintaining our research program when many thought direct targeting of EWS-FLI1 was not possible. The authors would like to thank Ms. Kerry Adam for editorial assistance.
Grant Support
Children’s Cancer Foundation of Baltimore, Maryland, Go4theGoal, Dani’s Foundation, Alex’s Lemonade Stand Foundation, Liddy Shriver Sarcoma Initiative, Amschwand Sarcoma Foundation, AFLAC Foundation, Burroughs Wellcome Clinical Scientist Award in Translational Research (J.A. Toretsky), and the NIH National Cancer Institute.
Footnotes
Disclosure of Potential Conflicts of Interest
A composition of matter and use patent application has been filed by author J.A. Toretsky for compound YK-4-279.
References
- 1.Ludwig JA. Ewing sarcoma: historical perspectives, current state-of-the-art, and opportunities for targeted therapy in the future. Curr Opin Oncol. 2008;20:412–8. doi: 10.1097/CCO.0b013e328303ba1d. [DOI] [PubMed] [Google Scholar]
- 2.Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors–a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med. 1994;331:294–9. doi: 10.1056/NEJM199408043310503. [DOI] [PubMed] [Google Scholar]
- 3.Riggi N, Suva ML, Stamenkovic I. Ewing’s sarcoma origin: from duel to duality. Expert Rev Anticancer Ther. 2009;9:1025–30. doi: 10.1586/era.09.81. [DOI] [PubMed] [Google Scholar]
- 4.Kovar H. Context matters: the hen or egg problem in Ewing’s sarcoma. Semin Cancer Biol. 2005;15:189–96. doi: 10.1016/j.semcancer.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Delattre O, Zucman J, Plougastel B, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–5. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 6.May WA, Lessnick SL, Braun BS, et al. The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol. 1993;13:7393–8. doi: 10.1128/mcb.13.12.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailly RA, Bosselut R, Zucman J, et al. DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t(11;22) translocation in Ewing sarcoma. Mol Cell Biol. 1994;14:3230–41. doi: 10.1128/mcb.14.5.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aryee DN, Sommergruber W, Muehlbacher K, Dockhorn-Dworniczak B, Zoubek A, Kovar H. Variability in gene expression patterns of Ewing tumor cell lines differing in EWS-FLI1 fusion type. Lab Invest. 2000;80:1833–44. doi: 10.1038/labinvest.3780194. [DOI] [PubMed] [Google Scholar]
- 9.Khan J, Wei JS, Ringner M, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–9. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uren A, Toretsky JA. Ewing’s sarcoma oncoprotein EWS-FLI1: the perfect target without a therapeutic agent. Future Oncol. 2005;1:521–8. doi: 10.2217/14796694.1.4.521. [DOI] [PubMed] [Google Scholar]
- 11.Knoop LL, Baker SJ. The splicing factor U1C represses EWS/FLI-mediated transactivation. J Biol Chem. 2000;275:24865–71. doi: 10.1074/jbc.M001661200. [DOI] [PubMed] [Google Scholar]
- 12.Uren A, Tcherkasskaya O, Toretsky JA. Recombinant EWS-FLI1 oncoprotein activates transcription. Biochemistry. 2004;43:13579–89. doi: 10.1021/bi048776q. [DOI] [PubMed] [Google Scholar]
- 13.Ng KP, Potikyan G, Savene RO, Denny CT, Uversky VN, Lee KA. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proc Natl Acad Sci U S A. 2007;104:479–84. doi: 10.1073/pnas.0607007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siligan C, Ban J, Bachmaier R, et al. EWS-FLI1 target genes recovered from Ewing’s sarcoma chromatin. Oncogene. 2005;24:2512–24. doi: 10.1038/sj.onc.1208455. [DOI] [PubMed] [Google Scholar]
- 15.Nielsen TO. Microarray analysis of sarcomas. Adv Anat Pathol. 2006;13:166–73. doi: 10.1097/00125480-200607000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Liu L, Westler WM, den Boon JA, et al. An amphipathic α-helix controls multiple roles of brome mosaic virus protein 1a in RNA replication complex assembly and function. PLoS Pathog. 2009;5:e1000351. doi: 10.1371/journal.ppat.1000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–71. [PubMed] [Google Scholar]
- 18.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–45. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Mittag T, Orlicky S, Choy WY, et al. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc Natl Acad Sci U S A. 2008;105:17772–7. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–46. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 21.Zhang WD, Xie CM, Mo YX, Li JY. [CT and MRI features of peripheral primitive neuroectodermal tumor] [Article in Chinese] Ai Zheng. 2007;26:643–6. [PubMed] [Google Scholar]
- 22.Watson DK, Robinson L, Hodge DR, Kola I, Papas TS, Seth A. FLI1 and EWS-FLI1 function as ternary complex factors and ELK1 and SAP1a function as ternary and quaternary complex factors on the Egr1 promoter serum response elements. Oncogene. 1997;14:213–21. doi: 10.1038/sj.onc.1200839. [DOI] [PubMed] [Google Scholar]
- 23.Kim S, Denny CT, Wisdom R. Cooperative DNA binding with AP-1 proteins is required for transformation by EWS-Ets fusion proteins. Mol Cell Biol. 2006;26:2467–78. doi: 10.1128/MCB.26.7.2467-2478.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arvand A, Welford SM, Teitell MA, Denny CT. The COOH-terminal domain of FLI-1 is necessary for full tumorigenesis and transcriptional modulation by EWS/FLI-1. Cancer Res. 2001;61:5311–7. [PubMed] [Google Scholar]
- 25.Bertolotti A, Melot T, Acker J, Vigneron M, Delattre O, Tora L. EWS, but not EWS-FLI-1, is associated with both TFIID and RNA polymerase II: interactions between two members of the TET family, EWS and hTAFII68, and subunits of TFIID and RNA polymerase II complexes. Mol Cell Biol. 1998;18:1489–97. doi: 10.1128/mcb.18.3.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petermann R, Mossier BM, Aryee DN, Khazak V, Golemis EA, Kovar H. Oncogenic EWS-Fli1 interacts with hsRPB7, a subunit of human RNA polymerase II. Oncogene. 1998;17:603–10. doi: 10.1038/sj.onc.1201964. [DOI] [PubMed] [Google Scholar]
- 27.Yang L, Chansky HA, Hickstein DD. EWS.Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. J Biol Chem. 2000;275:37612–8. doi: 10.1074/jbc.M005739200. [DOI] [PubMed] [Google Scholar]
- 28.Ramakrishnan R, Fujimura Y, Zou JP, et al. Role of protein-protein interactions in the antiapoptotic function of EWS-Fli-1. Oncogene. 2004;23:7087–94. doi: 10.1038/sj.onc.1207927. [DOI] [PubMed] [Google Scholar]
- 29.Nakatani F, Tanaka K, Sakimura R, et al. Identification of p21WAF1/CIP1 as a direct target of EWS-Fli1 oncogenic fusion protein. J Biol Chem. 2003;278:15105–15. doi: 10.1074/jbc.M211470200. [DOI] [PubMed] [Google Scholar]
- 30.Spahn L, Petermann R, Siligan C, Schmid JA, Aryee DN, Kovar H. Interaction of the EWS NH2 terminus with BARD1 links the Ewing’s sarcoma gene to a common tumor suppressor pathway. Cancer Res. 2002;62:4583–7. [PubMed] [Google Scholar]
- 31.Toretsky JA, Erkizan V, Levenson A, et al. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res. 2006;66:5574–81. doi: 10.1158/0008-5472.CAN-05-3293. [DOI] [PubMed] [Google Scholar]
- 32.Nakajima T, Uchida C, Anderson SF, et al. RNA helicase A mediates association of CBP with RNA polymerase II. Cell. 1997;90:1107–12. doi: 10.1016/s0092-8674(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 33.Hartman TR, Qian S, Bolinger C, Fernandez S, Schoenberg DR, Boris-Lawrie K. RNA helicase A is necessary for translation of selected messenger RNAs. Nat Struct Mol Biol. 2006;13:509–16. doi: 10.1038/nsmb1092. [DOI] [PubMed] [Google Scholar]
- 34.Erkizan HV, Kong Y, Merchant M, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med. 2009;15:750–6. doi: 10.1038/nm.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendiola M, Carrillo J, Garcia E, et al. The orphan nuclear receptor DAX1 is up-regulated by the EWS/FLI1 oncoprotein and is highly expressed in Ewing tumors. Int J Cancer. 2006;118:1381–9. doi: 10.1002/ijc.21578. [DOI] [PubMed] [Google Scholar]
- 36.Kinsey M, Smith R, Lessnick SL. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol Cancer Res. 2006;4:851–9. doi: 10.1158/1541-7786.MCR-06-0090. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Aragoncillo E, Carrillo J, Lalli E, et al. DAX1, a direct target of EWS/FLI1 oncoprotein, is a principal regulator of cell-cycle progression in Ewing’s tumor cells. Oncogene. 2008;27:6034–43. doi: 10.1038/onc.2008.203. [DOI] [PubMed] [Google Scholar]
- 38.Kinsey M, Smith R, Iyer AK, McCabe ER, Lessnick SL. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res. 2009;69:9047–55. doi: 10.1158/0008-5472.CAN-09-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knoop LL, B S. EWS/FLI alters 5′-splice site selection. J Biol Chem. 2001;276:22317–22. doi: 10.1074/jbc.M008950200. [DOI] [PubMed] [Google Scholar]
- 40.Chansky HA, Hu M, Hickstein DD, Yang L. Oncogenic TLS/ERG and EWS/Fli-1 fusion proteins inhibit RNA splicing mediated by YB-1 protein. Cancer Res. 2001;61:3586–90. [PubMed] [Google Scholar]
- 41.Sanchez G, Bittencourt D, Laud K, et al. Alteration of cyclin D1 transcript elongation by a mutated transcription factor up-regulates the oncogenic D1b splice isoform in cancer. Proc Natl Acad Sci U S A. 2008;105:6004–9. doi: 10.1073/pnas.0710748105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pahlich S, Quero L, Roschitzki B, Leemann-Zakaryan RP, Gehring H. Analysis of Ewing sarcoma (EWS)-binding proteins: interaction with hnRNP M, U, and RNA-helicases p68/72 within protein-RNA complexes. J Proteome Res. 2009;8:4455–65. doi: 10.1021/pr900235t. [DOI] [PubMed] [Google Scholar]
- 43.Klevernic IV, Morton S, Davis RJ, Cohen P. Phosphorylation of Ewing’s sarcoma protein (EWS) and EWS-Fli1 in response to DNA damage. Biochem J. 2009;418:625–34. doi: 10.1042/BJ20082097. [DOI] [PubMed] [Google Scholar]
- 44.Bachmaier R, Aryee DN, Jug G, et al. O-GlcNAcylation is involved in the transcriptional activity of EWS-FLI1 in Ewing’s sarcoma. Oncogene. 2009;28:1280–4. doi: 10.1038/onc.2008.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kauer M, Ban J, Kofler R, et al. A molecular function map of Ewing’s sarcoma. PLoS ONE. 2009;4:e5415–7. doi: 10.1371/journal.pone.0005415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riggi N, Suva ML, Suva D, et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008;68:2176–85. doi: 10.1158/0008-5472.CAN-07-1761. [DOI] [PubMed] [Google Scholar]
- 47.Richter GH, Plehm S, Fasan A, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuroectodermal differentiation. Proc Natl Acad Sci U S A. 2009;106:5324–9. doi: 10.1073/pnas.0810759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tirado OM, Mateo-Lozano S, Villar J, et al. Caveolin-1 (CAV1) is a target of EWS/FLI-1 and a key determinant of the oncogenic phenotype and tumorigenicity of Ewing’s sarcoma cells. Cancer Res. 2006;66:9937–47. doi: 10.1158/0008-5472.CAN-06-0927. [DOI] [PubMed] [Google Scholar]
- 49.Wakahara K, Ohno T, Kimura M, et al. EWS-Fli1 up-regulates expression of the Aurora A and Aurora B kinases. Mol Cancer Res. 2008;6:1937–45. doi: 10.1158/1541-7786.MCR-08-0054. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi A, Higashino F, Aoyagi M, et al. EWS/ETS fusions activate telomerase in Ewing’s tumors. Cancer Res. 2003;63:8338–44. [PubMed] [Google Scholar]
- 51.Deneen B, Hamidi H, Denny CT. Functional analysis of the EWS/ETS target gene uridine phosphorylase. Cancer Res. 2003;63:4268–74. [PubMed] [Google Scholar]
- 52.Luo W, Gangwal K, Sankar S, Boucher KM, Thomas D, Lessnick SL. GSTM4 is a microsatellite-containing EWS/FLI target involved in Ewing’s sarcoma oncogenesis and therapeutic resistance. Oncogene. 2009;28:4126–32. doi: 10.1038/onc.2009.262. [DOI] [PubMed] [Google Scholar]
- 53.Nozawa S, Ohno T, Banno Y, et al. Inhibition of platelet-derived growth factor-induced cell growth signaling by a short interfering RNA for EWS-Fli1 via down-regulation of phospholipase D2 in Ewing sarcoma cells. J Biol Chem. 2005;280:27544–51. doi: 10.1074/jbc.M411626200. [DOI] [PubMed] [Google Scholar]
- 54.Zwerner JP, Joo J, Warner KL, et al. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene. 2008;27:3282–91. doi: 10.1038/sj.onc.1210991. [DOI] [PubMed] [Google Scholar]
- 55.Beauchamp E, Bulut G, Abaan O, et al. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem. 2009;284:9074–82. doi: 10.1074/jbc.M806233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toretsky JA, Kalebic T, Blakesley V, LeRoith D, Helman LJ. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J Biol Chem. 1997;272:30822–7. doi: 10.1074/jbc.272.49.30822. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka K, Iwakuma T, Harimaya K, Sato H, Iwamoto Y. EWS-Fli1 antisense oligodeoxynucleotide inhibits proliferation of human Ewing’s sarcoma and primitive neuroectodermal tumor cells. J Clin Invest. 1997;99:239–47. doi: 10.1172/JCI119152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Toretsky JA, Connell Y, Neckers L, Bhat NK. Inhibition of EWS-FLI-1 fusion protein with antisense oligodeoxynucleotides. J Neurooncol. 1997;31:9–16. doi: 10.1023/a:1005716926800. [DOI] [PubMed] [Google Scholar]
- 59.Ouchida M, Ohno T, Fujimura Y, Rao VN, Reddy ES. Loss of tumorigenicity of Ewing’s sarcoma cells expressing antisense RNA to EWS-fusion transcripts. Oncogene. 1995;11:1049–54. [PubMed] [Google Scholar]
- 60.Kovar H, Aryee DN, Jug G, et al. EWS/FLI-1 antagonists induce growth inhibition of Ewing tumor cells in vitro. Cell Growth Differ. 1996;7:429–37. [PubMed] [Google Scholar]
- 61.Maksimenko A, Lambert G, Bertrand JR, Fattal E, Couvreur P, Malvy C. Therapeutic potentialities of EWS-Fli-1 mRNA-targeted vectorized antisense oligonucleotides. Ann N Y Acad Sci. 2003;1002:72–7. doi: 10.1196/annals.1281.017. [DOI] [PubMed] [Google Scholar]
- 62.Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ. Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res. 2005;65:8984–92. doi: 10.1158/0008-5472.CAN-05-0565. [DOI] [PubMed] [Google Scholar]
- 63.Grimm D, Streetz KL, Jopling CL, et al. Fatality in mice due to over-saturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–41. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 64.Grohar PJ, Woldemichael G, Griffin L, et al. Identification of transcription factor inhibitors: a method used to identify small molecules that block EWS-FLI1 transcription activity. Proceedings of the 100th Annual Meeting of the American Association for Cancer Research; Denver, CO. Philadelphia (PA): AACR; 2009. 2009. Abstract nr 3805. [Google Scholar]
- 65.Li R, Pei H, Watson DK. Regulation of Ets function by protein-protein interactions. Oncogene. 2000;19:6514–23. doi: 10.1038/sj.onc.1204035. [DOI] [PubMed] [Google Scholar]
- 66.Sillerud LO, Larson RS. Design and structure of peptide and peptidomimetic antagonists of protein-protein interaction. Curr Protein Pept Sci. 2005;6:151–69. doi: 10.2174/1389203053545462. [DOI] [PubMed] [Google Scholar]
- 67.Pagliaro L, Felding J, Audouze K, et al. Emerging classes of protein-protein interaction inhibitors and new tools for their development. Curr Opin Chem Biol. 2004;8:442–9. doi: 10.1016/j.cbpa.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 68.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–17. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 69.Murray JK, Gellman SH. Targeting protein-protein interactions: lessons from p53/MDM2. Biopolymers. 2007;88:657–86. doi: 10.1002/bip.20741. [DOI] [PubMed] [Google Scholar]
- 70.Maris JM, Morton CL, Gorlick R, et al. Initial testing of the aurora kinase a inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP) Pediatr Blood Cancer. 2010;55:26–34. doi: 10.1002/pbc.22430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Houghton PJ, Morton CL, Gorlick R, et al. Initial testing of a monoclonal antibody (IMC-A12) against IGF-1R by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2010;54:921–6. doi: 10.1002/pbc.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Manara MC, Landuzzi L, Nanni P, et al. Preclinical in vivo study of new insulin-like growth factor-I receptor–specific inhibitor in Ewing’s sarcoma. Clin Cancer Res. 2007;13:1322–30. doi: 10.1158/1078-0432.CCR-06-1518. [DOI] [PubMed] [Google Scholar]
- 73.Olmos D, Postel-Vinay S, Molife LR, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol. 2010;11:129–35. doi: 10.1016/S1470-2045(09)70354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]