

Abstract
We previously reported that in healthy mouse cerebral arteries, endothelial nitric oxide synthase (eNOS) produces H2O2, leading to endothelium-dependent dilation. In contrast, thromboxane A2 (TXA2), a potent pro-oxidant and pro-inflammatory endogenous vasoconstrictor, is associated with eNOS dysfunction. Our objectives were to elucidate whether (1) the cerebrovascular eNOS–H2O2 pathway was sensitive to oxidative stress associated with aging and dyslipidemia and (2) TXA2 contributed to cerebral eNOS dysfunction. Atherosclerotic (ATX= LDLR−/−; hApoB+/+) and wild-type (WT) control mice were used at 3 and 12 months old (m/o). Three-m/o ATX mice were treated with the cardio-protective polyphenol catechin for 9 months. Dilations to ACh and the simultaneous eNOS-derived H2O2 production were recorded in isolated pressurized cerebral arteries. The age-associated decrease in cerebral eNOS–H2O2 pathway observed in WT was premature in ATX mice, decreasing at 3 m/o and abolished at 12 m/o. Thromboxane synthase inhibition by furegrelate increased dilations at 12 months in WT and at 3 and 12 months in ATX mice, suggesting an anti-dilatory role of TXA2 with age hastened by dyslipidemia. In addition, the non-selective NADP(H) oxidase inhibitor apocynin improved the eNOS–H2O2 pathway only in 12-m/o ATX mice. Catechin normalized the function of this pathway, which became sensitive to L-NNA and insensitive to furegrelate or apocynin; catechin also prevented the rise in TXA2 synthase expression. In conclusion, the age-dependent cerebral endothelial dysfunction is precocious in dyslipidemia and involves TXA2 production that limits eNOS activity. Preventive catechin treatment reduced the impact of endogenous TXA2 on the control of cerebral tone and maintained eNOS function.
Keywords: Cerebral artery, Aging, Dyslipidemia, Endothelial function, Endothelium-derived constricting factors
Introduction
We previously reported that in mouse cerebral arteries, endothelial nitric oxide synthase (eNOS), cyclooxygenase (COX), and an endothelium-derived hyperpolarizing factor (EDHF) are important regulators of vascular tone in response to ACh and flow [7, 8]. We also reported that eNOS stimulation of healthy mouse cerebral arteries leads to the production of vasodilatory H2O2 and not the release of NO, showing that eNOS produces physiologically relevant levels of free radicals leading to H2O2-dependent dilation [7, 8]. This eNOS–H2O2 pathway represents a non-traditional mechanism for healthy vessels and is still a subject of ongoing debate. A similar vasodilatory pathway, however, has been described in peripheral mouse and human arteries [27, 33] and human coronary arterioles [25]. The generation by eNOS of H2O2 rather than NO is usually associated with eNOS uncoupling and endothelial dysfunction, and reported in a pathological situation [11]. With the brain being highly susceptible to oxidative damage [1], one would therefore expect the arterial eNOS–H2O2 pathway to be highly sensitive to the redox environment.
There is overwhelming evidence that a rise in oxidative stress is associated with aging and multiple cardiovascular diseases (CVD). Although an “apparent” protection of intracranial arteries from atherosclerosis has been reported based on the fact that atherosclerotic lesions are rarely observed in cerebral arteries from animal models [5, 49], it is now clear that endothelial dysfunction also occurs in the cerebral circulation: atherosclerosis in ApoE−/− mice is associated with NOS dysfunction and an increase in NADP (H) oxidase activity, leading to the rise in oxidative stress and cerebral endothelial damage [20, 31, 32]. In peripheral arteries, it has been proposed that the impairment of eNOS function by oxidative stress could promote TXA2 production, a potent vasoconstrictor with inflammatory properties, which would further contribute to the rise in ROS and endothelial dysfunction [13, 14, 57]. Serum levels of TXA2 and thromboxane receptor expression are elevated in patients with several vascular and ischemic diseases [37], including cerebral ischemia [2, 21, 43, 55]. In addition, a specific polymorphism in the TXA2 synthase-1 gene has been reported in some patients with cerebral infarction [40]. TXA2 is produced by brain tissues [9] and cerebral arteries, both in vitro [15] and in vivo [42], and the release of TXA2 in cerebral arteries is 10-fold higher than in coronary, mesenteric, or saphenous arteries [48]. Although reports on the contribution of TXA2 in cerebral arteries with atherosclerosis are scarce [10], they support a role of TXA2 in endothelial dysfunction and the pathological control of cerebrovascular tone [42].
Despite the demonstration that inhibition of TXA2 synthase [28] and COX activity [24] is neuroprotective in rodent models of stroke and neurodegenerative diseases, and that cardio-protective polyphenols with antioxidant properties reduce the incidence of ischemic stroke [23, 50], the exact link between cerebral eNOS dysfunction, a rise in oxidative stress, and TXA2 has not been defined. We hypothesized that in a pro-oxidant environment associated with aging and atherosclerosis, a dysfunction of cerebral eNOS function reduces the dilatory role of H2O2, increases the TXA2 synthase activity, thus perpetuating the vicious circle of oxidative stress and accelerating endothelial damage. To validate this hypothesis, chronic treatment with the polyphenol catechin [12, 50, 54] and the effects of acute pharmacological TXA2 synthase inhibition were tested in cerebral arteries isolated from middle-aged severely dyslipidemic mice. The results support the concept that with aging and dyslipidemia, eNOS dysfunction mirrors the rise in TXA2 synthase activity, promoting cerebral endothelial dysfunction. Long-term preventive catechin treatment minimizes the decrease in eNOS activity and the rise in TXA2 synthesis.
Materials and methods
The procedures and protocols were performed in accordance with our institutional guidelines and the Guide for the Care and Use of Laboratory Animals of Canada. We used 3- and 12-m/o C57Bl/6 male mice (WT, 29±1 g and 46±3 g, respectively, n=20; Charles River Laboratories, St-Constant, QC, Canada) and 3- and 12-m/o atherosclerotic mice (ATX, 25±1 g and 39±2 g, respectively, n=20). ATX mice are knockout for the low density lipoprotein receptor (LDLR) and express the human apolipoprotein B-100 gene (LDLR−/−; hApoB+/+ [6, 44]). Founders of the colony were generous gifts from Dr. H. Hobbs (University of Texas Southwestern, Dallas, TX, USA). An additional group of ATX mice received catechin (CAT, 98% purity; Sigma-Aldrich, Oakville, ON, Canada) for 9 months (from 3 to 12 m/o, n=20) in the drinking water (ATX+CAT; 30 mg/kg/day), as previously described [12] and to provide an appropriate dietary intake of ≈0.75 mg/day and per mouse of catechin ([18, 26, 34, 56]; see also comment in the supplementary data). All mice were fed with a normal standard diet (Harlan Laboratories, Tecklad 2014S, Montreal, QC, Canada) since ATX mice spontaneously develop atherosclerotic lesions.
Systemic pressure and plasma parameters
At the age of 3 and 12 months, mice were sacrificed by exsanguination under deep anesthesia (isoflurane) after monitoring heart rate and blood pressure using a Millar catheter inserted into the left carotid artery. Isoflurane anesthesia has no impact on vascular reactivity in isolated cerebral arteries (data not shown). Plasma was collected from blood sample and frozen at −80°C. Total cholesterol, LDL, and triglycerides were measured in the hospital biochemical laboratory. Plasma levels of the stable metabolite of TXA2 (11-dehydro-thromboxane B2, TXB2) were quantified using an ELISA according to the protocol provided by the manufacturer (Cayman Chemical, Ann Arbor, MI, USA).
Plaque area quantification
Thoracic aortas were removed from the mice and carefully dissected from surrounding tissues. The vessels were opened, pinned, and a picture was taken. Plaque area quantification was calculated with Photoshop software and expressed in percentage of total aortic area [6].
Quantification of oxidative stress
Concomitant with aortic plaque quantification, the oxidative fluorescent probe dihydroethidium (DHE, 5 μmol/l; Sigma-Aldrich Canada) was used to evaluate global in situ oxidative stress on 14-μm sections of unfixed frozen aorta [12]. In addition, brain sections containing the basilar artery were processed similarly to evaluate cerebrovascular oxidative stress [6]. DNA counterstaining was performed using ToPro-3 (2 μM; Molecular Probe, Burlington, ON, Canada). Computer-based analysis was performed using Image J software and data are expressed as average fluorescence per nucleus.
Experimental design for reactivity studies
The brain was rapidly removed from the cranial cavity and placed in ice-cold physiological salt solution (PSS) of the following composition (mM): NaCl 130, KCl 4.7, CaCl2 1.6, MgSO4 1.17, NaHCO3 14.9, KH2PO4 1.18, EDTA 0.026, and glucose 10. As previously described [6–8], cerebral arteries from the circle of Willis were isolated, cannulated at both ends (average internal diameter of 136±2 μm; n=60), and pressurized at 60 mmHg in no-flow condition in PSS oxygenated by a gas mixture containing 12% O2, 5% CO2, and 83% of N2, generating a pO2 of 150±10 mmHg. An equilibration period of 40 min was allowed before starting the experiments, time during which myogenic tone (MT) develops. The vessels were contracted with phenylephrine (PE; 10 to 30 μmol/l) to obtain a similar pre-constriction level (of 40–50%). Then, a single cumulative concentration–response curve to acetylcholine (ACh, 0.1 nM to 30 μM) was performed on each segment. In some protocols, the arteries were pre-treated for 30 min with (1) Nw-nitro-L-arginine (L-NNA; 10 μmol/l), a NOS inhibitor; (2) indomethacin (INDO; 10 μmol/l), a COX inhibitor; (3) furegrelate (FUR; 10 μmol/l), a potent TXA2 synthase inhibitor [13]; or (4) apocynin (APO; 10 μmol/l), a non-specific NADP(H) oxidase inhibitor. PE, ACh, INDO, FUR, APO, and L-NNA were purchased from Sigma-Aldrich Canada Ltd. The maximal diameter (Dmax) was determined by changing the PSS to a Ca2+-free PSS; MT and dilations were expressed as percentage of the Dmax [6–8]. Half-maximum effective concentration (EC50) of ACh was measured from each individual concentration–response curve using a logistic curve-fitting program (Allfit; Dr André De Léan, Université de Montreal, QC, Canada). Vascular sensitivity (pD2) values, the negative log of the EC50, were obtained.
Fluorescent measurement of eNOS-derived H2O2
We previously demonstrated that in mouse cerebral arteries, ACh-induced dilation is principally mediated by eNOS-derived H2O2 [8] and we reported the method to measure simultaneously the dilation and the production of H2O2, using a fluorescent probe, the 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (DCF-DA, Molecular Probe). In our model, DCF-DA is specific for H2O2 [7, 8]. Fluorescence intensities (λEx 492–495 nm) were measured at 520 nm with an IonOptix Acquire system (IonOptix, Milton, MA, USA). Before each experiment, basal fluorescence intensity was recorded. The increase in fluorescence associated with ACh stimulation was calculated as the difference between the intensity of DCF-DA fluorescence in the presence of ACh and the intensity of DCF-DA fluorescence in the presence of PE.
Western blots
Whole brain vessels were isolated as previously described [22]. Briefly, two brains from each group were pooled, homogenized with a loosely fitting Dounce tissue grinder in ice-cold phosphate buffer saline (PBS, pH 7.4), and centrifuged at 3,500 rpm for 5 min at 4°C. The pellet was then suspended in PBS, gently layered over 15% dextran solution (molecular mass, 43 kDa), and finally centrifuged at 5,000 rpm for 30 min at 4°C. Pellets containing blood vessels were collected over a 50-μm nylon mesh. Proteins were isolated from the vessels following 30-min incubation in a lysis buffer of the following composition (mM): Tris–HCl 50, β-glycerophosphate 20, NaF 20, EDTA 5, EGTA 10, Na3VO4 1, benzamidin 10, dithiothreitol 5, PMSF 0.5, leupeptin 0.02, microcystin LR 1, and Triton X-100 1% (v/v). For Western blot analysis, proteins (50 μg/lane) were subjected to SDS–PAGE, using a discontinuous buffer system (Laemmli), in 10–20% gradient gels and transferred to nitrocellulose. Blots were incubated with the primary antibodies [rabbit polyclonal anti-endothelial NOS (eNOS), 1:200, BD Transduction Laboratories, Franklin Lakes, NJ, USA; rabbit polyclonal anti-inducible NOS (iNOS), 1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA; rabbit polyclonal anti-COX-1, 1:1,000, Cayman Chemical, Arbor, MI, USA; rabbit polyclonal anti-COX-2, 1:1,000, Cayman Chemical; goat polyclonal anti-PGI2 synthase, 1:200, Santa Cruz Biotechnology; and rabbit polyclonal anti-TXA2 synthase, 1:1,000, Abcam, Cambridge, MA, USA]. Equal protein loading was established with GAPDH (mouse monoclonal anti-GAPDH, 1:100,000, Ambion, Streetville, ON, Canada) or with α smooth muscle actin (mouse monoclonal anti-α smooth muscle actin, α SMA, 1:100,000, Sigma-Aldrich Canada).
Statistics
“n” refers to the number of animals used in each protocol. Continuous variables are expressed as mean± standard error (SEM). One-way ANOVA followed by Tukey’s test as post hoc test was used to compare each parameter among the different groups of mice. Unpaired t test was performed to study the effects of L-NNA, FUR, APO, and INDO on ACh-induced dilations, MT, and H2O2-fluorescence increase. The results were considered to be statistically significant when the p value was <0.05.
Results
Heart rate and blood pressure
Heart rate was not affected by age in WT but it tended to decrease (p=0.06) in 12-m/o ATX mice (Table 1). Arterial blood pressure increased with age in both WT and ATX mice. At 3 months, however, ATX mice were already hypertensive compared to WT mice (Table 1). Catechin maintained blood pressure to values measured in 3-m/o WT without affecting heart rate at 12 months (Table 1). Arterial blood pressure was routinely measured and compared in animals under isoflurane anesthesia (Table 1) and also in non-anesthetized animals by the tail-cuff technique (6-m/o ATX mice were hypertensive when compared to WT mice, systolic blood pressure=128±2 versus 146±3 mmHg, WT versus ATX mice, n=8, p<0.05). In 6-m/o conscious mice, HR was not affected by age or dyslipidemia (666±10 versus 682±12 bpm, WT versus ATX mice, n=8, p>0.05). It is apparent that isoflurane anesthesia significantly depressed HR and blood pressure.
Table 1.
Effect of aging, atherosclerosis, and catechin treatment on hemodynamic parameters and plasma levels of cholesterol, triglycerides, and thromboxane B2 (TXB2)
| WT 3 m/o | WT 12 m/o | ATX 3 m/o | ATX 12 m/o | ATX+CAT 12 m/o | |
|---|---|---|---|---|---|
| Hemodynamic parameters | |||||
| Heart rate (bpm) | 377±19 | 375±29 | 371±22 | 304±18 | 300±17 |
| DAP (mmHg) | 59±3 | 73±1* | 73±2* | 81±2**, *** | 64±6**** |
| SAP (mmHg) | 94±2 | 111±3* | 106±3* | 122±3**, *** | 99±6**** |
| MAP (mmHg) | 71±3 | 86±2* | 84±3* | 95±2**, *** | 76±5**** |
| Plasma levels | |||||
| Cholesterol (mmol/l) | 2.1±0.1 | 4.5±0.2* | 16.6±2.0* | 23.3±3.4*** | 23.5±3.2 |
| LDL (mmol/l) | 0.5±0.1 | 2.5±0.2* | 10.3±1.6* | 14.2±2.1*** | 15.1±2.2 |
| HDL (mmol/l) | 1.3±0.1 | 1.8±0.1 | 1.7±0.1 | 2.1±0.2 | 1.9±0.1 |
| Triglycerides (mmol/l) | 0.7±0.1 | 0.4±0.1 | 6.2±0.5* | 9.5±0.9**, *** | 9.7±1.3 |
| TXB2 (pg/ml) | 19±2 | 20±3 | 22±9 | 20±2 | 4±1**** |
Data are mean±SEM of n=8 mice in each group
WT wild-type mice, ATX atherosclerotic mice, bpm beats per minute, DAP diastolic arterial pressure, SAP systolic arterial pressure, MAP mean arterial pressure, LDL low-density lipoprotein, HDL high-density lipoprotein, TXB2 11-dehydro-thromboxane B2, the stable metabolite of TXA2
p<0.05 versus 3-m/o WT,
p<0.05 versus 3-m/o ATX,
p<0.05 versus 12-m/o WT,
p<0.05 versus 12-m/o ATX
Plasma parameters
ATX mice were severely dyslipidemic at both 3 and 12 months of age compared to WT mice (Table 1); as expected, catechin did not modify cholesterol and triglycerides levels (Table 1). Of interest, cholesterol rose with age in WT mice, essentially due to a rise in LDL cholesterol (Table 1). Plasma thromboxane B2 (the stable metabolite of TXA2) levels were unaffected by age or dyslipidemia (Table 1). Following catechin treatment, however, TXB2 levels were reduced by 80% in the plasma of ATX mice (Table 1).
Plaque area and oxidative stress
WT mice did not develop atherosclerotic plaque with age, in contrast to ATX mice (Supplementary Fig. S1). Catechin significantly reduced plaque formation by 35% (Fig. S1). Atherosclerotic lesions were also present in the carotid arteries from middle-aged ATX mice. In contrast, lesions or fatty deposits were observed only occasionally in cerebral arteries from ATX mice (data not shown). Global vascular oxidative stress, assessed by aortic DHE staining, was significantly higher in 12-m/o compared to 3-m/o WT mice (Fig. 1a). DHE staining was already greater at 3 months in ATX than in WT mice but did not increase further with age. Catechin normalized oxidative stress in 12-m/o ATX mice to 3-m/o WT values (Fig. 1a). In cerebral arteries, DHE staining was similar between 3-m/o WT and ATX mice (Fig. 1b), as previously reported [6]. In contrast to aortic-DHE staining, superoxide staining in cerebral arteries decreased in 12-m/o ATX mice and was not affected by catechin (Fig. 1b).
Fig. 1.

Catechin treatment reduces aortic oxidative stress. Effect of 9-month catechin treatment on a aortic and b cerebrovascular superoxide production quantified by DHE staining (n=4, in triplicate). Data are mean±SEM. *p<0.05 versus 3-m/o WT, †p<0.05 versus 3-m/o ATX, ‡p<0.05 versus 12-m/o WT, §p<0.05 versus 12-m/o ATX
Cerebral myogenic tone: basal eNOS dysfunction and up-regulation of TXA2 with atherosclerosis
The physiological response of resistance arteries to a rise in intra-luminal pressure is an increase in MT, i.e., a reduction in diameter. The MT of cerebral arteries induced by a pressure of 60 mmHg was greater in ATX than in WT mice at both ages, while aging alone had no effect (Fig. 2a). L-NNA increased MT in cerebral arteries isolated from WT mice only, suggesting an impaired basal eNOS activity in ATX mice (Fig. 2a). The increased MT in arteries isolated from ATX mice was eliminated by INDO or FUR, while these agents had no effect on MT in WT mice, suggesting a role for TXA2 in the elevated MT in ATX mice. After 9 months of treatment with catechin, MT of vessels isolated from ATX mice was normalized to WT values, sensitive to L-NNA and unaffected by INDO or FUR (Fig. 2a), demonstrating a reversal of the eNOS dysfunction and a reduction in TXA2-dependent regulation of cerebrovascular MT.
Fig. 2.

Cerebral eNOS dysfunction with aging and atherosclerosis. a Effect of eNOS, COX, and TXA2 synthase inhibition with L-NNA, indomethacin (INDO), and furegrelate (FUR), respectively, on cerebral myogenic tone (%) in 3- and 12-m/o WT and ATX mice treated or not with catechin (CAT). Effect of aging, atherosclerosis (ATX), and catechin treatment for 9 months (ATX+CAT) on b maximal endothelial-dependent dilations to ACh (10 μM) and c the associated eNOS–H2O2 production in cerebral arteries isolated from 3- and 12-m/o WT and ATX mice. Data are mean±SEM (n=8). *p< 0.05 versus 3-m/o WT, †p<0.05 versus 12-m/o WT, ‡p<0.05 versus 12-m/o ATX, §p<0.05 versus MT
Decline in cerebral eNOS function and up-regulation of TXA2 with age and atherosclerosis
In cerebral arteries isolated from 3-m/o WT mice, ACh induced endothelium-dependent dilations (pD2 of 7.0±0.2 and Emax of 48±4%; Table 2) and we previously reported that eNOS-derived H2O2 production is a key endothelium-derived relaxing factor in young and healthy mouse cerebral arteries [8]. We therefore investigated eNOS function by simultaneous recording ACh-mediated dilations and H2O2 production (Figs. 2b–c, 3, and 4). With age, efficacy to ACh decreased by ≈50% in arteries from WT mice and dilations were almost abolished in middle-aged ATX mice (Fig. 2b, Table 2). Accordingly, H2O2 production was reduced in 12-m/o WT mice and in ATX mice when compared to 3-m/o WT mice (Fig. 2c). Chronic catechin treatment prevented the accelerated decline in endothelium-dependent dilations with age in ATX mice and improved eNOS–H2O2 production (Fig. 2b–c, Table 2).
Table 2.
Effect of L-NNA, indomethacin, furegrelate, and apocynin, used acutely to inhibit eNOS, COX, TXA2 synthase, and NADP(H) oxidase, respectively, on the sensitivity (pD2) and the efficacy (Emax) to ACh in cerebral arteries
| Control | L-NNA | Indomethacin | Furegrelate | Apocynin | |
|---|---|---|---|---|---|
| 3-m/o WT | |||||
| pD2 | 7.0±0.2 | 7.4±0.2 | 7.8±0.1* | 7.5±0.1 | 6.8±0.2 |
| Emax (%) | 48.0±4.2 | 23.7±2.2* | 25.5±4.1* | 42.8±2.4 | 40.2±1.9 |
| 12-m/o WT | |||||
| pD2 | 7.2±0.3 | ND | 6.9±0.2** | 7.4±0.2 | 7.9±0.2** |
| Emax (%) | 20.8±2.5** | 5.3±1.5*, ** | 38.1±2.2*, ** | 36.7±2.7* | 25.3±1.6** |
| 3-m/o ATX | |||||
| pD2 | 7.5±0.2 | 6.4±0.3*, ** | 7.2±0.1 | 6.7±0.2** | 7.4±0.3 |
| Emax (%) | 26.6±1.3** | 14.1±2.1*, **, **** | 44.6±0.8*, ** | 47.3±1.3*, **** | 26.2±1.6** |
| 12-m/o ATX | |||||
| pD2 | ND | ND | 6.9±0.3** | 7.4±0.2*** | 7.2±0.4 |
| Emax (%) | 6.7±1.9**, *** | 9.8±2.5** | 20.8±2.3*, ***, **** | 26.2±2.1*, **, ***, **** | 27.2±2.7*, ** |
| 12-m/o ATX+CAT | |||||
| pD2 | 7.5±0.3 | ND | 7.2±0.3 | 7.4±0.2 | 7.0±0.1 |
| Emax (%) | 23.9±4.7**, ***** | 4.8±1.0*, **, *** | 27.0±2.5*** | 28.5±2.3**, *** | 33.8±1.7**** |
Data are mean±SEM of n=8 mice in each group
WT wild-type mice, ATX atherosclerotic mice, ATX+CAT ATX mice treated with catechin for 9 months, ND not determined (values too low)
p<0.05 versus control,
p<0.05 versus 3-m/o WT,
p<0.05 versus 3-m/o ATX,
p<0.05 versus 12-m/o WT,
p<0.05 versus 12-m/o ATX
Fig. 3.

Aging is associated with cerebral endothelial dysfunction. Effect of N-nitro-L-arginine (L-NNA; 10 μmol/l), indomethacin (INDO; 10 μmol/l), L-NNA combined with INDO, furegrelate (FUR; 10 μmol/l), and apocynin (APO; 10 μmol/l) on a dose–response curve to ACh, b maximal endothelium-dependent dilations to ACh, and c the associated eNOS-derived H2O2 production in cerebral arteries isolated from 3- and 12-m/o WT mice (n=6). Data are mean±SEM. ap<0.05 versus control, *p<0.05 versus 3-m/o WT
Fig. 4.

Catechin treatment prevents cerebral endothelial dysfunction associated with aging and dyslipidemia. Effect of N-nitro-L-arginine (L-NNA; 10 μmol/l), indomethacin (INDO; 10 μmol/l), L-NNA combined with INDO, furegrelate (FUR; 10 μmol/l), apocynin (APO; 10 μmol/l), and catechin (CAT) treatment on a dose–response curve to ACh, b maximal endothelium-dependent dilations to ACh, and c the associated eNOS-derived H2O2 production in cerebral arteries isolated from 3- and 12-m/o ATX mice treated or not with CAT. Data are mean±SEM (n=6). ap<0.05 versus control, *p<0.05 versus 3-m/o WT, †p<0.05 versus 3-m/o ATX, ‡p<0.05 versus 12-m/o WT, §p<0.05 versus 12-m/o ATX
In 3-m/o WT mice, ACh-induced dilations were equally reduced in the presence of L-NNA or INDO, while FUR or APO had no effect (Fig. 3a–b, Table 2), suggesting a dilatory role for eNOS and prostacyclin. Accordingly, eNOS–H2O2 production was abolished by L-NNA but unaffected by INDO, FUR, or APO (Fig. 3c). In 12-m/o WT mice, the inhibitory effect of L-NNA was lower when compared to 3-m/o WT mice, suggesting the development of eNOS dysfunction with age (Fig. 3). In addition, both INDO and FUR increased ACh dilations and H2O2-associated fluorescence in arteries from 12-m/o WT mice (Fig. 3, Table 2), suggesting an up-regulation of TXA2 with age, counteracting the dilatory effect of eNOS.
Premature eNOS dysfunction in cerebral arteries from 3-m/o ATX mice was illustrated by the lower inhibitory effect of L-NNA on both dilation and H2O2 production when compared to 3-m/o WT mice (Fig. 4, Table 2). This was associated with an early deleterious role of TXA2, as shown by the improvement of ACh dilations in the presence of INDO or FUR (Fig. 4a–b, Table 2). Interestingly, INDO and FUR also improved eNOS–H2O2 production (Fig. 4c), suggesting that TXA2 inhibits eNOS function.
In 12-m/o ATX mice, eNOS function was abolished since ACh dilations and H2O2-associated fluorescence were insensitive to L-NNA (Fig. 4, Table 2). ACh dilations in 12-m/o ATX mice were increased by FUR or INDO, although to a lesser extent than in 3-m/o ATX mice (Fig. 4a–b, Table 2). Neither INDO alone nor its combination with L-NNA altered H2O2 production (Fig. 4c). In addition, the fluorescent signal measured in the combined presence of L-NNA and INDO was associated with a significant vasodilation (Fig. 4b), suggesting the implication of an EDHF [8]. Thus, the rise in H2O2-associated fluorescence induced by ACh was eNOS independent; it was, however, abolished by APO, a non-specific NADP(H) oxidase inhibitor, which had no effect in the other groups (Fig. 4c). This is in accordance with the effects of APO on ACh-induced dilations, which only improved dilations in arteries isolated from 12-m/o ATX mice (Fig. 4, Table 2), suggesting that NADP(H) oxidase may contribute to endothelial dysfunction in these middle-aged atherosclerotic mice.
Following the chronic treatment with catechin, eNOS function was preserved: in cerebral arteries from 12-m/o ATX mice, both ACh dilations and H2O2-associated fluorescence became sensitive to L-NNA and were affected neither by INDO, nor FUR or APO (Fig. 4), suggesting eNOS preservation and TXA2 down-regulation. eNOS dysfunction and TXA2 up-regulation observed with age and atherosclerosis are summarized in Fig. 5.
Fig. 5.

Evolution of the contribution of eNOS–H2O2 as a dilatory factor and TXA2 as an anti-dilatory factor with age and severe dyslipidemia. The contribution of eNOS activity to the dilation induced by ACh (10 μM) is reported as the amplitude of the control dilation (%) minus the amplitude of the L-NNA-sensitive dilation (%); the inhibitory effect of TXA2 on the dilatory response induced by ACh is reported as the amplitude of the control dilation (%) minus the amplitude of the FUR-sensitive dilation (%). Data are mean±SEM (n=6). *p<0.05 versus 3-m/o WT, §p<0.05 versus 12-m/o ATX
Protein expression: decline in PGI2 synthase and rise in TXA2 synthase with atherosclerosis
PGI2 synthase protein expression was high in cerebral vessels isolated from 3-m/o WT mice only (Fig. 6a), supporting the role of PGI2 as an EDRF in these mice only. In ATX mice, the expression of the enzyme was significantly lower at 3 months of age compared to WT and was not further reduced with aging (Fig. 6a). Catechin treatment did not reverse PGI2 synthase protein level, in agreement with the functional data reported in Fig. 5, where INDO had no inhibitory effect on the endothelium-dependent dilations. In contrast, TXA2 synthase increased dramatically in middle-aged ATX mice (Fig. 6b), an effect that was fully prevented by 9 months of treatment with catechin. This is in accordance with the functional data (Fig. 4) where catechin reversed the effects of FUR on eNOS function. Similarly, COX-1 and COX-2 expression rose with age in WT mice, and further in ATX mice (Supplementary Fig. S2); catechin prevented the rise in COX-1 expression and normalized the rise in COX-2 expression to 3-m/o ATX level (Supplementary Fig. S2). Aging, atherosclerosis, or catechin affected the expression of neither eNOS nor iNOS protein in cerebral vessels (Supplementary Fig. S3). Total expression of eNOS may, however, not be truly representative of eNOS function, unlike eNOS dimer/monomer ratio. We indeed previously showed that in mouse cerebral arteries, monomeric eNOS is the preferred state for the production of H2O2, although addition of BH4 did not significantly increase the dimerization of eNOS, while it strongly increased NO production [8].
Fig. 6.

Catechin treatment prevents cerebral up-regulation of TXA2 synthase. Effect of aging, atherosclerosis, and catechin (CAT) treatment on whole brain vessels protein expression of a PGI2 synthase and b TXA2 synthase of wild-type (WT) and atherosclerotic (ATX) mice. Protein expressions are normalized to GAPDH protein expression (PGI2 synthase) or to α smooth muscle actin protein expression (TXA2 synthase) (n=5). Data are mean±SEM. *p<0.05 versus 3-m/o WT, †p<0.05 versus 12-m/o WT, ‡p<0.05 versus 3-m/o ATX, §p<0.05 versus 12-m/o ATX
Discussion
In the present study, we demonstrate for the first time that the age-related cerebral endothelial dysfunction in WT mice occurs prematurely in ATX mice. This endothelial dysfunction is associated with an up-regulation of TXA2, counter-acting eNOS activity and the cerebrovascular dilation. This is accompanied by hypertension and severe peripheral conductance artery atheroma. Our data demonstrate that chronic preventive catechin treatment, a dietary cardio-protective polyphenol with antioxidant properties, reduces plaque size, normalizes blood pressure, and, most importantly, prevents the rise in TXA2 synthase activity maintaining cerebral artery eNOS function.
In mouse cerebral arteries, we reported that the normal physiological function of eNOS is to generate dilatory H2O2—and not NO—in response to ACh [8] or to increases in intra-luminal flow [7]. In cerebral arteries of 12-m/o WT mice, endothelial dysfunction developed and eNOS-associated dilation and H2O2 release were both reduced by ≈50%. PGI2 synthase protein levels were decreased (Fig. 6a), but INDO and FUR restored the normal dilatation. Therefore, in the cerebral arteries of middle-aged WT mice, H2O2 and prostacyclin production decline due to a rise in TXA2 synthase activity, as illustrated in Fig. 5. This contrasts with the report that impaired cerebral dilations in aged rats are insensitive to INDO and thus not related to production of COX-derived constrictor metabolites [30]. In cerebral vessels isolated from 3-m/o ATX, the amplitude of the dilation associated with eNOS-derived H2O2 was also reduced by ≈50%; both INDO and FUR restored the dilation and eNOS activity, while PGI2 synthase expression was very low. Therefore, a similar deleterious profile is measured in young ATX and in middle-aged WT mice.
The early and reversible cerebral endothelial dysfunction observed in 3-m/o ATX mice is in sharp contrast with the almost complete loss of ACh-induced dilations in arteries isolated from 12-m/o ATX mice. In the latter, INDO and FUR only slightly improved dilations to ACh, but the production of H2O2 was insensitive to L-NNA; this demonstrates that combined with severe dyslipidemia, aging leads to irreversible damages. Interestingly, in the presence of ACh, the rise in H2O2-associated fluorescence—but not the dilatory response—was abolished by apocynin, suggesting that in cerebral arteries isolated from 12-m/o ATX mice (unlike in any other groups), the NADP(H) oxidase is sensitive to muscarinic receptor activation. It has been shown that NADP(H) oxidase was partly responsible for flow-mediated dilation in rat brain arteries in vivo [39], a mechanism absent in isolated mouse cerebral arteries [7]. Further studies including the measurement of NAD(P)H oxidase subunits expression are needed to clarify the exact role of this enzyme in the endothelial dysfunction observed in 12-m/o ATX mice.
The rise in TXA2 synthase and the potentially augmented NADP(H) oxidase activity in middle-aged ATX mice must contribute to the change in the redox environment. The increase in cerebrovascular NADP(H) oxidase activity in animal models of hypercholesterolemia was recently reported [20, 32]. Furthermore, it has been shown that aging limits the ability of antioxidant enzymes to up-regulate in response to an acute and short-term pro-atherogenic challenge [3]. In our experimental conditions, however, this imbalance towards pro-oxidation was likely the result of a long-term pro-atherogenic stimulus leading to chronic damage [53]. This was indeed completely reversed by a chronic and long-term preventive treatment with catechin, a scavenger of superoxide anions [41, 50]. Catechin is, however, a non-specific antioxidant that displays large cardio-protective properties [45], including anti-inflammatory properties [35, 52]. To achieve significant beneficial effects against cardiovascular diseases, frequent and habitual consumption of catechins present in green tea is required [35, 38, 54]. In the present study, beneficial functional effects of long-term chronic catechin treatment were associated with reduced aortic plaque formation and aortic oxidative stress (but not cerebral oxidative stress), as we previously reported [12], and normalization of blood pressure in ATX mice. The catechin treatment preserved ACh-induced endothelium-dependent dilations to the level of arteries isolated from age-matched WT mice. This was associated with a lack of TXA2 production or a potential contribution of NADP(H) oxidase during ACh stimulation, leading to the prevention of eNOS dysfunction. Catechins have been shown to modulate NOS activities, i.e., to inhibit iNOS and to stimulate eNOS [35, 50, 51]. There are numerous reports showing that antioxidants in general preserve or improve eNOS function either directly or by increasing NO bioavailability, and reduce the development of atherosclerosis and inflammation in mice models of dyslipidemia [4, 16, 58]. The beneficial role of antioxidants on the cerebral circulation, particularly in the context of atherosclerosis, is, however, less studied [20, 31, 32].
Although previous studies suggested an association between TXA2 and stroke or cerebral ischemia [2, 21, 43, 55], the role of TXA2 in cerebrovascular dysfunction associated with severe dyslipidemia and atherosclerosis is less clear [10]. Previous studies suggested that TXA2 not only competes with but also lowers eNOS activity via an increase in ROS following receptor activation in renal arteries [13], in cultured endothelial cells [57], and in cerebral arteries [36]. In the present study, FUR increased endothelium-dependent dilations and reduced MT, suggesting a major role for TXA2 in cerebral arteries from 12-m/o ATX mice.
In ATX mice, the reduction in TXA2 synthase expression by catechin was accompanied by a strong reduction in plasma TXB2 levels. Accordingly, TXA2 production in platelets was reported to be inhibited by polyphenols [29]. The phenolic antioxidant AGI-1067 also decreased TXB2 plasma levels without affecting PGI2 production in patients with multiple risk factors for cardiovascular diseases [47]. It has also been previously reported that green tea extract was neuroprotective in rats exposed to ischemia/reperfusion by lowering eicosanoids brain concentration, including TXA2 [19].
Our study has some limitations: catechin and apocynin have potential non-specific effects that should also be considered. Catechin, used for 9 months, likely displays effects other than antioxidative properties [35]. Apocynin is a non-specific inhibitor of the NADP(H) oxidase, which may also act as a ROS scavenger [17, 46]. In addition, although used acutely in the present study, furegrelate, a specific TXA2 synthase inhibitor, may affect the level of other prostaglandins besides TXA2. Finally, oxidative stress was assessed only by DHE staining, but the quantification of isoprostane levels could have been a more reliable index.
In conclusion, in our murine model of severe human dyslipidemia, atherosclerosis led to premature endothelial aging via eNOS dysfunction, an increase in TXA2 production and oxidative stress (see Fig. 7 for summary). The impairment of eNOS function by oxidative stress could promote TXA2 production, which would further contribute to the rise in ROS and endothelial dysfunction. Catechin, used in prevention, slowed the progression of atherosclerosis and the endothelial dysfunction via a reduction of the role of the TXA2 pathway and thus the protection of the cerebrovascular eNOS function.
Fig. 7.
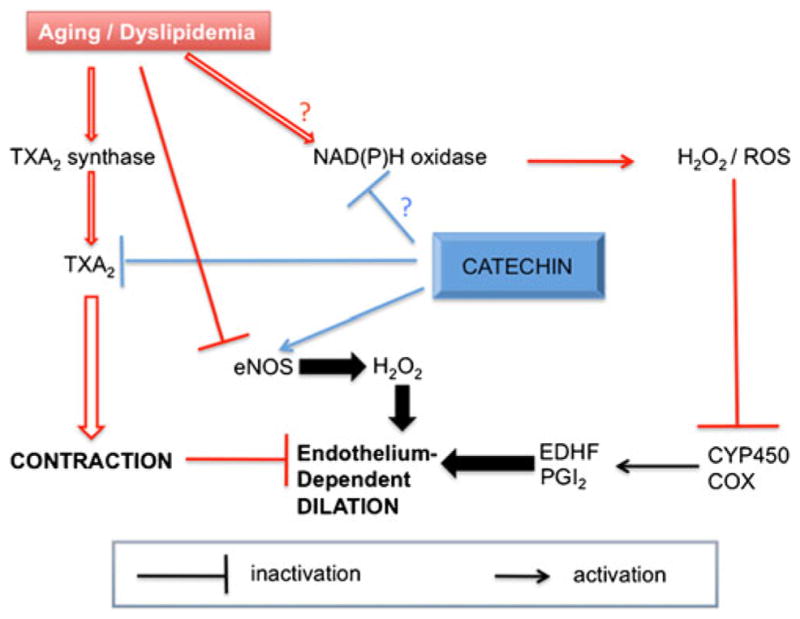
Schematic representation of the effects of age and severe dyslipidemia on the regulation of the cerebrovascular function and the preventive effects of catechin. Both aging and dyslipidemia favor the contribution of contractile TXA2 which compromises endothelium-dependent cerebral dilation mediated through eNOS-derived H2O2. Aging and dyslipidemia are also associated with a rise in ROS, potentially originating from NADP(H) oxidase, which further impairs endothelial-dependent dilation via inhibition of the non-NO and non-PGI2 component of the dilation, likely cytochrome P450-derived EDHF. Chronic treatment with the cardio-protective polyphenol catechin preserves eNOS–H2O2 pathway, limits the implications of TXA2, and potentially that of NADP(H) oxidase
Acknowledgments
This work was supported in part by the Foundation of the Montreal Heart Institute, the Heart and Stroke Foundation of Quebec, and the Canadian Institutes of Health Research (MOP87388). A. Drouin and N. Farhat hold the Frederick Banting and Charles Best Canada Graduate Scholarships—Doctoral Award in association with the Canadian Institutes of Health Research.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00424-011-0973-y) contains supplementary material, which is available to authorized users.
Ethical standards The experiments comply with the current laws of Canada in which they were performed.
Conflict of interest The authors declare that they have no conflict of interest.
Contributor Information
Annick Drouin, Faculty of Medicine, Department of Physiology, Université de Montréal, Montreal, QC, Canada, Montreal Heart Institute Research Center, Montreal, QC, Canada.
Nada Farhat, Faculty of Medicine, Department of Pharmacology, Université de Montréal, Montreal, QC, Canada, Montreal Heart Institute Research Center, Montreal, QC, Canada.
Virginie Bolduc, Faculty of Medicine, Department of Pharmacology, Université de Montréal, Montreal, QC, Canada, Montreal Heart Institute Research Center, Montreal, QC, Canada.
Nathalie Thorin-Trescases, Montreal Heart Institute Research Center, Montreal, QC, Canada.
Marc-Antoine Gillis, Montreal Heart Institute Research Center, Montreal, QC, Canada.
Louis Villeneuve, Montreal Heart Institute Research Center, Montreal, QC, Canada.
Albert Nguyen, Faculty of Medicine, Department of Pharmacology, Université de Montréal, Montreal, QC, Canada, Montreal Heart Institute Research Center, Montreal, QC, Canada.
Eric Thorin, Faculty of Medicine, Department of Surgery, Université de Montréal, Montreal, QC, Canada, Montreal Heart Institute Research Center, Montreal, QC, Canada, Institut de Cardiologie de Montréal, centre de recherche, 5000, rue Bélanger, Montréal, QC H1T 1C8, Canada.
References
- 1.Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4:461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 2.Ansar S, Larsen C, Maddahi A, Edvinsson L. Subarachnoid hemorrhage induces enhanced expression of thromboxane A2 receptors in rat cerebral arteries. Brain Res. 2010;1316:163–172. doi: 10.1016/j.brainres.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 3.Collins AR, Lyon CJ, Xia X, Liu JZ, Tangirala RK, Yin F, Boyadjian R, Bikineyeva A, Pratico D, Harrison DG, Hsueh WA. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ Res. 2009;104:e42–e54. doi: 10.1161/CIRCRESAHA.108.188771. [DOI] [PubMed] [Google Scholar]
- 4.Crawford RS, Kirk EA, Rosenfeld ME, LeBoeuf RC, Chait A. Dietary antioxidants inhibit development of fatty streak lesions in the LDL receptor-deficient mouse. Arterioscler Thromb Vasc Biol. 1998;18:1506–1513. doi: 10.1161/01.atv.18.9.1506. [DOI] [PubMed] [Google Scholar]
- 5.Didion SP, Heistad DD, Faraci FM. Mechanisms that produce nitric oxide-mediated relaxation of cerebral arteries during atherosclerosis. Stroke. 2001;32:761–766. doi: 10.1161/01.str.32.3.761. [DOI] [PubMed] [Google Scholar]
- 6.Drouin A, Bolduc V, Thorin-Trescases N, Belanger E, Fernandes P, Baraghis E, Lesage F, Gillis MA, Villeneuve L, Hamel E, Ferland G, Thorin E. Catechin treatment improves cerebrovascular flow-mediated dilation and learning abilities in atherosclerotic mice. Am J Physiol Heart Circ Physiol. 2011;300:H1032–H1043. doi: 10.1152/ajpheart.00410.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drouin A, Thorin E. Flow-induced dilation is mediated by Akt-dependent activation of endothelial nitric oxide synthase-derived hydrogen peroxide in mouse cerebral arteries. Stroke. 2009;40:1827–1833. doi: 10.1161/STROKEAHA.108.536805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drouin A, Thorin-Trescases N, Hamel E, Falck JR, Thorin E. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007;73:73–81. doi: 10.1016/j.cardiores.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Ellis EF, Nies AS, Oates JA. Cerebral arterial smooth muscle contraction by thromboxane A2. Stroke. 1977;8:480–483. doi: 10.1161/01.str.8.4.480. [DOI] [PubMed] [Google Scholar]
- 10.Faraci FM, Williams JK, Breese KR, Armstrong ML, Heistad DD. Atherosclerosis potentiates constrictor responses of cerebral and ocular blood vessels to thromboxane in monkeys. Stroke. 1989;20:242–247. doi: 10.1161/01.str.20.2.242. [DOI] [PubMed] [Google Scholar]
- 11.Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 12.Gendron ME, Theoret JF, Mamarbachi AM, Drouin A, Nguyen A, Bolduc V, Thorin-Trescases N, Merhi Y, Thorin E. Late chronic catechin antioxidant treatment is deleterious to the endothelial function in aging mice with established atherosclerosis. Am J Physiol Heart Circ Physiol. 2010;298:H2062–H2070. doi: 10.1152/ajpheart.00532.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gendron ME, Thorin E. A change in the redox environment and thromboxane A2 production precede endo-thelial dysfunction in mice. Am J Physiol Heart Circ Physiol. 2007;293:H2508–H2515. doi: 10.1152/ajpheart.00352.2007. [DOI] [PubMed] [Google Scholar]
- 14.Gendron ME, Thorin-Trescases N, Villeneuve L, Thorin E. Aging associated with mild dyslipidemia reveals that COX-2 preserves dilation despite endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2007;292:H451–H458. doi: 10.1152/ajpheart.00551.2006. [DOI] [PubMed] [Google Scholar]
- 15.Hagen AA, White RP, Robertson JT. Synthesis of prostaglandins and thromboxane B2 by cerebral arteries. Stroke. 1979;10:306–309. doi: 10.1161/01.str.10.3.306. [DOI] [PubMed] [Google Scholar]
- 16.Hayek T, Fuhrman B, Vaya J, Rosenblat M, Belinky P, Coleman R, Elis A, Aviram M. Reduced progression of atherosclerosis in apolipoprotein E-deficient mice following consumption of red wine, or its polyphenols quercetin or catechin, is associated with reduced susceptibility of LDL to oxidation and aggregation. Arterioscler Thromb Vasc Biol. 1997;17:2744–2752. doi: 10.1161/01.atv.17.11.2744. [DOI] [PubMed] [Google Scholar]
- 17.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 18.Hishikawa K, Nakaki T, Fujita T. Oral flavonoid supplementation attenuates atherosclerosis development in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:442–446. doi: 10.1161/01.ATV.0000148404.24271.fc. [DOI] [PubMed] [Google Scholar]
- 19.Hong JT, Ryu SR, Kim HJ, Lee JK, Lee SH, Kim DB, Yun YP, Ryu JH, Lee BM, Kim PY. Neuroprotective effect of green tea extract in experimental ischemia–reperfusion brain injury. Brain Res Bull. 2000;53:743–749. doi: 10.1016/s0361-9230(00)00348-8. [DOI] [PubMed] [Google Scholar]
- 20.Kitayama J, Faraci FM, Lentz SR, Heistad DD. Cerebral vascular dysfunction during hypercholesterolemia. Stroke. 2007;38:2136–2141. doi: 10.1161/STROKEAHA.107.481879. [DOI] [PubMed] [Google Scholar]
- 21.Koudstaal PJ, Ciabattoni G, van Gijn J, Nieuwenhuis HK, de Groot PG, Sixma JJ, Patrono C. Increased thromboxane biosynthesis in patients with acute cerebral ischemia. Stroke. 1993;24:219–223. doi: 10.1161/01.str.24.2.219. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Geary GG, Gonzales RJ, Krause DN, Duckles SP. Effect of estrogen on cerebrovascular prostaglandins is amplified in mice with dysfunctional NOS. Am J Physiol Heart Circ Physiol. 2004;287:H588–H594. doi: 10.1152/ajpheart.01176.2003. [DOI] [PubMed] [Google Scholar]
- 23.Liang W, Lee AH, Binns CW, Huang R, Hu D, Zhou Q. Tea consumption and ischemic stroke risk: a case–control study in southern China. Stroke. 2009;40:2480–2485. doi: 10.1161/STROKEAHA.109.548586. [DOI] [PubMed] [Google Scholar]
- 24.Liang X, Wu L, Wang Q, Hand T, Bilak M, McCullough L, Andreasson K. Function of COX-2 and prostaglandins in neurological disease. J Mol Neurosci. 2007;33:94–99. doi: 10.1007/s12031-007-0058-8. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res. 2011;108:566–573. doi: 10.1161/CIRCRESAHA.110.237636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loke WM, Proudfoot JM, Hodgson JM, McKinley AJ, Hime N, Magat M, Stocker R, Croft KD. Specific dietary poly-phenols attenuate atherosclerosis in apolipoprotein E-knockout mice by alleviating inflammation and endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2010;30:749–757. doi: 10.1161/ATVBAHA.109.199687. [DOI] [PubMed] [Google Scholar]
- 27.Matoba T, Shimokawa H. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Pharmacol Sci. 2003;92:1–6. doi: 10.1254/jphs.92.1. [DOI] [PubMed] [Google Scholar]
- 28.Matsuo Y, Izumiyama M, Onodera H, Kurosawa A, Kogure K. Effect of a novel thromboxane A2 receptor antagonist, S-1452, on postischemic brain injury in rats. Stroke. 1993;24:2059–2064. doi: 10.1161/01.str.24.12.2059. discussion 2064–2055. [DOI] [PubMed] [Google Scholar]
- 29.Mattiello T, Trifiro E, Jotti GS, Pulcinelli FM. Effects of pomegranate juice and extract polyphenols on platelet function. J Med Food. 2009;12:334–339. doi: 10.1089/jmf.2007.0640. [DOI] [PubMed] [Google Scholar]
- 30.Mayhan WG, Faraci FM, Baumbach GL, Heistad DD. Effects of aging on responses of cerebral arterioles. Am J Physiol. 1990;258:H1138–H1143. doi: 10.1152/ajpheart.1990.258.4.H1138. [DOI] [PubMed] [Google Scholar]
- 31.Miller AA, Budzyn K, Sobey CG. Vascular dysfunction in cerebrovascular disease: mechanisms and therapeutic intervention. Clin Sci (Lond) 2010;119:1–17. doi: 10.1042/CS20090649. [DOI] [PubMed] [Google Scholar]
- 32.Miller AA, De Silva TM, Judkins CP, Diep H, Drummond GR, Sobey CG. Augmented superoxide production by Nox2-containing NADPH oxidase causes cerebral artery dysfunction during hypercholesterolemia. Stroke. 2010;41:784–789. doi: 10.1161/STROKEAHA.109.575365. [DOI] [PubMed] [Google Scholar]
- 33.Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–e40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- 34.Miura Y, Chiba T, Tomita I, Koizumi H, Miura S, Umegaki K, Hara Y, Ikeda M, Tomita T. Tea catechins prevent the development of atherosclerosis in apoprotein E-deficient mice. J Nutr. 2001;131:27–32. doi: 10.1093/jn/131.1.27. [DOI] [PubMed] [Google Scholar]
- 35.Moore RJ, Jackson KG, Minihane AM. Green tea (Camellia sinensis) catechins and vascular function. Br J Nutr. 2009;102:1790–1802. doi: 10.1017/S0007114509991218. [DOI] [PubMed] [Google Scholar]
- 36.Neppl RL, Lubomirov LT, Momotani K, Pfitzer G, Eto M, Somlyo AV. Thromboxane A2-induced bi-directional regulation of cerebral arterial tone. J Biol Chem. 2009;284:6348–6360. doi: 10.1074/jbc.M807040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogletree ML. Overview of physiological and pathophysiological effects of thromboxane A2. Fed Proc. 1987;46:133–138. [PubMed] [Google Scholar]
- 38.Oyama J, Maeda T, Kouzuma K, Ochiai R, Tokimitsu I, Higuchi Y, Sugano M, Makino N. Green tea catechins improve human forearm endothelial dysfunction and have antiatherosclerotic effects in smokers. Circ J. 2010;74:578–588. doi: 10.1253/circj.cj-09-0692. [DOI] [PubMed] [Google Scholar]
- 39.Paravicini TM, Miller AA, Drummond GR, Sobey CG. Flow-induced cerebral vasodilatation in vivo involves activation of phosphatidylinositol-3 kinase, NADPH-oxidase, and nitric oxide synthase. J Cereb Blood Flow Metab. 2006;26:836–845. doi: 10.1038/sj.jcbfm.9600235. [DOI] [PubMed] [Google Scholar]
- 40.Park SA, Park BL, Park JH, Lee TK, Sung KB, Lee YK, Chang HS, Park CS, Shin HD. Association of polymorphisms in thromboxane A2 receptor and thromboxane A synthase 1 with cerebral infarction in a Korean population. BMB Rep. 2009;42:200–205. doi: 10.5483/bmbrep.2009.42.4.200. [DOI] [PubMed] [Google Scholar]
- 41.Robak J, Gryglewski RJ. Flavonoids are scavengers of superoxide anions. Biochem Pharmacol. 1988;37:837–841. doi: 10.1016/0006-2952(88)90169-4. [DOI] [PubMed] [Google Scholar]
- 42.Rosenblum WI, Bryan D. Evidence that in vivo constriction of cerebral arterioles by local application of tert-butyl hydroperoxide is mediated by release of endogenous thromboxane. Stroke. 1987;18:195–199. doi: 10.1161/01.str.18.1.195. [DOI] [PubMed] [Google Scholar]
- 43.Saloheimo P, Juvela S, Riutta A, Pyhtinen J, Hillbom M. Thromboxane and prostacyclin biosynthesis in patients with acute spontaneous intracerebral hemorrhage. Thromb Res. 2005;115:367–373. doi: 10.1016/j.thromres.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 44.Sanan DA, Newland DL, Tao R, Marcovina S, Wang J, Mooser V, Hammer RE, Hobbs HH. Low density lipoprotein receptor-negative mice expressing human apolipoprotein B-100 develop complex atherosclerotic lesions on a chow diet: no accentuation by apolipoprotein(a) Proc Natl Acad Sci USA. 1998;95:4544–4549. doi: 10.1073/pnas.95.8.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schini-Kerth VB, Auger C, Kim JH, Etienne-Selloum N, Chataigneau T. Nutritional improvement of the endothelial control of vascular tone by polyphenols: role of NO and EDHF. Pflugers Arch. 2010;459:853–862. doi: 10.1007/s00424-010-0806-4. [DOI] [PubMed] [Google Scholar]
- 46.Schluter T, Steinbach AC, Steffen A, Rettig R, Grisk O. Apocynin-induced vasodilation involves Rho kinase inhibition but not NADPH oxidase inhibition. Cardiovasc Res. 2008;80:271–279. doi: 10.1093/cvr/cvn185. [DOI] [PubMed] [Google Scholar]
- 47.Serebruany V, Malinin A, Qiu FH, Xu XC, Kunsch C, Scott R. Selective thromboxane inhibition after vascular protectant AGI-1067: results of assessment of lipoprotein profiles (ALPS) biomarkers in vitro and in vivo substudy. J Thromb Thrombolysis. 2009;27:438–446. doi: 10.1007/s11239-008-0233-y. [DOI] [PubMed] [Google Scholar]
- 48.Shirahase H, Fujiwara M, Usui H, Kurahashi K. A possible role of thromboxane A2 in endothelium in maintaining resting tone and producing contractile response to acetylcholine and arachidonic acid in canine cerebral arteries. Blood Vessels. 1987;24:117–119. doi: 10.1159/000158682. [DOI] [PubMed] [Google Scholar]
- 49.Stewart-Lee AL, Burnstock G. Changes in vasoconstrictor and vasodilator responses of the basilar artery during maturation in the Watanabe heritable hyperlipidemic rabbit differ from those in the New Zealand white rabbit. Arterioscler Thromb. 1991;11:1147–1155. doi: 10.1161/01.atv.11.5.1147. [DOI] [PubMed] [Google Scholar]
- 50.Sutherland BA, Rahman RM, Appleton I. Mechanisms of action of green tea catechins, with a focus on ischemia-induced neurodegeneration. J Nutr Biochem. 2006;17:291–306. doi: 10.1016/j.jnutbio.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 51.Sutherland BA, Shaw OM, Clarkson AN, Jackson DN, Sammut IA, Appleton I. Neuroprotective effects of (−)-epigallocatechin gallate following hypoxia–ischemia-induced brain damage: novel mechanisms of action. FASEB J. 2005;19:258–260. doi: 10.1096/fj.04-2806fje. [DOI] [PubMed] [Google Scholar]
- 52.Suzuki J, Isobe M, Morishita R, Nagai R. Tea polyphenols regulate key mediators on inflammatory cardiovascular diseases. Mediat Inflamm. 2009;2009:494928. doi: 10.1155/2009/494928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thorin E, Thorin-Trescases N. Vascular endothelial ageing, heartbeat after heartbeat. Cardiovasc Res. 2009;84:24–32. doi: 10.1093/cvr/cvp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Unno K, Takabayashi F, Kishido T, Oku N. Suppressive effect of green tea catechins on morphologic and functional regression of the brain in aged mice with accelerated senescence (SAMP10) Exp Gerontol. 2004;39:1027–1034. doi: 10.1016/j.exger.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 55.van Kooten F, Ciabattoni G, Koudstaal PJ, Grobbee DE, Kluft C, Patrono C. Increased thromboxane biosynthesis is associated with poststroke dementia. Stroke. 1999;30:1542–1547. doi: 10.1161/01.str.30.8.1542. [DOI] [PubMed] [Google Scholar]
- 56.Waddington E, Puddey IB, Croft KD. Red wine polyphenolic compounds inhibit atherosclerosis in apolipoprotein E-deficient mice independently of effects on lipid peroxidation. Am J Clin Nutr. 2004;79:54–61. doi: 10.1093/ajcn/79.1.54. [DOI] [PubMed] [Google Scholar]
- 57.Zhang M, Song P, Xu J, Zou MH. Activation of NAD (P)H oxidases by thromboxane A2 receptor uncouples endothelial nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2011;31:125–132. doi: 10.1161/ATVBAHA.110.207712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang WJ, Bird KE, McMillen TS, LeBoeuf RC, Hagen TM, Frei B. Dietary alpha-lipoic acid supplementation inhibits atherosclerotic lesion development in apolipoprotein E-deficient and apolipoprotein E/low-density lipoprotein receptor-deficient mice. Circulation. 2008;117:421–428. doi: 10.1161/CIRCULATIONAHA.107.725275. [DOI] [PubMed] [Google Scholar]