

Abstract
Insufficient neovascularization, characterized by poor endothelial cell (EC) growth, contributes to the pathogenesis of ischemic heart disease and limits cardiac tissue preservation and regeneration. The E2F family of transcription factors are critical regulators of the genes responsible for cell-cycle progression and growth; however, the specific roles of individual E2Fs in ECs are not well understood. Here we investigated the roles of E2F2 and E2F3 in EC growth, angiogenesis, and their functional impact on myocardial infarction (MI). An endothelial-specific E2F3-deficient mouse strain VE-Cre; E2F3fl/fl was generated, and MI was surgically induced in VE-Cre; E2F3fl/fl and E2F2-null (E2F2 KO) mice and their wild-type (WT) littermates, VE-Cre; E2F3+/+ and E2F2 WT, respectively. The cardiac function, infarct size, and vascular density were significantly better in E2F2 KO mice and significantly worse in VE-Cre; E2F3fl/fl mice than in their WT littermates. The loss of E2F2 expression was associated with an increase in the proliferation of ECs both in vivo and in vitro, while the loss of E2F3 expression led to declines in EC proliferation. Thus, E2F3 promotes while E2F2 suppresses ischemic cardiac repair through corresponding changes in EC proliferation; and differential targeting of specific E2F members may provide a novel strategy for therapeutic angiogenesis of ischemic heart disease.
Introduction
Ischemic heart disease (IHD) represents one of the largest epidemics facing the aging population. Insufficient neovascularization, characterized by poor vessel growth and survival, contributes to the pathogenesis of IHD and limits cardiac tissue preservation and regeneration; thus enhancement of cardiac neovascularization is a therapeutic goal.
Following tissue ischemia, vascular injury recruits endothelial cells (ECs) required for neovascularization to restore blood perfusion [1]–[5]. Neovascularization is a complex sequence of events involving coordinated vascular cell proliferation, migration and tube formation [6]–[10]. Accumulating evidence indicates that neovascularization is critically dependent on the appropriate regulation of EC cell cycle [11], [12]; a deficient endothelial proliferative response to ischemia can result in tissues necrosis [13]. Additionally, ECs can contribute to cardiac repair by mediating favorable cell:cell interactions and secreting paracrine factors that protect the function and survival of cardiomyocytes [14].
The E2F family of transcription factors, with eight members identified, play a central role in regulating the expression of genes responsible for cell-cycle control and provide an ideal target for therapeutic modulation of vascular growth [15]–[20]. However, the PREVENT trial, targeting E2F activity in the vascular smooth muscle cells in the autologous vein grafts of CABG surgery to prevent graft overgrowth and failure, generated negative results presumably due to the non-specific inhibition of both activating and repressive E2F species [21], [22]; therefore, it is imperative to elucidate the specific function of individual E2F members in the vascular biology [20]. The E2F1, 2 and 3, in particular, are considered “transactivators” that activate transcription of target genes for DNA replication and G1/S transition, thus cell proliferation [23]–[26]. However, we have recently found that E2F1 inhibits ischemic angiogenesis by suppressing the expression of angiogenic factors, VEGF and PlGF [27]. The functions of E2F2 and E2F3 in ECs during ischemic disease are largely unknown.
In this study, we sought to investigate the roles of E2F2 and E2F3 in EC proliferation and their functional impact on myocardial infarction (MI). We found that E2F3 is essential for EC growth, neovascularization, and preservation of cardiac function, while E2F2 plays a contrasting role. Thus, individual E2F factors, rather than E2F family as a whole, may provide more specific molecular targets for therapeutic neovascularization of IHD.
Materials and Methods
Mice
The E2F3fl/fl and E2F2+/− mice were obtained from Dr. Gustavo Leone’s lab (Ohio State University) [23]. VE-cadherin-Cre (VE-Cre) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). VE-Cre; E2F3fl/fl mice and littermate VE-Cre; E2F3+/+ mice were generated by crossing VE-Cre; E2F3fl/+ male and E2F3fl/+ female parents. E2F2−/− (E2F2 KO) and littermate E2F2+/+ (E2F2 WT) mice were generated by crossing E2F2+/− parents as previously described [28]. All these mice were on C57BL/6 background. Mouse genotypes were determined via polymerase chain reaction (PCR) with tail DNA. All the animal work presented in this report was approved by the Institutional Animal Care and Use Committee of Northwestern University and performed in the barrier acilities of the Center for Comparative Medicine of the university.
Surgical MI Model and Echocardiographic Assessments of Left Ventricular (LV) Function
MI was induced in 8 week-old male VE-Cre; E2F3fl/fl, VE-Cre; E2F3+/+, E2F2 KO, and E2F2 WT mice by permanent ligation in the middle of the left anterior descending (LAD) coronary artery as described previously [29], [30]. Mice were anesthetized by inhaling Isoflurane™ delivered at 2–4% throughout the surgical procedure, and were injected subcutaneously with Metacam (1 mg/kg) as analgesic immediately after the surgery and then daily for the next 2 to 3 days. Trans-thoracic 2-dimensional echocardiographic measurements were performed before MI (baseline) and at 7, 14 and 28 days post-MI by using a commercially available high resolution system (VEVO 770™, VisualSonics Inc., Toronto, Canada) equipped with a 30-MHz transducer. M-mode tracings were used to measure LV wall thickness, end-systolic diameter (LVESD), and end diastolic diameter (LVEDD). Systolic and diastolic LV areas were determined by M-mode in long-axis configuration and fractional shortening (FS) was measured at the mid-ventricular level. The LV chamber volumes in diastole and systole were derived from their respective measured 2D areas using a LV volume algorithm within the Vevo770 echo software. Cardiac ejection fraction (EF) was determined offline by the equation: EF = (Diastolic Volume - Systolic Volume/Diastolic Volume) x100.
Histological Assessments of Infarct Size, Vascular Density, and in vivo EC Proliferation
Fourteen and 28 days after surgically induced MI, the vasculature was labeled by injecting 50 µL BS Lectin I (Vector laboratories, Inc., Burlingame, CA) into the tail vein, and mice were euthanized 10 min later by CO2 inhalation (primary method) and cervical dislocation (secondary method). A portion of animals also received Bromodeoxyuridine (BrdU) (30 mg/kg IP) for 48 h (Q12H×4) before euthanasia, which permits identification of proliferating cells. Cardiac tissues were fixed in 4% paraformaldehyde for 4 h, incubated overnight in 30% sucrose, embedded in OCT compound (Sakura Finetek U.S.A., Inc., Torrance, CA), snap-frozen in liquid nitrogen, and cut into 5 µm sections. Serial cryosectioning was performed starting at 1 mm below the suture (used to ligate the LAD) moving toward the apex, with three consecutive sections per 1 mm to allow for quantitative pathohistological analysis at each level. To evaluate infarct area, the Masson Trichrome elastic tissue staining was performed as described previously [30], [31]. Infarct size was reported as the ratio of the length of fibrotic area to the length of the LV inner circumference. Immunohistochemical staining was performed with fluorescent anti- BS Lectin 1 (Vector Laboratories, Inc.), anti-CD31 (Santa Cruz), and anti-BrdU (Abcam, Cambridge, MA, USA) antibodies to evaluate vascular density and proliferating cells as described previously [27], [29], [30]; 3 sections per ischemic heart and 6 fields per section were examined.
Isolation of Mouse Primary Cardiac ECs and Adenoviral Vector Transduction
Primary ECs were isolated from mouse heart tissues as described previously [32]. Briefly, tissues were minced and digested with collagenase and dispase, then the mixture was passed through a 100-µm cell strainer to obtain single cell suspensions. Cell debris were removed via density centrifugation with Histopaque-1.083 (Sigma), then the ECs were immunostained with CD31-PE, FACS sorted to ≥95% purity, and cultured in EBM-2 medium (Lonza, Walkersville, MD). The cells were used before passage 5. The ECs were infected by adenovirus-Cre (Vector BioLabs, Philadelphia, PA) by following manufacturer’s instructions.
EdU Incorporation Assay
Proliferation of primary ECs was measured with a commercially available Click-iT™ EdU (5-ethynyl-2′-deoxyuridine) Flow Cytometry Assay kit (Invitrogen) by following the manufacturer’s instructions. Prior to the addition of EdU, subconfluent ECs were synchronized by incubation in EBM-2 medium with 0.1% FBS for 24 h and stimulated to proliferate by addition of EBM-2 supplemented with 5% FBS.
Statistical Analysis
All values are expressed as mean ± SEM. Comparison between two means was performed with an unpaired Student’s t test, whereas ANOVA with Fisher’s protected least significant differences and Bonferroni–Dunn post hoc analysis were used for comparisons of more than two means.
Results
Functional Recovery of the Infarcted Heart is Enhanced by the Loss of E2F2 Expression and Impaired by the Loss of Endothelial E2F3 Expression
We induced MI by surgical ligation of LAD coronary artery in VE-Cre; E2F3fl/fl and E2F2 KO mice and their WT littermates, VE-Cre; E2F3+/+ and E2F2 WT mice, respectively. E2F2 KO mice exhibited a greater EF and FS and a smaller LV systolic and diastolic volume as compared with E2F2 WT mice (Figure 1). The significantly better cardiac function in E2F2 KO mice was observed as early as day 7 and persisted till day 28 post-MI. In contrast, VE-Cre; E2F3fl/fl exhibited a worsened heart function as compared with VE-Cre; E2F3+/+ mice at days 14 and 28 post-MI (Figure 1). These results suggest that E2F3 improved while E2F2 impairs cardiac function in response to ischemic injury.
Figure 1. Functional recovery of the infarcted heart is enhanced by the loss of E2F2 expression and impaired by the loss of endothelial E2F3 expression.

MI was surgically induced in VE-Cre; E2F3fl/fl and E2F2 KO mice and their WT littermates, VE-Cre; E2F3+/+ and E2F2 WT, respectively, and the heart function was assessed with echocardiography at the indicated time points for (A) LV ejection fraction, (B) fractional shortening, (C) end-systolic and (D) end-diastolic volumes. n = 12 mice per group. *P<0.05 vs. VE-Cre; E2F3+/+, #P<0.05 vs. E2F2 WT.
Infarct Size and Vessel Density are Improved by the Loss of E2F2 Expression and Worsened by the Loss of Endothelial E2F3 Expression
At day 28 post-MI, infarct size was significantly smaller in E2F2 KO and significantly larger in VE-Cre; E2F3fl/fl mice, than in their littermates with WT levels of E2F2 and endothelial E2F3 expression (Figure 2). The loss of E2F2 expression in E2F2 KO mice also led to improvements in vessel density, while the endothelial deletion of E2F3 in VE-Cre; E2F3fl/fl mice was associated with a decline in vessel density (Figure 3). Thus, E2F3 enhance while E2F2 suppresses cardiac neovascularization.
Figure 2. The infarct size is smaller in E2F2 KO mice and larger in endothelial specific E2F3 KO mice than in their WT littermates.

Masson Trichrome staining was performed in heart samples 28 days after MI surgery. (A) Representative microphotographs and (B) Quantification of the infarct size. n = 12 mice per group; *P<0.05 versus VE-Cre; E2F3+/+, #P<0.05 versus E2F2 WT; Scale bar = 100 µm.
Figure 3. The vascular density at infarct border zone is greater in E2F2 KO mice and lower in endothelial E2F3 KO mice than in their WT littermates.
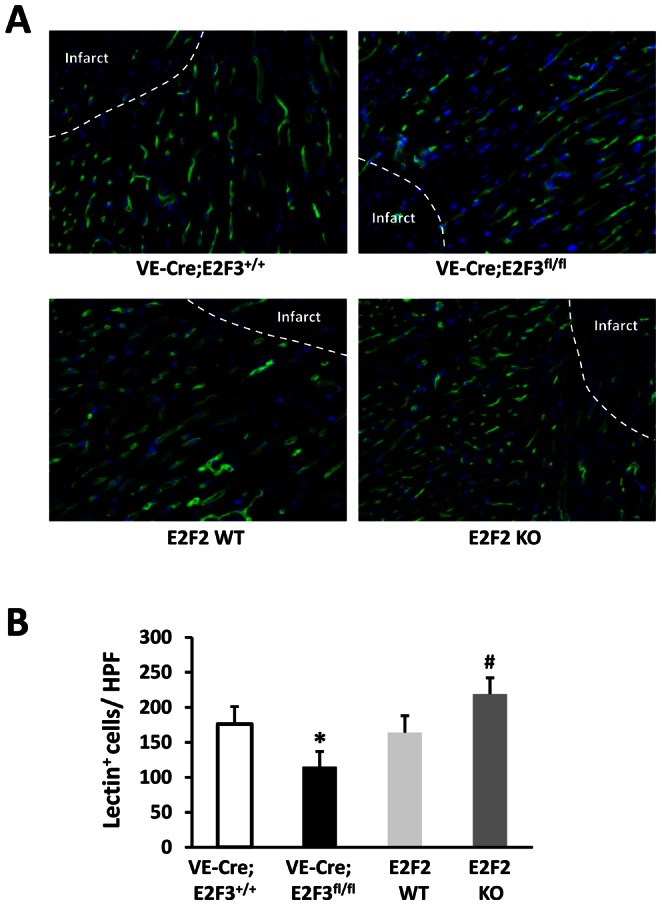
Blood vessels were stained with BS lectin 1 (green), and nuclei were counterstained with DAPI (blue). (A) Representative immunofluorescence images. (B) Quantification of vascular density at the infarct border zone. n = 12 mice per group; *P<0.05 vs. VE-Cre; E2F3+/+, #P<0.05 vs. E2F2 WT; HPF, high power field.
EC Proliferation is Increased by Declines in E2F2 Expression and Reduced by Declines in E2F3 Expression
Because E2F2 and E2F3 are cell cycle regulators, we assessed EC proliferation in the ischemic tissue (i.e., the infarct border zone) at day 14 post-MI with CD31 and BrdU double staining. The frequency of proliferating ECs was significantly higher in E2F2 KO mice and significant lower in VE-Cre; E2F3fl/fl mice, than in their WT littermates (Figure 4A–B).
Figure 4. Proliferation is enhanced in E2F2 KO ECs and impaired in E2F3-deleted ECs.

(A–B) Immunofluorescent double staining was performed in the ischemic heart sections for CD31 (green) and BrdU (red) to identify ECs (green), proliferating cells (red), and proliferating ECs (yellow). (A) Representative immunofluorescence images and (B) quantification of proliferating ECs in the infarct border zone. n = 12 mice per group; *P<0.05 vs. VE-Cre; E2F3+/+, #P<0.05 vs. E2F2 WT; HPF, high power field. (C) Primary ECs were isolated from the hearts of E2F2 KO, E2F2 WT, and E2F3fl/fl mice, and the E2F3fl/fl cells were subsequently transduced with Adenovirus-Cre/GFP or Adenovirus-GFP. EdU incorporation based flow cytometry analyses were performed to assess DNA synthesis. Shown is representative of 3 independent experiments.
To determine whether the contrasting roles of E2F2 and E2F3 in EC proliferation in vivo are cell autonomous, we isolated primary EC from the hearts of E2F2 KO, E2F2 WT, and E2F3fl/fl mice and the E2F3fl/fl ECs were subsequently transduced with a vector coding for GFP alone or with a vector coding for both GFP and Cre expression to knockout the expression of E2F3. Cells were starved to quiescence by serum deprivation and then re-stimulated by the addition of serum. The EdU incorporation assays showed that the proliferating (i.e., EdU+) ECs were more frequent in the E2F2 KO population and much less frequent in the E2F3 KO population, than in cells with cells with WT levels of E2F2 and E2F3 expression (Figure 4C). These results indicate that E2F2 suppresses and E2F3 enhances EC proliferation.
Discussion
E2F1, 2, and 3 are classified as a subgroup of “activating” E2Fs that promote cell proliferation [33]–[37]. However, here we found that EC growth, neovascularization, and cardiac function post-MI are improved by the loss of E2F2 expression and impaired by the loss of endothelial E2F3 expression, which suggest that E2F2 and E2F3 play contrasting roles. Data from this current study and the results we have recently reported on E2F1 [27], collectively, suggest diverse but specific roles of these E2F species in the regulation of vascular growth.
Our data clearly show that E2F3 is essential for EC proliferation and ischemic angiogenesis, which cannot be compensated by other “activating” E2Fs (i.e., E2F1 and 2). Although consistent with reports in the literature documenting a critical role of E2F3 for cell cycle progression, this role appears to be greater in ECs than in other cell types [25], [38], [39]. Whether this role of E2F3 is specific to ECs or E2F3 plays a similar role in other vascular cells such as VSMCs remains to be investigated.
The finding that E2F2 suppresses EC proliferation is somewhat unexpected. It is in contrast to the observations made in several other cell types, in which overexpression of E2F2 activates cell cycle progression [40]. While the molecular mechanism underlying the enhanced growth of E2F2 KO ECs is yet to be determined, other lab did report that E2F2 KO mice display hyper-proliferation of immune cells [41]. Given the strong transactivity of E2F3 in ECs, it is tempting to speculate that in the absence of E2F2 (i.e., in E2F2 KO cells), the regulatory DNA elements normally bound by E2F2 may be occupied by E2F3 instead, thereby exerting a stronger transactivity for the expression of these genes. This hypothesis, however, remains to be tested in our future study.
In summary, our study revealed that E2F2 and E2F3, two members in the “activating” E2F subfamily exert contrasting roles in the regulation of EC growth and neovascularization and in the preservation of cardiac tissues from ischemic injury. Thus, individual E2Fs, rather than E2F family as a whole, may provide more specific targets for therapeutic angiogenesis of IHD.
Acknowledgments
We thank Dr. Gustavo Leone (The Ohio State University) for providing E2F2 knockout and E2F3 floxed mice.
Funding Statement
This work was supported by the National Institute of Health Grants (HL093439 and HL113541 to G.Q.) and American Heart Association Grants (0430135N to G.Q. and 10POST4360009 to J.Z.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Adams RH, Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8: 464–478. [DOI] [PubMed] [Google Scholar]
- 2. Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438: 932–936. [DOI] [PubMed] [Google Scholar]
- 3. Simons M (2005) Angiogenesis: where do we stand now? Circulation 111: 1556–1566. [DOI] [PubMed] [Google Scholar]
- 4. Robich MP, Chu LM, Oyamada S, Sodha NR, Sellke FW (2011) Myocardial therapeutic angiogenesis: a review of the state of development and future obstacles. Expert Review of Cardiovascular Therapy 9: 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Potente M, Gerhardt H, Carmeliet P (2011) Basic and Therapeutic Aspects of Angiogenesis. Cell 146: 873–887. [DOI] [PubMed] [Google Scholar]
- 6. Chung AS, Ferrara N (2011) Developmental and Pathological Angiogenesis. Annual Review of Cell and Developmental Biology 27: 563–584. [DOI] [PubMed] [Google Scholar]
- 7. Fraisl P, Mazzone M, Schmidt T, Carmeliet P (2009) Regulation of Angiogenesis by Oxygen and Metabolism. Developmental Cell 16: 167–179. [DOI] [PubMed] [Google Scholar]
- 8. Weis SM, Cheresh DA (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 17: 1359–1370. [DOI] [PubMed] [Google Scholar]
- 9. Herbert SP, Stainier DYR (2011) Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol 12: 551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lähteenvuo J, Rosenzweig A (2012) Effects of Aging on Angiogenesis. Circulation Research 110: 1252–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Semenza GL (2011) Hypoxia. Cross talk between oxygen sensing and the cell cycle machinery. American Journal of Physiology - Cell Physiology 301: C550–C552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geudens I, Gerhardt H (2011) Coordinating cell behaviour during blood vessel formation. Development 138: 4569–4583. [DOI] [PubMed] [Google Scholar]
- 13. Toutain CE, Brouchet L, Raymond-Letron I, Vicendo P, Bergès H, et al. (2009) Prevention of Skin Flap Necrosis by Estradiol Involves Reperfusion of a Protected Vascular Network. Circulation Research 104: 245–254. [DOI] [PubMed] [Google Scholar]
- 14. Robich MP, Chu LM, Oyamada S, Sodha NR, Sellke FW (2011) Myocardial therapeutic angiogenesis: a review of the state of development and future obstacles. Expert Rev Cardiovasc Ther 9: 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Helin K (1998) Regulation of cell proliferation by the E2F transcription factors. Curr Opin Genet Dev 8: 28–35. [DOI] [PubMed] [Google Scholar]
- 16.Dyson N (1998) The regulation of E2F by pRB-family proteins. Genes Dev: 2245–2262. [DOI] [PubMed]
- 17. Wenzel PL, Chong J-L, Sáenz-Robles MT, Ferrey A, Hagan JP, et al. (2011) Cell proliferation in the absence of E2F1-3. Developmental Biology 351: 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giangrande PH, Zhang J, Tanner A, Eckhart AD, Rempel RE, et al. (2007) Distinct roles of E2F proteins in vascular smooth muscle cell proliferation and intimal hyperplasia. Proc Natl Acad Sci U S A 104: 12988–12993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spyridopoulos I, Principe N, Krasinski KL, Xu S, Kearney M, et al. (1998) Restoration of E2F expression rescues vascular endothelial cells from tumor necrosis factor-alpha-induced apoptosis. Circulation 98: 2883–2890. [DOI] [PubMed] [Google Scholar]
- 20.Qin G, Losordo DW (2008) E2F transcription factors in cardiovascular physiology. In: Yoshida K, editor. Control of Cellular Physiology by E2F Transciption Factors. Kerala: Research Signpost. 341–366.
- 21. PREVENT IV Investigators (2005) Efficacy and Safety of Edifoligide, an E2F Transcription Factor Decoy, for Prevention of Vein Graft Failure Following Coronary Artery Bypass Graft Surgery: PREVENT IV: A Randomized Controlled Trial. JAMA 294: 2446–2454. [DOI] [PubMed] [Google Scholar]
- 22. Alexander JH, Hafley G, Harrington RA, Peterson ED, Ferguson TB Jr, et al. (2005) Efficacy and safety of edifoligide, an E2F transcription factor decoy, for prevention of vein graft failure following coronary artery bypass graft surgery: PREVENT IV: a randomized controlled trial. JAMA 294: 2446–2454. [DOI] [PubMed] [Google Scholar]
- 23. Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, et al. (2001) The E2F1–3 transcription factors are essential for cellular proliferation. Nature 414: 457–462. [DOI] [PubMed] [Google Scholar]
- 24. Nevins J (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ 9: 585–593. [PubMed] [Google Scholar]
- 25. Saavedra HI, Wu L, de Bruin A, Timmers C, Rosol TJ, et al. (2002) Specificity of E2F1, E2F2, and E2F3 in Mediating Phenotypes Induced by Loss of Rb. Cell Growth Differ 13: 215–225. [PubMed] [Google Scholar]
- 26. Chen H-Z, Tsai S-Y, Leone G (2009) Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 9: 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qin G, Kishore R, Dolan CM, Silver M, Wecker A, et al. (2006) Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc Natl Acad Sci U S A 103: 11015–11020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou J, Zhu Y, Cheng M, Dinesh D, Thorne T, et al. (2009) Regulation of vascular contractility and blood pressure by the E2F2 transcription factor. Circulation 120: 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tang YL, Zhu W, Cheng M, Chen L, Zhang J, et al. (2009) Hypoxic Preconditioning Enhances the Benefit of Cardiac Progenitor Cell Therapy for Treatment of Myocardial Infarction by Inducing CXCR4 Expression. Circ Res 104: 1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qin G, Ii M, Silver M, Wecker A, Bord E, et al. (2006) Functional disruption of alpha4 integrin mobilizes bone marrow-derived endothelial progenitors and augments ischemic neovascularization. J Exp Med 203: 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tang YL, Zhu W, Cheng M, Chen L, Zhang J, et al. (2009) Hypoxic preconditioning enhances the benefit of cardiac progenitor cell therapy for treatment of myocardial infarction by inducing CXCR4 expression. Circ Res 104: 1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou J, Zhu Y, Cheng M, Dinesh D, Thorne T, et al. (2009) Regulation of Vascular Contractility and Blood Pressure by the E2F2 Transcription Factor. Circulation 120: 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DeGregori J, Johnson D (2006) Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis Current Molecular Medicine. 6: 739–748. [DOI] [PubMed] [Google Scholar]
- 34. Trimarchi JM, Lees JA (2002) Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 3: 11–20. [DOI] [PubMed] [Google Scholar]
- 35. Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, et al. (2001) The E2F1–3 transcription factors are essential for cellular proliferation. Nature 414: 457–462. [DOI] [PubMed] [Google Scholar]
- 36. Helin K (1998) Regulation of cell proliferation by the E2F transcription factors. Current Opinion in Genetics & Development 8: 28–35. [DOI] [PubMed] [Google Scholar]
- 37. Tao Y, Kassatly RF, Cress WD, Horowitz JM (1997) Subunit composition determines E2F DNA-binding site specificity. Molecular and Cellular Biology 17: 6994–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Field SJ, Tsai F-Y, Kuo F, Zubiaga AM, Kaelin Jr WG, et al. (1996) E2F-1 Functions in Mice to Promote Apoptosis and Suppress Proliferation. Cell 85: 549–561. [DOI] [PubMed] [Google Scholar]
- 39. DeGregori J (2002) The genetics of the E2F family of transcription factors: shared functions and unique roles. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1602: 131–150. [DOI] [PubMed] [Google Scholar]
- 40. Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, et al. (2001) E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev 15: 267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Murga M, Fernandez-Capetillo O, Field SJ, Moreno B, Borlado LR, et al. (2001) Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity 15: 959–970. [DOI] [PubMed] [Google Scholar]