

SUMMARY
We developed a new screening method for orphan receptor ligands, in which cell-surface proteins are expressed in Drosophila embryos from GAL4-dependent insertion lines and ligand candidates identified by the presence of ectopic staining with receptor fusion proteins. Stranded at second (Sas) binds to the receptor tyrosine phosphatase Ptp10D in embryos and in vitro. Sas and Ptp10D can interact in trans when expressed in cultured cells. Interactions between Sas and Ptp10D on longitudinal axons are required to prevent them from abnormally crossing the midline. Sas is expressed on both neurons and glia, while Ptp10D is restricted to CNS axons. We conducted epistasis experiments by overexpressing Sas in glia and examining how the resulting phenotypes are changed by removal of Ptp10D from neurons. We find that neuronal Ptp10D restrains signaling by overexpressed glial Sas, which would otherwise produce strong glial and axonal phenotypes.
INTRODUCTION
Receptor tyrosine phosphatases (RPTPs) are single-span transmembrane proteins that reverse reactions catalyzed by tyrosine kinases (TKs). A major problem in the phosphotyrosine signaling field is to identify and characterize ligands and coreceptors that interact with the extracellular (XC) domains of RPTPs and regulate their functions in vivo.
The IIb, IIa, and III subtypes, comprising 11 of the 19 human RPTPs, have XC regions containing immunoglobulin-like (Ig) domains and fibronectin type III (FN3) repeats, which are found in cell adhesion molecules (CAMs) (reviewed by (Tonks, 2006)). Type IIb RPTPs are homophilic CAMs that regulate cadherin-mediated adhesion (Aricescu et al., 2007). Type IIa (Lar-like) RPTPs bind to heparan sulfate (HSPG) and chondroitin sulfate (CSPG) proteoglycans (Aricescu et al., 2002; Coles et al., 2011; Fox and Zinn, 2005; Johnson et al., 2006). The HSPGs Syndecan (Sdc) and Dallylike (Dlp) are in vivo ligands and coreceptors for Drosophila Lar (Fox and Zinn, 2005; Johnson et al., 2006). The Type III RPTP PTPRB interacts with VE-cadherin in cis (Nawroth et al., 2002), and PTPRJ can bind to a fragment of the Syndecan-2 protein (Whiteford et al., 2011).
Dimeric placental alkaline phosphatase (AP) fusion proteins have been used to visualize ligand binding in situ for many receptors (Flanagan and Cheng, 2000). RPTP-AP probes derived from the XC domains of four Drosophila RPTPs (Ptp10D, Ptp69D, Ptp99A, and Lar), all stain CNS axons in live-dissected late stage 16 embryos. Lar-AP also stains muscle attachment sites. This provided the basis for a screen of deletion (Df) mutations in which we screened the Bloomington Stock Center Df ‘kit’, found a Df for which homozygotes lacked muscle attachment site staining with Lar-AP, and identified Sdc as the Lar ligand/coreceptor encoded within the deleted region (Fox and Zinn, 2005).
This Df screen had two problems. First, homozygotes for many Dfs in the kit had severe CNS defects that preclude screening. To alleviate this, we replaced Dfs for which development failed with smaller Dfs that allow better development, creating a ‘phenotype Df kit’ with which >80% of the genome can be screened for ligands (http://fly.bio.indiana.edu/Browse/df/zinn.php). This kit can also be used for systematic genome screening for any embryonic phenotype, and such a screen requires analysis of less than 400 lines (Wright et al., 2010).
Second, if multiple RPTP ligands are expressed in overlapping patterns, deletion of a gene encoding one ligand might not produce a decrease in staining that can be unambiguously scored. For Lar-AP, deletion of Sdc only slightly reduces axonal staining. A Lar-AP mutant that cannot bind to HSPGs stained CNS axons with equal intensity in wild-type and Sdc embryos, indicating that Lar has non-HSPG axonal binding partners (Fox and Zinn, 2005).
In this paper, we describe a new screen that addresses the second problem. GAL4-dependent expression of Sdc on muscles conferred bright staining with Lar-AP (Fox and Zinn, 2005). This suggested that if we ectopically expressed many cell-surface and secreted (CSS) proteins, potential ligands could be identified by ectopic staining with RPTP-AP probes. We had assembled a collection of lines bearing P element insertions with GAL4 binding sites upstream of 410 CSS protein genes, in order to screen for genes involved in targeting of motor axons to muscle fibers (Kurusu et al., 2008). These lines were later used to find genes affecting axonal and dendritic targeting in the antennal lobe (Hong et al., 2012; Hong et al., 2009).
Screening the CSS insertion lines for ectopic staining with Ptp10D-AP (10D-AP) identified a binding partner for Ptp10D, Stranded at second (Sas). The sas gene encodes two large cell-surface proteins of unknown function that are expressed on the apical surfaces of epithelially derived cells (Schonbaum et al., 1992).
Ptp10D is a Type III RPTP. It is orthologous to a group of four mammalian RPTPs that negatively regulate receptor TKs (RTKs) by direct dephosphorylation (reviewed by (Matozaki et al., 2010)). PTPRJ (DEP-1/CD148), which corresponds to the Suppressor of colon cancer 1 (Scc1) gene (Ruivenkamp et al., 2002) dephosphorylates the EGFR, VEGFR, and CSF1R/Met growth factor RTKs (Chabot et al., 2009; Palka et al., 2003; Tarcic et al., 2009). PTPRB (VE-PTP) regulates Tie-2 (Winderlich et al., 2009), and PTPRO regulates TrkC and Eph RTKs (Hower et al., 2009; Shintani et al., 2006). Type III RPTPs can be tyrosine-phosphorylated on a C-terminal YxNΦ (Φ=hydrophobic) motif, and this causes activation of Src-family tyrosine kinases (SFKs) (Murata et al., 2010).
Ptp10D is expressed on CNS axons (Tian et al., 1991; Yang et al., 1991) and in the tracheal system (Jeon and Zinn, 2009). In the tracheae, it negatively regulates several growth factor RTK orthologs, and it associates with EGFR in cultured cells (Jeon et al., 2012; Jeon and Zinn, 2009). It also has the C-terminal YxNΦ motif.
Ptp10D single mutants have no known embryonic phenotypes, but Ptp10D genetically interacts with other Rptp genes. The analysis of Ptp10D Ptp69D double mutants demonstrated that Ptp10D is involved in the control of axon guidance across the midline of the CNS (Sun et al., 2000; Sun et al., 2001). Double mutants lacking both Ptp10D and the other Type III RPTP, Ptp4E, have unique defects in tracheal tube formation in which unicellular tubes are converted into spherical cysts (Jeon and Zinn, 2009).
In this paper, we show that Ptp10D binds to Sas in embryos and in vitro. Loss-of-function (LOF) genetic interaction studies show that Sas and Ptp10D act together in neurons to control longitudinal axon guidance. Signaling by Sas in glia is negatively regulated by its interactions with Ptp10D on neurons.
RESULTS
Identification of Sas as a Ptp10D binding partner using an ectopic expression screen
To find potential ligands and coreceptors for Ptp10D, we used 10D-AP to stain embryos that ectopically expressed CSS proteins. The set of lines for CSS protein expression was originally assembled by creating a database of 976 CSS proteins that includes more than 80 types of XC domains, then collecting lines bearing insertions of UAS-containing (EP-like) elements in the 5′ ends of 410 of the genes (Kurusu et al., 2008).
311 of these lines, representing all genes encoding plausible ligand candidates (Table S1), were crossed to a line bearing two GAL4 drivers, Elav-GAL4 (pan-neuronal) and 24B-GAL4 (pan-muscle). Live-dissected stage 16 embryos resulting from these crosses were incubated with 10D-AP, followed by fixation and antibody staining (Fox and Zinn, 2005; Lee et al., 2009)(Figure 1A).
Figure 1. Identification of Sas in an ectopic expression screen for binding to 10D-AP.
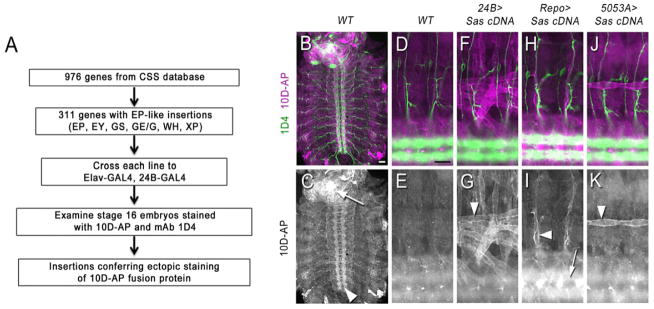
(A) Flowchart of the screen.
(B–K) Staining of live-dissected late stage 16 embryos with 10D-AP. The top row of images shows double-labeling of CNS longitudinal tracts and motor axons with mAb 1D4 (Alexa 488, green) and 10D-AP (Alexa 568, magenta); the bottom row shows 10D-AP labeling alone, in white.
(B–C) A whole filleted embryo. Note that 10D-AP prominently stains VNC axons (arrowhead) and the brain (white spot; arrow).
(D–K) Each image shows two hemisegments, including the CNS and the ventral and lateral body walls. Anterior is to the left, and dorsal is up.
(D–E) Wild-type (yw). The axon tracts of the CNS (out of focus) are stained by 10D-AP, but there is no clear pattern in the body wall.
(F–G) 24B>Sas. The ventral, ventrolateral, and lateral muscle fibers are brightly stained. Arrowhead indicates muscle 12.
(H–I) Repo>Sas. Peripheral glia surrounding the motor and sensory axon tracts are stained (arrowhead). The CNS is brighter due to staining of CNS glia (out of focus; arrow).
(J–K) 5053A>Sas. This driver expresses only in muscle 12, which is brightly labeled (arrowhead).
Scale bars in (A) and (D), 20 μm.
In wild-type stage 16 embryos, 10D-AP brightly stains axons in the ventral nerve cord (VNC) and brain (Figure 1C), but body wall staining is weak and has no clear pattern (Figures 1D–E). We searched for insertion lines that produced embryos displaying strong staining of muscle fibers and/or increased staining of cell bodies and axons in the VNC. One line, GE24911, reproducibly conferred strong ectopic staining of muscle fibers and increased VNC staining intensity. This line has an insertion of an EP-like GE element 226 bp 5′ to the transcription start site of sas (Schonbaum et al., 1992).
Analysis of ectopic Sas expression showed that 24B-GAL4 is weaker in the double driver line than as a single driver, suggesting that we might have missed some genes due to insufficient ectopic expression. Accordingly, we repeated the entire screen using a strong pancellular driver, tubulin (tub)-GAL4, and identified 9 other candidate binding partners (Figure S1). We do not yet know if these are genuine Ptp10D ligands or coreceptors, as their binding to Ptp10D has not been examined by ‘reverse’ staining of embryos (see below) or by in vitro binding. Interestingly, one is Ptp10D itself, indicating that the Ptp10D XC domain may have homophilic binding activity. This was also observed in a cell aggregation assay (see below).
sas encodes two cell-surface proteins of 1693 and 1348 amino acids (aa). The mRNA for the larger isoform includes an alternatively spliced exon encoding a 345 aa sequence that is inserted at aa 929. Sas proteins have two XC domains with defined structures: a von Willebrand factor type C (VWC) domain (aa ~750–825) and two FN3 repeats (aa ~1400–1600). There is a single predicted transmembrane domain, and a short cytoplasmic domain of 37 aa (Schonbaum et al., 1992). Western blotting of Sas from embryos reveals multiple bands between 100 kD and 220 kD, suggesting that there are extracellular Sas proteins that lack transmembrane and cytoplasmic sequences. Sas does not have an obvious vertebrate ortholog, but there are numerous vertebrate proteins with VWC domains and FN3 repeats homologous to those in Sas.
To confirm that Sas was responsible for ectopic binding of 10D-AP to embryos from GE24911 crosses, we made transgenic lines containing a UAS-linked cDNA construct encoding the large Sas isoform. When these were crossed to a variety of GAL4 driver lines, we observed ectopic 10D-AP staining in patterns that corresponded to the expression patterns of the drivers. Ventrolateral and ventral muscles were brightly labeled by 10D-AP in embryos expressing Sas from the 24B-GAL4 driver (Figures 1F–G). Embryos expressing Sas in glia from Repo-GAL4 showed increased 10D-AP staining along peripheral nerves (Figures 1H–I). Embryos expressing Sas from 5053A-GAL4, which is selectively expressed in muscle 12, displayed ectopic 10D-AP staining only on that muscle (Figures 1J–K). We compared 10D-AP and anti-Sas staining in ectopic expression embryos, and found that the two patterns were superimposable (Figure S2).
Although ectopic 10D-AP staining is observed wherever Sas is overexpressed, Sas is not required for axonal staining by 10D-AP. In embryos homozygous for a sas point mutation or a Df that removes sas, a normal axonal pattern of 10D-AP staining was observed (data not shown). This indicates that Ptp10D has other binding partners (see Figure S1).
A Sas fusion protein binds to Ptp10D in embryos
The fact that ectopic Sas expression confers ectopic staining with an exogenous Ptp10D fusion protein does not prove that Sas itself binds to Ptp10D. To determine whether exogenous Sas can interact with endogenous Ptp10D, we performed ‘reverse staining’ experiments, incubating embryos with purified Sas-Fc, a dimeric human Fc fusion protein containing the XC domain of the large Sas isoform. In live-dissected wild-type embryos, we observed only weak staining with Sas-Fc, and we could not see changes in the staining pattern when we ectopically expressed Ptp10D using the Ptp10DEP1172 insertion line. We then made the surprising observation that genetic removal of the other Drosophila Type III RPTP, Ptp4E, which is widely expressed in embryos (Jeon et al., 2008; Oon et al., 1993), allowed visualization of ectopic Ptp10D by staining with Sas-Fc. The simplest explanation for this effect is that heterodimerization of Ptp4E with Ptp10D blocks Sas binding, but we have no evidence that such heterodimers exist.
Figures 2A–H show double staining of the VNC with Sas-Fc and an anti-Ptp10D monoclonal antibody (mAb). In wild-type embryos, faint staining of CNS axons with Sas-Fc was observed (Figure 2B). This was increased in intensity in a Ptp4E mutant (Figure 2D). In a Ptp4E Ptp10D double mutant, no Ptp10D protein is present (Figure 2E), and the intensity of staining with Sas-Fc was reduced relative to the Ptp4E single mutant (Figure 2F), suggesting that Ptp10D is one of the major binding partners for Sas-Fc on CNS axons. When Ptp10D was overexpressed on all neurons in a Ptp4E mutant background using the Elav-GAL4 driver, Ptp10D and Sas-Fc staining intensities were increased (Figures 2G–H).
Figure 2. Sas-Fc binds to Ptp10D in embryos.
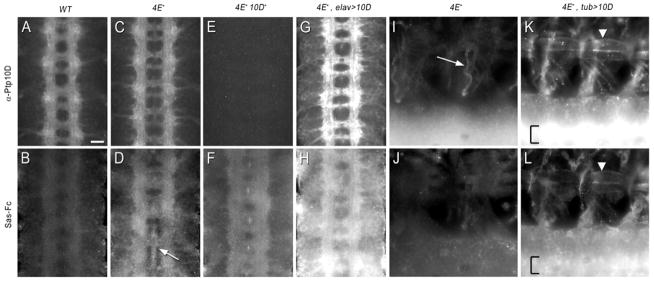
Live-dissected late stage 16 embryos double-stained with anti-Ptp10D mAb (top row) and Sas-Fc (bottom row) using immunofluorescence for detection (Alexa 488 for Ptp10D, Alexa 568 for Sas-Fc). In (A–H), four segments of the CNS are shown in each panel; anterior is up. In (I–L), two segments of the CNS, ventral, and ventrolateral muscles are shown in each panel; anterior is to the left and dorsal is up.
(A–B) Wild-type. Sas-Fc weakly stains CNS axons.
(C–D) Ptp4E1. Sas-Fc staining intensity is increased relative to (B). CNS axons are labeled, in addition to staining at the midline (arrow) and on cell bodies, which are located on the sides of the axon ladder.
(E–F) Ptp4E1 Ptp10D1. No Ptp10D staining is observed in (E). Sas-Fc staining intensity is reduced relative to (D).
(G–H) Ptp4E1, Elav>Ptp10D. The intensity of Ptp10D axonal staining is increased relative to (C), and some cell body staining is observed. Sas-Fc staining intensity is increased relative to (D), and both axons and cell bodies are stained.
(I–J) Ptp4E1. Anti-Ptp10D stains ganglionic tracheal branches (arrow). Sas-Fc labeling has no clear pattern in the periphery.
(K–L) Ptp4E1, Tub>Ptp10D. The edges of the ventral and ventrolateral muscles are brightly labeled by anti-Ptp10D in (K) and by Sas-Fc in (L). Arrowhead indicates muscle 6. One brightly labeled, out-of-focus CNS longitudinal tract (bracket) is seen in each panel. This tract is much brighter in (K) vs. (I), and in (L) vs. (J), reflecting Ptp10D overexpression and increased Sas-Fc staining on axons.
Scale bar in (A): 10 μm.
Sas-Fc stained body walls weakly in Ptp4E mutant embryos (Figure 2J). However, when Ptp10D was pancellularly expressed in the Ptp4E mutant background using tub-GAL4, bright Sas-Fc staining (and anti-Ptp10D staining) was observed on muscle fibers, and VNC staining intensity was increased (Figures 2K–L). These data show that Sas and Ptp10D can interact with each other in embryos, but do not demonstrate that they can bind to each other in the absence of other cofactors. To evaluate this issue, we analyzed interactions between Sas and Ptp10D in vitro.
Sas and Ptp10D bind to each other in vitro
We examined binding in vitro between Sas and Ptp10D using a modified ELISA assay and the AlphaScreen (Perkin-Elmer Co.), which measures interactions between proteins bound to beads. For studies of interactions between cell surface proteins, these methods are superior to assays such as two-hybrid, GST pulldown, cell aggregation, and cell adhesion-to-substrate. Two-hybrid and GST pulldown assays produce unreliable results with XC domains because they lack normal disulfide bond formation and glycosylation when expressed within yeast or bacterial cells, and cell aggregation and cell adhesion-to-substrate assays cannot demonstrate that proteins interact in the absence of cellular cofactors.
The modified ELISA assay was developed for analysis of Dscam interactions (Wojtowicz et al., 2007). While we obtained robust signals with Dscam positive controls, our initial attempts to measure binding of Sas to Ptp10D produced weak signals. We made some modifications to the assay that increased its signal-to-noise ratio by about 10-fold (see Experimental Procedures), and were then able to readily demonstrate selective Sas-Ptp10D interactions.
Figure 3A shows the results of an experiment in which purified Sas-Fc was used alone (blank) or mixed at a ~1:1 molar ratio with three different RPTP XC domain fusion proteins: 10D-AP, Ptp69D-AP (69D-AP) or Lar-AP. All proteins were made using the baculovirus system. Eight identical replicates of each binding reaction were assessed. Binding was observed only for the Sas::Ptp10D pair. The mean signal value for Sas::Ptp10D was 30 to 43-fold higher than for any of the three controls, and these differences were highly statistically significant (p<.0001).
Figure 3. Sas-Ptp10D interactions in vitro and in cultured cells.

The bar graphs show the absorbance at 370 nm obtained using the modified ELISA assay for the indicated pairs of proteins. The height of each bar represents the mean of 8 replicate assays.
(A) Sas-Fc binds to 10D-AP, but not to 69D-AP or Lar-AP. Actual values for the four pairs are: Sas-Fc::10D-AP: 0.340±0.040 (S.D.); Sas-Fc::Lar-AP: 0.007±0.001; Sas-Fc::69D-AP: 0.010±0.002; Sas-Fc::blank: 0.017±0.004. The limit of quantification (LOQ) is given by: LOQ = (mean blank signal) + (10 × S.D. for the blank). If a signal is above the LOQ it is very unlikely that the difference between the signal and the blank arose by chance. Here LOQ is 0.053, signal is 0.340, and signal/LOQ is >6.
(B) 10D-AP binds to Sas-Fc, but not to Unc5-Fc or FasII-Fc. Actual values are: Sas-Fc::10D-AP: 0.375±0.104; Sas-Fc::Lar-AP: 0.010±0.004; Unc5-Fc::10D-AP: 0.012±0.002; FasII-Fc::10D-AP: 0.028±0.008. LOQ for Unc5-Fc::10D-AP is 0.031, and signal/LOQ is 12; LOQ for FasII-Fc::10D-AP is 0.111, and signal/LOQ is 3.4.
(C) A field of Ptp10D-expressing stably transfected S2 cells (magenta). A typical cluster is circled (dotted outline). Scale bar, 20 μm.
(D) A field of Sas-expressing stably transfected S2 cells (green). No clusters form when these cells are incubated alone.
(E) A field of a mixture of Ptp10D and Sas-expressing cells. The outlined cluster of Ptp10D-expressing cells has several green Sas cells associated with it.
These data show that Sas binds to Ptp10D and not to other RPTP-AP fusion proteins, but do not address the possibility that 10D-AP is ‘sticky’ and can bind nonselectively to any Fc fusion protein. To evaluate this, we conducted the experiment in Figure 3B, in which 10D-AP was mixed at a ~1:1 molar ratio with two other Fc fusion proteins, Unc5-Fc and Fasciclin II (FasII)-Fc, with one of the controls from the Figure 3A experiment, Sas-Fc::Lar-AP, serving as the blank. Eight identical replicates of each binding reaction were assessed. As in Figure 3A, binding was observed only between Ptp10D and Sas. The mean signal value for Sas::Ptp10D was 13 to 32-fold higher than for the two Fc controls, and the differences were highly statistically significant (p<.0001 for both the Unc5::10D and FasII::10D controls).
The AlphaScreen relies on a proximity-dependent chemical reaction to measure binding between two proteins bound to ‘donor’ and ‘acceptor’ beads. This assay can work well for low-affinity interactions, because the high concentrations of protein on the beads facilitate bead-bead interactions via avidity effects. Cell-cell interactions mediated by adhesion molecules are strongly influenced by avidity. Indeed, the AlphaScreen worked extremely well for Dscam, which is a homophilic adhesion molecule. The signal due to homophilic binding of the 7.13.25 isoform was 178-fold higher than that for heterophilic binding of 7.13.25 to 7.8.25, which differs only in exon 3 (see (Wojtowicz et al., 2007) for nomenclature). We were also able to detect concentration-dependent binding of Sas to Ptp10D using the AlphaScreen. However, the signal-to-background ratio (Sas-Fc::10D-AP vs. Sas-Fc::69D-AP or Sas-Fc::blank) was only about 6-fold (Figure S3), so this assay was inferior to the ELISA for this ligand-receptor pair.
These results show that Sas and Ptp10D selectively interact with each other, but do not define whether their in vivo interactions are likely to be in cis (between proteins on the same cell surface), in trans (between proteins on different cell surfaces), or both. We used a cell aggregation assay to evaluate whether Ptp10D and Sas can interact in trans. This was done by making stable Schneider 2 (S2) cell lines expressing full-length Ptp10D and a Sas-mCD8-GFP fusion protein. Ptp10D-expressing cells formed small clusters (Figure 3C), consistent with the observation that 10D-AP binds to ectopically expressed Ptp10D in embryos (Figure S1). Sas-expressing cells did not aggregate (Figure 3D). When the cell lines were mixed, we observed Ptp10D-expressing cell clusters that were associated with one to several Sas-expressing cells (Figure 3E). These cells sometimes formed straight interfaces with Ptp10D cells that are suggestive of adhesive interactions (Figure S3). Large cell clusters were seldom observed, suggesting that these interactions are weak or limited by unknown factors.
Sas and Ptp10D are both expressed on CNS axons and tracheal cells
Within the CNS, Ptp10D is expressed only on axons (Sun et al., 2000; Tian et al., 1991; Yang et al., 1991)(Figure 4A). It is also expressed on the apical surfaces of tracheal cells (Jeon and Zinn, 2009)(Figure 4C).
Figure 4. Expression patterns of Ptp10D and Sas at late stage 16.

Embryos were double-stained with anti-Ptp10D mAb and rabbit anti-Sas, and visualized by immunofluorescence.
(A) Ptp10D. Note that CNS axons are brightly stained, and cell outlines are almost invisible.
(B) Sas. The intersegmental (arrowhead) and segmental (double arrowhead) nerve roots are brightly stained. Two longitudinal bundles express Sas at higher levels within each longitudinal tract (arrows). Cell outlines are clearly visible (ring around asterisk).
(C) Ptp10D. The apical surfaces of tracheal branches are brightly labeled. Arrowheads in (C) and (D) indicate the dorsal trunk.
(D) Sas. The tracheal pattern of Sas expression is essentially identical to that of Ptp10D. Scale bars in (A) and (C): 10 μm.
Antibody against Sas has been widely used as a selective marker for apical cell surfaces (Schonbaum et al., 1992; Wodarz et al., 1995). Sas is probably expressed at some level on the apical surfaces of all cells of epithelial origin, including neurons and glia. Most or all cell bodies and axons in the VNC appear to stain with anti-Sas. Sas is expressed at higher levels on some axons than on others, in particular on the intersegmental and segmental nerve roots and on two longitudinal bundles on each side of the CNS (Figure 4B). Figure S4 demonstrates that ensheathing cells on the dorsal surface of the CNS express Sas. These are the perineurial, subperineurial, interface, and channel glia (Ito et al., 1995). In the tracheal system, the patterns of staining with anti-Ptp10D and anti-Sas are essentially identical (Figures 4C, D)(Jeon and Zinn, 2009).
Sas is involved in formation of longitudinal axon tracts
Although lethal ethyl methane sulfonate (EMS) mutations in sas were isolated many years ago, little is known about the functions of the sas gene. sas mutants do not exhibit embryonic lethality, but their post-embryonic growth is stunted and they usually die as second instar larvae (hence the name). Third instar escapers have convoluted tracheae (Schonbaum et al., 1992), but we were unable to find any tracheal defects in embryos.
To evaluate whether sas mutants have CNS phenotypes, we examined embryos homozygous for a strong EMS mutation, sas15, or transheterozygotes bearing this mutation over Df(3R)ED5221, which removes the entire sas gene. We sequenced the sas gene on the sas15 chromosome, and found that sas15 introduces a stop codon after aa 642, which is N-terminal to all conserved Sas domains. These data indicate that sas15 is likely to be a null mutation.
To visualize CNS axons, we used mAbs BP102 and 1D4. BP102 labels most or all CNS axons, producing a ladder-like pattern, with two commissural tracts in each segment and two longitudinal tracts extending the length of the embryo (Seeger et al., 1993)(Figures 5B, D, F). 1D4 recognizes the transmembrane form of FasII (Vactor et al., 1993), and stains pioneer axons at stage 14 (Figure 5A). By late stage 16, 1D4 staining within the CNS is restricted to three distinct longitudinal axon bundles on each side of the VNC, and no staining is seen on commissural axon bundles that cross the CNS midline (Figure 5E).
Figure 5. sas LOF CNS phenotypes.
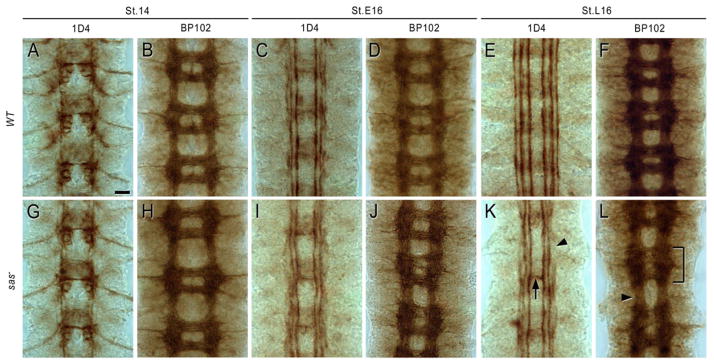
Embryos were stained with mAb 1D4 or mAb BP102, and HRP immunohistochemistry was used for detection. Each panel shows three segments of the VNC, which was dissected out of the embryos. (A, B, G, H): stage 14. (C, D, I, J): early stage 16. (E, F, K, L): late stage 16.
(G, H) sas15/Df transheterozygotes are indistinguishable from wild-type (yw) (A, B) at stage 14 using either marker.
(I, J) Slight irregularities in the 1D4 and BP102 patterns are observed at early stage 16 in sas15/Df.
(E) In late stage 16 wild-type embryos, 1D4 labels three longitudinal bundles on each side of the CNS, and does not stain commissural axons.
(K) In sas15/Df at late stage 16, the outer 1D4 bundle is interrupted (arrowhead), and there are occasional axon bundles crossing the midline (arrow).
(F) In late stage 16 wild-type embryos, a regular BP102 ladder is observed.
(L) In sas15/Df at late stage 16, the ladder is slightly irregular and narrowed. One segment has an abnormal anterior commissure (bracket).
sas15/Df transheterozygotes appear normal at stage 14 (Figures 5G, H), but display weak longitudinal axon defects at stage 16. The outer 1D4 axon bundle is interrupted and the other bundles appear slightly irregular (Figures 5I, K). Complete breaks of a longitudinal tract are rare, but do occasionally occur. 1D4 axons ectopically cross the midline in about 15% of segments. Irregularities in the BP102 axon ladder are observed in at least one segment of most embryos (Figures 5J, L). sas15 homozygotes have identical phenotypes, consistent with sas15 being a null mutation (data not shown).
Ptp10D and Sas regulate axon guidance across the midline
To examine whether Sas is required for Ptp10D signaling using LOF genetics requires examination of double mutant phenotypes, because Ptp10D single null mutant embryos have no known phenotypes. Most relevant to this study, Ptp10D Ptp69D double-null mutants have strong CNS phenotypes in which 1D4-positive longitudinal axons that would normally remain on one side instead cross the midline (Sun et al., 2000). At late stage 16, most segments have a thick 1D4-positive commissural tract with several distinct bundles, oriented perpendicular to the longitudinal tracts. The inner longitudinal 1D4 bundle is present, but the outer two bundles are missing or fused with the inner bundle (compare Figure 6C to 6A). BP102 staining shows that the anterior and posterior commissures are fused into a single commissural tract (compare Figure 6H to 6F). The Ptp10D Ptp69D double mutation affects a unique subset of axons, and is quite different from other phenotypes in which 1D4 axons cross the midline. For example, in roundabout (robo) mutants, pioneer axons that normally extend in the inner 1D4 bundle instead follow curving pathways across the midline, creating distinctive circular patterns. The outer two 1D4 bundles are still present, although they are often interrupted (Seeger et al., 1993).
Figure 6. Ptp10D and Sas work together in neurons to regulate axon guidance across the midline.

1D4 (A–E) and BP102 (F–J) staining at late stage 16 is shown.
(B, G) Ptp10D, sas. The 1D4 and BP102 patterns are like that of sas (Figure 5) with occasional axon bundles crossing the midline (arrow) and interruptions in the outer 1D4 bundle (arrowhead). The BP102 ladder has an abnormal anterior commissure (arrowhead).
(C, H) Ptp10D, Ptp69D. In (C), thick axon bundles cross the midline in each segment (bracket), and there is only one distinct longitudinal bundle. In (H), the commissures are completely fused in all three segments (bracket), and the longitudinal tracts are thinned (arrowhead).
(D, I) sas, Ptp69D. In (D), two bundles cross the midline in each segment (bracket), and the outer longitudinal tract is often missing (arrowhead). In (I), commissural organization is disrupted (bracket), and longitudinal tracts are thinned (arrowhead).
(E, J) sas, Ptp69D, FasII>Sas. The 1D4 and BP102 patterns are almost like wild-type. Mutant genotypes shown in panels are: Ptp10D1, sas15/Df(3R)ED5221 (B, G); Ptp10D1, Ptp69D1/Df(3L)8ex25 (C, H); sas15/sas15, Ptp69D1/Df(3L)8ex25 (D); sas15/Df(3R)ED5221, Ptp69D1/Df(3L)8ex25 (I); sas15/Df(3R)ED5221, Ptp69D1/Df(3L)8ex25, FasII-GAL4Mz507, UAS-Sas cDNA (E, J).
(K) Bar graph quantitating the % of segments exhibiting ectopic midline crossing of 1D4 axons. Brown bar, Ptp10D Ptp69D; gray bars, sas15 genotypes; black bars, sas15/Df genotypes. n, number of segments scored (n=162 for wt (yw); 128 for Ptp10D; 738 for Ptp69D; 864 for sas; 585 for sas/Df; 702 for Ptp10D, sas; 522 for Ptp10D, sas/Df; 225 for Ptp10D Ptp69D; 324 for sas, Ptp69D; 685 for sas/Df, Ptp69D, 567 for sas, Ptp69D, FasII>Sas; 284 for sas/Df, Ptp69D, FasII>Sas). p<.00001 for the differences in penetrance between sas/Df and sas/Df Ptp69D, and between sas/Df Ptp69D and sas/Df Ptp69D, FasII>Sas.
The existence of the distinctive Ptp10D Ptp69D double mutant phenotype allows us to ask whether Sas is important for Ptp10D signaling, by determining if loss of Sas together with Ptp69D produces the same phenotypes as loss of Ptp10D together with Ptp69D. Ptp10D and Ptp69D single mutants have almost no midline crossing defects (0% in Ptp10D, 1.4% in Ptp69D) (Sun et al., 2000). sas15/Df transheterozygotes and sas15 homozygotes have 1D4-positive axon bundles that cross the midline in 11–15% of segments, and this penetrance is increased to 22–27% in Ptp10D sas double mutants (Figures 6B, K). However, in sas Ptp69D double mutants, 63–74% of segments have 1D4 bundles that cross the midline (Figures 6D, K). This phenotype is almost as strong as that of Ptp10D Ptp69D double mutants, in which 1D4 bundles cross the midline in 76% of segments (Figures 6C, K).
The sas Ptp69D 1D4 phenotype (Figure 6D) has many of the distinctive features of the Ptp10D Ptp69D phenotype (Figure 6C). Multiple axon bundles cross the midline in each segment, and these are perpendicular to the longitudinal tracts, not curving as in robo mutants. The inner longitudinal 1D4 bundle is intact, but one or both of the outer longitudinal bundles are missing. Ptp10D Ptp69D and sas Ptp69D embryos display similar fused commissure phenotypes, as revealed by BP102 staining (Figures 6H, I).
These results show that Sas is necessary for Ptp10D’s functions in preventing longitudinal axons from crossing the midline. However, the sas Ptp69D and Ptp10D Ptp69D 1D4 phenotypes are somewhat different. Fewer 1D4-positive bundles cross the midline in each segment in sas Ptp69D than in Ptp10D Ptp69D, and there are more distinct bundles remaining in the longitudinal tracts (compare Figure 6C to 6D). There are also complete breaks in the 1D4-positive longitudinal tracts in sas Ptp69D (Figure 6D), which are not observed in Ptp10D Ptp69D.
We further analyzed these phenotypes by staining with other markers for specific neurons and axons. Apterous (Ap)-GAL4 is expressed in a small number of neurons whose axons extend within a single longitudinal bundle (Garbe et al., 2007; Lundgren et al., 1995). Surprisingly, in both Ptp10D Ptp69D and sas Ptp69D these neurons do not extend axons at all, or have short processes that project in the wrong direction (Figure S5). Anti-Connectin stains two longitudinal bundles that are distinct from the 1D4-positive bundles. Ptp10D Ptp69D and sas Ptp69D display similar phenotypes in which the outer Connectin bundle is missing and there are occasional breaks in the inner bundle (Figure S5).
To evaluate whether Sas is likely to act together with Ptp10D in 1D4-positive axons to prevent them from crossing the midline, we expressed the Sas cDNA construct from the FasII-GAL4Mz507 driver in the sas Ptp69D double mutant background, in order to restore Sas selectively to the same subset of CNS neurons whose phenotype is scored through analysis of the 1D4 staining pattern. We observed that Sas expression in FasII-positive neurons almost completely rescued the sas Ptp69D CNS phenotypes (Figures 6E, J, K).
To test whether Sas must be expressed in FasII neurons in order to signal to Ptp10D, we then overexpressed Sas in glia using the Gcm-GAL4 driver in the sas Ptp69D double mutant background. This also rescued the phenotype (Figure S5). These data, however, do not necessarily indicate that phenotypic rescue is the result of interactions between neuronal Ptp10D and Sas on glial cell surfaces. When Sas is expressed in small clusters of cells (e.g., Apterous neurons), anti-Sas antibody staining spreads out from the cell bodies in a pattern suggesting that non-membrane-bound Sas proteins are deposited in the extracellular matrix (ECM) (Figure S5). ECM-bound (‘soluble’) Sas may be able to interact with Ptp10D in the same manner whether it is expressed on neurons or in glia.
Communication between glia and neurons via interactions between Sas and Ptp10D
We further characterized genetic interactions between sas and Ptp10D by examining their epistatic relationships, asking whether sas gain-of-function (GOF) phenotypes are modified by LOF mutations in Ptp10D (Ptp10D has no known GOF phenotypes). We overexpressed Sas cDNA in wild-type and Ptp10D mutant backgrounds, using three drivers: the strong pan-neuronal driver Elav-GAL4, the midline glial driver Sim-GAL4, and Repo-GAL4, which drives expression in all glia except midline and perineurial glia. In a wild-type background, all three driver crosses produced weak phenotypes in which the CNS axon ladder had a normal morphology, but axons in the inner 1D4 longitudinal bundle occasionally crossed the midline (Figure 7C; Figure S6). In Elav>Sas embryos, ~14% of segments had 1D4-positive axons crossing the midline, and this phenotype was not enhanced or suppressed when Ptp10D was genetically removed (Figure S6).
Figure 7. Interactions between glial Sas and axonal Ptp10D affect glial organization and axon guidance.

(A–D) 1D4 staining at late stage 16. In (A–C), a normal 1D4 pattern is observed, although the 1D4 bundles are slightly wavy in Repo>Sas (C), and one segment has an axon bundle crossing the midline (arrow).
(D) In Ptp10D1, Repo>Sas, a strong axon guidance phenotype is observed. The inner 1D4 bundle crosses the midline in all three segments (arrow), and the longitudinal bundles are partially fused and disrupted (arrowhead).
(E) Bar graph quantitating the % of segments exhibiting ectopic midline crossing of 1D4 axons. n=162 for wt; 128 for Ptp10D−; 414 for Repo>Sas; 424 for Ptp10D−, Repo>Sas. p<.00001 for the difference in penetrance between Repo>Sas and Ptp10D−, Repo>Sas.
(F–I) Glial nuclei, visualized with anti-dsRed. Repo-GAL4 was used to drive RedStinger (a nuclear version of dsRed) in the indicated genotypes. The focal plane is at the level of the interface glia. Note the organized glial lattice in (F–H), with a double vertical row of nuclei along each longitudinal tract and a band of nuclei crossing the midline in each segment. The vertical rows are slightly irregular in (H).
(I) In Ptp10D1, Repo>Sas, the glial lattice is disrupted. The vertical rows have become aggregations of varying width, and there is no regular pattern of glial nuclei near the midline.
The Sim-GAL4 and Repo-GAL4 crosses, in which Sas is overexpressed in cells that do not express endogenous Ptp10D, behaved differently. ~5% of segments had 1D4-positive axons crossing the midline with each driver (Figures 7C, E; Figure S6). When Ptp10D was genetically removed, these phenotypes were enhanced, suggesting that signaling by overexpressed glial Sas is negatively regulated by neuronal Ptp10D. For Sim>Sas, 11% of segments displayed ectopic midline crossing in the Ptp10D background, but the overall structure of the CNS was unchanged (Figure S6). For Repo>Sas, however, loss of Ptp10D produced a phenotype in which the entire pattern of 1D4-positive axons was dramatically altered, and >50% of segments had ectopic midline crossing (Figures 7D–E). We asked whether negative regulation of glial Sas signaling by Ptp10D requires that Ptp10D be expressed on neurons by driving both Sas and Ptp10D in glia in a Ptp10D mutant background. This was able to rescue the midline crossing phenotype (Figure S6).
Although the Ptp10D, Repo>Sas phenotype, like the Ptp10D Ptp69D and sas Ptp69D double mutant phenotypes, is quantitatively analyzed by scoring ectopic midline crossing, it is qualitatively a different phenotype. In Ptp10D, Repo>Sas embryos, the inner 1D4 bundle crosses the midline, but the outer bundles, which cross in Ptp10D Ptp69D, usually do not (Figure 7D; compare to Figures 6C and 6D). The phenotype is of variable strength, and has similarities to those of mutants with defects in Slit-Robo pathway components (Bashaw et al., 2000; Seeger et al., 1993).
The axon guidance phenotype seen in Ptp10D−, Repo>Sas embryos is an indirect consequence of overexpression of Sas in glia. To examine whether the glia themselves are visibly altered by Sas overexpression, we crossed in a UAS-nuclear dsRed construct and visualized Repo-GAL4-expressing nuclei using anti-dsRed antibodies. In Figures 7F–I, we show the focal plane containing the nuclei of the interface glia, which lie just dorsal to the axon ladder and are required for normal axon guidance. Nuclei of nerve root glia and some of the channel and subperineurial glia are also visible within this focal plane (Ito et al., 1995).
CNS glia migrate extensively between stages 13 and 16, so that glial nuclear patterns undergo rapid changes (reviewed by (Hidalgo and Griffiths, 2004)). At late stage 16, glial nuclei in the focal plane of Figure 7F are organized in a semi-regular lattice in wild-type embryos, with two rows of nuclei extending along each longitudinal tract and a segmentally repeated pattern of nuclei closer to the midline. The analysis of this pattern is complicated by the large numbers of glia, their distribution through multiple focal planes, and the dynamic nature of the pattern. Even in wild-type, there is substantial variation between glial lattices in different embryos that appear to be at the same stage of development. This is in contrast to the essentially invariant nature of the pattern of 1D4-positive axons in late stage 16 wild-type embryos (compare Figures 5E, 6A, and 7A).
Despite these caveats, it is clear that wild-type, Ptp10D, and Repo>Sas embryos all have organized glial lattices with a similar overall appearance, although the Repo>Sas lattice is somewhat irregular (Figures 7F–H). By contrast, in Ptp10D, Repo>Sas embryos the pattern of glial nuclei is quite disorganized (Figure 7I). The double row of nuclei along each longitudinal tract is replaced by aggregations of varying width, and there is no segmentally repeated pattern of nuclei near the midline.
These results show that when Sas is overexpressed in glia in embryos lacking the neuron-specific Ptp10D protein, it generates a phenotype characterized by glial disorganization and altered CNS axon guidance. Interactions between neuronal Ptp10D and glial Sas appear to hold Sas activity in check when it is overexpressed, thereby preserving the relatively normal glial and axonal patterns seen in Repo>Sas embryos.
DISCUSSION
We devised a screen for orphan receptor ligands in which CSS proteins are ectopically expressed in embryos and ligand candidates are identified by the presence of ectopic staining with receptor-AP fusion proteins. We performed a screen of GAL4-dependent insertion lines for 311 CSS protein genes (Table S1) with the XC domain of the Ptp10D RPTP, and identified a gene encoding the cell-surface protein Sas (Schonbaum et al., 1992) (Figures 1 and 2). We used a modified ELISA assay to show that Sas and Ptp10D selectively bind to each other in the absence of other cofactors, and demonstrated that Sas and Ptp10D-expressing cells can aggregate with each other (Figure 3).
Ptp10D is selectively expressed on CNS axons (Sun et al., 2000; Tian et al., 1991; Yang et al., 1991)(Figure 4A), and functions redundantly with Ptp69D to prevent a specific subset of longitudinal axons from crossing the midline (Sun et al., 2000). Ptp10D Ptp69D and sas Ptp69D double mutants both have strong ectopic midline crossing phenotypes, suggesting that Sas is required for Ptp10D signaling in the context of longitudinal axon guidance (Figures 6 and 8A).
Figure 8. Models for signaling by Ptp10D and Sas.
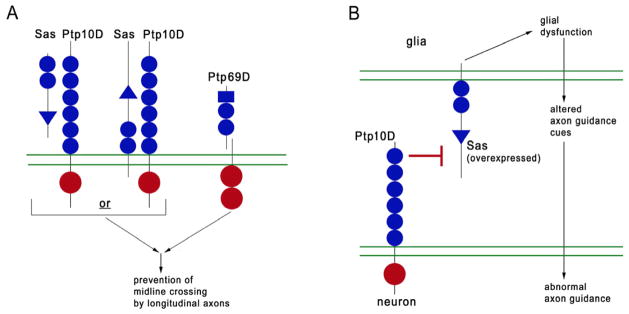
Triangles, VWC domains; blue circles, FN3 repeats (Ptp10D actually has 12); rectangles, Ig domains; red circles, PTP domains; double green lines, cell membranes. (A) Models for signaling in neurons in wild-type. The sas Ptp69D phenotype is rescued by Sas expression in longitudinal tract neurons, suggesting that either: 1) Sas interacts with Ptp10D in the same cell, or, 2) ECM-bound or transmembrane Sas on other longitudinal axons interacts with Ptp10D. Ptp10D/Sas and Ptp69D may both affect the activity of a common downstream signaling protein(s) that helps prevent longitudinal axons from crossing the midline. Ptp69D is cleaved as indicated.
(B) Model for signaling between glia and neurons in embryos overexpressing Sas in glia. Overexpressed Sas produces a signal in glia (top) that alters their presentation of axon guidance cues to neurons (bottom). Generation of the signal is suppressed (⊥ symbol) by interactions between neuronal Ptp10D and glial Sas.
Sas appears to be expressed on all cells in the CNS, including glia (Figures 4 and S4). We conducted genetic epistasis experiments by overexpressing Sas in glia and asking whether loss of the neuron-specific Ptp10D modifies the resulting phenotype. The results suggest that overexpressed Sas can produce a signal that alters glia and affects their communication with neurons (Figure 7). Interactions between Ptp10D and Sas suppress the production of this signal (Figure 8B).
Ptp10D/Sas signaling in neurons controls guidance of longitudinal axons
RPTP signaling controls the decisions by axonal growth cones to choose longitudinal vs. commissural pathways, because in a quadruple Rptp mutant (Ptp10D Lar Ptp69D Ptp99A), all 1D4 (FasII)-positive longitudinal axons are diverted into the commissures and the longitudinal bundles are absent. Ptp10D and Ptp69D are key to these guidance decisions, because triple Rptp mutants in which either Ptp10D or Ptp69D is wild-type have a relatively normal 1D4 pattern, but any mutant combination that includes both Ptp10D and Ptp69D mutations has thick 1D4-positive commissures (Sun et al., 2001). This suggests that Ptp10D and Ptp69D share some critical substrate(s) or interacting protein(s) that controls these decisions.
sas Ptp69D double mutants also have strong ectopic midline crossing phenotypes that are rescued by selective expression of Sas in FasII neurons (Figure 6). The simplest model to explain these findings is that Ptp10D forms a complex with Sas in FasII-expressing longitudinal tract neurons in order to activate the downstream signaling pathway(s) that it shares with Ptp69D (Figure 8A). However, the axons of FasII neurons bundle together, so Sas on one axon could contact Ptp10D on another axon. The sas Ptp69D phenotype can also be rescued by expression of Sas in glia, and Sas protein(s) appear to be deposited in the ECM (Figure S5). Thus, signaling interactions relevant to midline crossing might also be mediated by binding of soluble Sas to Ptp10D on axons.
Signaling by overexpressed glial Sas is negatively regulated by interactions with neuronal Ptp10D
Longitudinal axon guidance and interface glial development are intertwined processes (for review see (Hidalgo and Griffiths, 2004)). Perturbation of interface glia can cause longitudinal axons to cross the midline (Kinrade and Hidalgo, 2004). Conversely, the fates of longitudinal glia, which are a subset of the interface glia (Ito et al., 1995), are controlled by signals from neurons (Griffiths and Hidalgo, 2004; Thomas and van Meyel, 2007).
The analysis of glial-neuronal interactions provides an excellent system in which to examine whether signaling through Sas can be regulated by interaction with Ptp10D. Ptp10D is only on axons, while Sas is expressed on glia (Figures 4 and S4). Driving Sas overexpression in glia with Repo-GAL4 produces only subtle phenotypes. However, genetic removal of Ptp10D from Repo>Sas embryos generates strong ectopic midline crossing phenotypes. These phenotypes are accompanied by disorganization of interface glia (Figure 7). Glial mispositioning might be sufficient to affect axon guidance. However, given the severity of the axonal phenotype, we think it more likely that the disruption of the glial lattice is reflective of changes in gene expression that cause the glia to send abnormal axon guidance signals to the neurons (Figure 8B).
The 37 aa Sas cytoplasmic domain interacts with Numb in the yeast two-hybrid assay (Chien et al., 1998). Numb is an inhibitor of Notch signaling, and both elevation and loss of Notch signaling affect longitudinal glia (Griffiths et al., 2007; Griffiths and Hidalgo, 2004; Kato et al., 2011; Thomas and van Meyel, 2007). We suggest that when Sas is overexpressed in glia and is not restrained by Ptp10D binding, it might sequester Numb, thereby increasing Notch signaling.
Comparison to other ligand-receptor systems involved in axon guidance
Binding of Sas to Ptp10D on longitudinal axons facilitates Ptp10D’s functions in regulation of CNS axon guidance. In glia, overexpressed Sas produces a signal that is suppressed by interactions with neuronal Ptp10D (Figure 8). Other receptors involved in axon guidance exhibit interactions with ligands that produce different signaling outcomes depending on whether the ligands and receptors are expressed on the same or on different cells. In retinal ganglion cells and spinal motor neurons, Eph RTKs interact with Ephrin ligands both on other cells (in trans) and on the same cells (in cis), and cis interactions attenuate the responses of the RTKs to ligand presented in trans ((Carvalho et al., 2006); reviewed by (Dudanova and Klein, 2011)).
Like Sas, Ephrins and type III neuregulin 1, a ligand for ErbB RTKs, also generate ‘reverse’ signals in the cells that express them that are important for axon pathfinding (Hancock et al., 2011). However, Ephrin and neuregulin signals are produced upon engagement of the ligands with their receptors, not blocked by receptor engagement as in the case of Sas and Ptp10D.
EXPERIMENTAL PROCEDURES
Genetics
FasII-GAL4Mz507 (B. Altenheim) was from Hermann Aberle. Ptp10D is on the X, and is examined as a hemizygote. Ptp10D1, which is a P element excision mutation that deletes N-terminal coding sequence, eliminates all detectable protein expression (Jeon and Zinn, 2009; Sun et al., 2000). For Ptp69D, transheterozygotes between 5′ (Ptp69D1) and 3′ (Df(3L)8ex25) excision mutations, both of which remove coding sequence, are used in order to generate a null phenotype affecting only Ptp69D (Desai et al., 1996). Crosses and embryo collections were performed at room temperature. For the screen, embryos were shifted to 29°C for 60 min utes prior to dissection. For Sas overexpression experiments, embryos were shifted to 29°C for 90 min. prior to fixation and staining. To express Sas and Ptp10D together on glia in a Ptp10D background (Figure S6), we used the Ptp10DEP1172 allele, which reduces Ptp10D expression and produces a phenotype when combined with Ptp69D (Sun et al., 2000), but also confers GAL4-dependent Ptp10D expression.
Immunohistochemistry
Procedures for live dissection and RPTP-AP staining were described previously (Fox and Zinn, 2005; Lee et al., 2009). For whole-mount antibody staining of embryo collections, we modified procedures described by (Patel, 1994). The following antibodies were used: mAb 1D4 ((Vactor et al., 1993), used at 1:3), mAb BP102 ((Seeger et al., 1993), used at 1:20), mAb 8B2 against Ptp10D ((Kurusu and Zinn, 2008), used at 1:5); rabbit-anti-Sas ((Schonbaum et al., 1992), used at 1:2000, gift of D. Cavener (Penn State University)); rabbit anti-AP (Serotec, used at 1:600); rabbit-anti-RFP (dsRed) (Rockland, used at 1:1000); rabbit anti-GFP (Invitrogen, used at 1:1000); Rhodamine-conjugated phalloidin (Invitrogen, used at 1:2000); AlexaFluor 488 anti-mouse and AlexaFluor 568 anti-rabbit (Invitrogen, used at 1:1000); HRP-conjugated goat-anti-mouse and goat anti-rabbit (Jackson Immunoresearch, used at 1:150).
Molecular Biology
The Sas cDNA construct was made using a full-length cDNA clone for the 1693 aa protein. This was inserted into pUAST-attB, and site-specific X and 2nd chromosome transgenics were made by Rainbow Genetics, Inc. Expression of RPTP-AP proteins using baculovirus was described by (Fox and Zinn, 2005). Sas-Fc was made by inserting the entire XC domain sequence of Sas into an S2 expression vector containing the human Fc sequence with a His tag (Wojtowicz et al., 2007). Then, the entire Sas-Fc coding region, minus the signal sequence, was amplified by PCR from this plasmid and inserted into a baculovirus vector, pAcGP67A. Sas-Fc was purified from supernatants of cells infected with the virus derived from this vector, using Ni-NTA agarose.
We PCR-amplified exons of the sas15 gene from sas15 homozygote larvae and sequenced multiple clones from multiple amplifications to ensure that observed changes were due to mutation and not to PCR errors. We observed changes from wild-type as follows: exon 2, pos. 3530, noncoding; exon 2, pos. 3590, noncoding (5 bp deletion); exon 2, pos. 3784, missense; exon 6, pos. 17890, nonsense (stop codon mutation at aa 642); exon 6, pos. 18024, missense; exon 9, pos. 20000, missense.
ELISA method
Each well of Nunc Immunosorb 96-well plates was incubated overnight at 4° C with 50 μl (3 μg/ml antibody) of unpurified ascites fluid containing the IgG2A anti-AP mAb 8B6 (Sigma) in 1× PBS, pH 7.4. Wells were washed 5 times for 1–3 min at room temperature with 150 μl 1× PBS, pH 7.4 + 0.05% Tween-20 (PBST). Wells were incubated for 1–2 hr at room temperature with 150 μl 1% casein in 1× PBS, pH 7.4, on a rocking platform. The 1% casein block was removed. This was followed by the addition of 20 μl Fc fusion protein (Sas-Fc (5 ng/μl), Unc5-Fc (5 ng/μl) or FasII-Fc (5 ng/μl)) and 20 μl AP fusion protein (10D-AP (8.5 ng/μl), Lar-AP (8.5ng/ μl), 69D-AP(8.5ng/μl), or blank culture medium), for a total volume of 40 μl. The AP fusion protein dilutions also contained HRP-conjugated mouse anti-human IgG1 (2 μg/ml; Serotec). Plates were covered and incubated overnight at room temperature protected from light. The next day, wells were washed 5 times for 1–3 min at room temperature with 150ul PBST. 1-Step Ultra TMB-ELISA HRP substrate (100 μl; Pierce cat 34028) equilibrated to room temperature was added and plates were incubated for 1 hr at room temperature. Absorbance at both 370nm and 652nm wavelengths was measured using an ND-1000 spectrophotometer.
Supplementary Material
HIGHLIGHTS.
A new method to find genes encoding ligands for orphan receptors.
A Type III receptor tyrosine phosphatase, Ptp10D, binds to Sas, a cell surface protein.
Sas and Ptp10D work together in longitudinal axons to regulate midline crossing.
Glia and neurons can communicate via interactions between Sas and Ptp10D.
Acknowledgments
We thank Woj Wojtowicz (University of California, Berkeley) for Dscam fusion proteins, DNA constructs, antibodies, and advice on ELISAs; Engin Ozkan and Chris Garcia (Stanford) for Unc5-Fc and FasII-Fc; Douglas Cavener (Penn State) for anti-Sas, and Hermann Aberle for FasII-GAL4. We thank Violana Nesterova for help with figure preparation. Protein expression was done at the Caltech Protein Expression Center. This work was supported by an NIH RO1 grant to K.Z., NS28182.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aricescu AR, McKinnell IW, Halfter W, Stoker AW. Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase sigma. Mol Cell Biol. 2002;22:1881–1892. doi: 10.1128/MCB.22.6.1881-1892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aricescu AR, Siebold C, Choudhuri K, Chang VT, Lu W, Davis SJ, van der Merwe PA, Jones EY. Structure of a tyrosine phosphatase adhesive interaction reveals a spacer-clamp mechanism. Science. 2007;317:1217–1220. doi: 10.1126/science.1144646. [DOI] [PubMed] [Google Scholar]
- Bashaw GJ, Kidd T, Murray D, Pawson T, Goodman CS. Repulsive axon guidance: Abelson and Enabled play opposing roles downstream of the roundabout receptor. Cell. 2000;101:703–715. doi: 10.1016/s0092-8674(00)80883-1. [DOI] [PubMed] [Google Scholar]
- Carvalho RF, Beutler M, Marler KJ, Knoll B, Becker-Barroso E, Heintzmann R, Ng T, Drescher U. Silencing of EphA3 through a cis interaction with ephrinA5. Nat Neurosci. 2006;9:322–330. doi: 10.1038/nn1655. [DOI] [PubMed] [Google Scholar]
- Chabot C, Spring K, Gratton JP, Elchebly M, Royal I. New role for the protein tyrosine phosphatase DEP-1 in Akt activation and endothelial cell survival. Mol Cell Biol. 2009;29:241–253. doi: 10.1128/MCB.01374-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien CT, Wang S, Rothenberg M, Jan LY, Jan YN. Numb-associated kinase interacts with the phosphotyrosine binding domain of Numb and antagonizes the function of Numb in vivo. Mol Cell Biol. 1998;18:598–607. doi: 10.1128/mcb.18.1.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles CH, Shen Y, Tenney AP, Siebold C, Sutton GC, Lu W, Gallagher JT, Jones EY, Flanagan JG, Aricescu AR. Proteoglycan-specific molecular switch for RPTPsigma clustering and neuronal extension. Science. 2011;332:484–488. doi: 10.1126/science.1200840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai CJ, Gindhart JG, Jr, Goldstein LS, Zinn K. Receptor tyrosine phosphatases are required for motor axon guidance in the Drosophila embryo. Cell. 1996;84:599–609. doi: 10.1016/s0092-8674(00)81035-1. [DOI] [PubMed] [Google Scholar]
- Dudanova I, Klein R. The axon’s balancing act: cis- and trans-interactions between Ephs and ephrins. Neuron. 2011;71:1–3. doi: 10.1016/j.neuron.2011.06.030. [DOI] [PubMed] [Google Scholar]
- Flanagan JG, Cheng HJ. Alkaline phosphatase fusion proteins for molecular characterization and cloning of receptors and their ligands. Methods in enzymology. 2000;327:198–210. doi: 10.1016/s0076-6879(00)27277-7. [DOI] [PubMed] [Google Scholar]
- Fox AN, Zinn K. The heparan sulfate proteoglycan syndecan is an in vivo ligand for the Drosophila LAR receptor tyrosine phosphatase. Curr Biol. 2005;15:1701–1711. doi: 10.1016/j.cub.2005.08.035. [DOI] [PubMed] [Google Scholar]
- Garbe DS, Das A, Dubreuil RR, Bashaw GJ. beta-Spectrin functions independently of Ankyrin to regulate the establishment and maintenance of axon connections in the Drosophila embryonic CNS. Development. 2007;134:273–284. doi: 10.1242/dev.02653. [DOI] [PubMed] [Google Scholar]
- Griffiths RC, Benito-Sipos J, Fenton JC, Torroja L, Hidalgo A. Two distinct mechanisms segregate Prospero in the longitudinal glia underlying the timing of interactions with axons. Neuron Glia Biol. 2007;3:75–88. doi: 10.1017/S1740925X07000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths RL, Hidalgo A. Prospero maintains the mitotic potential of glial precursors enabling them to respond to neurons. Embo J. 2004;23:2440–2450. doi: 10.1038/sj.emboj.7600258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock ML, Nowakowski DW, Role LW, Talmage DA, Flanagan JG. Type III neuregulin 1 regulates pathfinding of sensory axons in the developing spinal cord and periphery. Development. 2011;138:4887–4898. doi: 10.1242/dev.072306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo A, Griffiths R. Coupling glial numbers and axonal patterns. Cell Cycle. 2004;3:1118–1120. [PubMed] [Google Scholar]
- Hong W, Mosca TJ, Luo L. Teneurins instruct synaptic partner matching in an olfactory map. Nature. 2012;484:201–207. doi: 10.1038/nature10926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W, Zhu H, Potter CJ, Barsh G, Kurusu M, Zinn K, Luo L. Leucine-rich repeat transmembrane proteins instruct discrete dendrite targeting in an olfactory map. Nat Neurosci. 2009;12:1542–1550. doi: 10.1038/nn.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hower AE, Beltran PJ, Bixby JL. Dimerization of tyrosine phosphatase PTPRO decreases its activity and ability to inactivate TrkC. J Neurochem. 2009;110:1635–1647. doi: 10.1111/j.1471-4159.2009.06261.x. [DOI] [PubMed] [Google Scholar]
- Ito K, Urban J, Technau GM. Distribution, Classification, and Development of Drosophila Glial-Cells in the Late Embryonic and Early Larval Ventral Nerve Cord. Roux Arch Dev Biol. 1995;204:284–307. doi: 10.1007/BF02179499. [DOI] [PubMed] [Google Scholar]
- Jeon M, Nguyen H, Bahri S, Zinn K. Redundancy and compensation in axon guidance: genetic analysis of the Drosophila Ptp10D/Ptp4E receptor tyrosine phosphatase subfamily. Neural development. 2008;3:3. doi: 10.1186/1749-8104-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M, Scott MP, Zinn K. Interactions between Type III receptor tyrosine phosphatases and growth factor receptor tyrosine kinases regulate tracheal tube formation in Drosophila. Biology Open. 2012;1:548–558. doi: 10.1242/bio.2012471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M, Zinn K. Receptor tyrosine phosphatases control tracheal tube geometries through negative regulation of Egfr signaling. Development. 2009;136:3121–3129. doi: 10.1242/dev.033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KG, Tenney AP, Ghose A, Duckworth AM, Higashi ME, Parfitt K, Marcu O, Heslip TR, Marsh JL, Schwarz TL, et al. The HSPGs Syndecan and Dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development. Neuron. 2006;49:517–531. doi: 10.1016/j.neuron.2006.01.026. [DOI] [PubMed] [Google Scholar]
- Kato K, Forero MG, Fenton JC, Hidalgo A. The glial regenerative response to central nervous system injury is enabled by pros-notch and pros-NFkappaB feedback. PLoS Biol. 2011;9:e1001133. doi: 10.1371/journal.pbio.1001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinrade EF, Hidalgo A. Lateral neuron--glia interactions steer the response of axons to the Robo code. Neuron Glia Biol. 2004;1:101–112. doi: 10.1017/s1740925x04000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurusu M, Cording A, Taniguchi M, Menon K, Suzuki E, Zinn K. A screen of cell-surface molecules identifies leucine-rich repeat proteins as key mediators of synaptic target selection. Neuron. 2008;59:972–985. doi: 10.1016/j.neuron.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurusu M, Zinn K. Receptor tyrosine phosphatases regulate birth orderdependent axonal fasciculation and midline repulsion during development of the Drosophila mushroom body. Molecular and cellular neurosciences. 2008;38:53–65. doi: 10.1016/j.mcn.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Wright AP, Zinn K. Live dissection of Drosophila embryos: streamlined methods for screening mutant collections by antibody staining. J Vis Exp. 2009 doi: 10.3791/1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren SE, Callahan CA, Thor S, Thomas JB. Control of neuronal pathway selection by the Drosophila LIM homeodomain gene apterous. Development. 1995;121:1769–1773. doi: 10.1242/dev.121.6.1769. [DOI] [PubMed] [Google Scholar]
- Matozaki T, Murata Y, Mori M, Kotani T, Okazawa H, Ohnishi H. Expression, localization, and biological function of the R3 subtype of receptor-type protein tyrosine phosphatases in mammals. Cell Signal. 2010;22:1811–1817. doi: 10.1016/j.cellsig.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Murata Y, Mori M, Kotani T, Supriatna Y, Okazawa H, Kusakari S, Saito Y, Ohnishi H, Matozaki T. Tyrosine phosphorylation of R3 subtype receptor-type protein tyrosine phosphatases and their complex formations with Grb2 or Fyn. Genes Cells. 2010;15:513–524. doi: 10.1111/j.1365-2443.2010.01398.x. [DOI] [PubMed] [Google Scholar]
- Nawroth R, Poell G, Ranft A, Kloep S, Samulowitz U, Fachinger G, Golding M, Shima DT, Deutsch U, Vestweber D. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. Embo J. 2002;21:4885–4895. doi: 10.1093/emboj/cdf497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oon SH, Hong A, Yang X, Chia W. Alternative splicing in a novel tyrosine phosphatase gene (DPTP4E) of Drosophila melanogaster generates two large receptor-like proteins which differ in their carboxyl termini. J Biol Chem. 1993;268:23964–23971. [PubMed] [Google Scholar]
- Palka HL, Park M, Tonks NK. Hepatocyte growth factor receptor tyrosine kinase met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J Biol Chem. 2003;278:5728–5735. doi: 10.1074/jbc.M210656200. [DOI] [PubMed] [Google Scholar]
- Patel NH. Imaging neuronal subsets and other cell types in whole-mount Drosophila embryos and larvae using antibody probes. Methods Cell Biol. 1994;44:445–487. doi: 10.1016/s0091-679x(08)60927-9. [DOI] [PubMed] [Google Scholar]
- Ruivenkamp CA, van Wezel T, Zanon C, Stassen AP, Vlcek C, Csikos T, Klous AM, Tripodis N, Perrakis A, Boerrigter L, et al. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet. 2002;31:295–300. doi: 10.1038/ng903. [DOI] [PubMed] [Google Scholar]
- Schonbaum CP, Organ EL, Qu S, Cavener DR. The Drosophila melanogaster stranded at second (sas) gene encodes a putative epidermal cell surface receptor required for larval development. Dev Biol. 1992;151:431–445. doi: 10.1016/0012-1606(92)90183-h. [DOI] [PubMed] [Google Scholar]
- Seeger M, Tear G, Ferres-Marco D, Goodman CS. Mutations affecting growth cone guidance in Drosophila: genes necessary for guidance toward or away from the midline. Neuron. 1993;10:409–426. doi: 10.1016/0896-6273(93)90330-t. [DOI] [PubMed] [Google Scholar]
- Shintani T, Ihara M, Sakuta H, Takahashi H, Watakabe I, Noda M. Eph receptors are negatively controlled by protein tyrosine phosphatase receptor type O. Nat Neurosci. 2006;9:761–769. doi: 10.1038/nn1697. [DOI] [PubMed] [Google Scholar]
- Sun Q, Bahri S, Schmid A, Chia W, Zinn K. Receptor tyrosine phosphatases regulate axon guidance across the midline of the Drosophila embryo. Development. 2000;127:801–812. doi: 10.1242/dev.127.4.801. [DOI] [PubMed] [Google Scholar]
- Sun Q, Schindelholz B, Knirr M, Schmid A, Zinn K. Complex genetic interactions among four receptor tyrosine phosphatases regulate axon guidance in Drosophila. Molecular and cellular neurosciences. 2001;17:274–291. doi: 10.1006/mcne.2000.0939. [DOI] [PubMed] [Google Scholar]
- Tarcic G, Boguslavsky SK, Wakim J, Kiuchi T, Liu A, Reinitz F, Nathanson D, Takahashi T, Mischel PS, Ng T, et al. An unbiased screen identifies DEP-1 tumor suppressor as a phosphatase controlling EGFR endocytosis. Curr Biol. 2009;19:1788–1798. doi: 10.1016/j.cub.2009.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GB, van Meyel DJ. The glycosyltransferase Fringe promotes Delta-Notch signaling between neurons and glia, and is required for subtype-specific glial gene expression. Development. 2007;134:591–600. doi: 10.1242/dev.02754. [DOI] [PubMed] [Google Scholar]
- Tian SS, Tsoulfas P, Zinn K. Three receptor-linked protein-tyrosine phosphatases are selectively expressed on central nervous system axons in the Drosophila embryo. Cell. 1991;67:675–685. doi: 10.1016/0092-8674(91)90063-5. [DOI] [PubMed] [Google Scholar]
- Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nature reviews. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- Vactor DV, Sink H, Fambrough D, Tsoo R, Goodman CS. Genes that control neuromuscular specificity in Drosophila. Cell. 1993;73:1137–1153. doi: 10.1016/0092-8674(93)90643-5. [DOI] [PubMed] [Google Scholar]
- Whiteford JR, Xian X, Chaussade C, Vanhaesebroeck B, Nourshargh S, Couchman JR. Syndecan-2 is a novel ligand for the protein tyrosine phosphatase receptor CD148. Mol Biol Cell. 2011;22:3609–3624. doi: 10.1091/mbc.E11-02-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winderlich M, Keller L, Cagna G, Broermann A, Kamenyeva O, Kiefer F, Deutsch U, Nottebaum AF, Vestweber D. VE-PTP controls blood vessel development by balancing Tie-2 activity. J Cell Biol. 2009;185:657–671. doi: 10.1083/jcb.200811159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodarz A, Hinz U, Engelbert M, Knust E. Expression of crumbs confers apical character on plasma membrane domains of ectodermal epithelia of Drosophila. Cell. 1995;82:67–76. doi: 10.1016/0092-8674(95)90053-5. [DOI] [PubMed] [Google Scholar]
- Wojtowicz WM, Wu W, Andre I, Qian B, Baker D, Zipursky SL. A vast repertoire of Dscam binding specificities arises from modular interactions of variable Ig domains. Cell. 2007;130:1134–1145. doi: 10.1016/j.cell.2007.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AP, Fox AN, Johnson KG, Zinn K. Systematic screening of Drosophila deficiency mutations for embryonic phenotypes and orphan receptor ligands. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XH, Seow KT, Bahri SM, Oon SH, Chia W. Two Drosophila receptorlike tyrosine phosphatase genes are expressed in a subset of developing axons and pioneer neurons in the embryonic CNS. Cell. 1991;67:661–673. doi: 10.1016/0092-8674(91)90062-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.