

Abstract
Background:
Chordomas of the skull base are rare locally aggressive neoplasms with a predilection for encapsulating critical neurovascular structures, bony destruction and irregular growth patterns, and from which patients succumb to recurrence and treatment failures.
Methods:
A review of the medical literature is performed, using standard search engines and identifying articles related to skull base chordomas, surgery, radiation therapy, chemotherapy, molecular genetics, and prospective trials.
Results:
A synthesis of the literature is presented, including sections on pathology, treatment, molecular genetics, challenges, and future directions.
Conclusion:
Beyond an understanding of the current treatment paradigms for skull base chordomas, the reader gains insight into the collaborative approach applied to orphan diseases, of which chordomas is a prime exemplar.
Keywords: Chordomas, cell lines, radiation therapy, review, skull base neoplasms, surgery
INTRODUCTION
Chordomas are rare, locally aggressive neoplasms thought to arise from notochordal remnants in the axial skeleton. In the skull base, they destroy bone, encapsulate critical neurovascular structures, and have irregular patterns of growth, rendering safe maximal surgical removal and effective radiation therapy challenging. In many ways, however, skull base chordomas are a paradigm for advancing progress in orphan diseases due to multi-institutional collaboration. In this article, we review the current treatment paradigms for cranial base chordomas, outline the molecular biology and potential therapeutic targets, and discuss future directions for such collaboration and research.
OVERVIEW
Chordomas occur with an annual age-adjusted rate of 0.02/100,000 person years and account for 0.1% of all reported brain tumors in the United States, 2004-2007.[13] The median survival based on Surveillance Epidemiology and End Results (SEER) data is 6.29 years,[49] although updated epidemiologic data suggest a significantly longer median survival longer in 1987 and later period compared with pre-1987.[48] Weighted mean 5-year overall and progression-free survival (PFS) are 78.4% and 50.8%, respectively, based on literature meta-analysis data.[23] Sacral and cranial locations are the most frequent and occur with a roughly equal proportion.[49]
Virchow in 1846 is credited with first describing the physaliphorous cell, however, he believed these tumors were derived from a cartilaginous overgrowth at the base of the skull, and termed them “ecchondrosis physaliphora spheno-occipitalis”.[95] It was Heinrich Müller in 1858 who rejected Virchow's theory of cartilaginous origin, and asserted correctly that chordomas arose from remnants of the primitive notochord, and termed these tumors “ecchordosis physaliphora spheno-occipitalis”.[51] There have since been a number of indirect lines of evidence linking chordomas to the primitive notochord. Salisbury et al. noted morphologic patterns of arrangement of the cranial and caudal ends of the notochord, which they postulated could predispose to failure of involution and development of chordoma.[72] Similarly, intraosseous benign notochordal cell tumors follow a similar distribution as classic chordomas, suggesting a common origin.[20,98] Finally, transcription factor T, which is crucial for notochordal development, frequently demonstrates copy number variations in both sporadic and familial chordomas.[65,102]
Histologically, three types of chordoma are recognized by the World Health Organization: Classic chordoma, chondroid chordoma, and dedifferentiated chordoma.[50] Whereas classic chordoma displays typical physaliphorous cells [Figure 1], chondroid chordomas exhibit typical chordoma architecture with areas of cartilaginous hyaline stroma,[34] but with positive cytokeratin and epithelial membrane antigen immunoreactivity. Dedifferentiated chordomas are very rare and have been reported primarily in sacrococcygeal chordomas and in pediatric patients;[90] their histologic appearance is that of a high-grade spindle cell sarcoma.[15]
Figure 1.
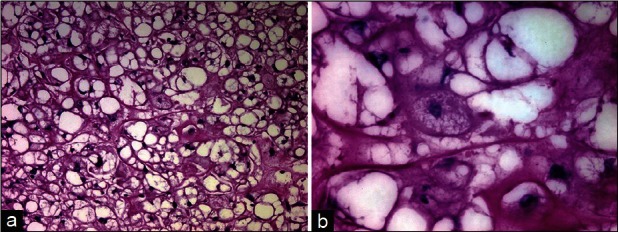
Histologic specimen stained with hematoxylin and eosin stain demonstrating the classic lobular architecture of chordoma, ×10 (a), composed of physaliphorous cells, ×60 (b)
Systemic metastases may occur in as many as 30% of patients,[14,26] although they tend to be more frequent in sacral than skull base chordomas.[94] The frequency of metastasis from all chordomas is substantially higher in the very young.[6] Drop metastases are also known to occur.[47,93] In the skull base, iatrogenic seeding of chordoma tissue along the surgical trajectory, including at the site of abdominal fat graft harvest, has been well-documented.[3]
TREATMENT
Surgery
There is compelling evidence that en bloc surgical excision of chordomas confers long-term recurrence-free survival, based on experience with spinal column tumors.[7,99] Boriani et al. documented in their series that all patients undergoing intralesional resection in spinal chordomas recurred within 2 years, compared with 12/18 patients with en bloc resection who remained disease-free after a mean follow-up of 8 years.[7] In a systematic review of the literature, 119 primary chordomas of the spine undergoing en bloc surgical resection had a 5-year recurrence-free survival of 58.5%. The median time to recurrence was 94 months with negative surgical margins, compared with 50 months if the margins were positive.[16]
The vast majority of cranial base chordomas are not amenable to strict en bloc oncologic resection; instead an intralesional resection toward normal appearing bony margins is typically performed in order to avoid neurovascular complications. Nevertheless, maximal safe resection of skull base chordomas is generally advocated, although there has been limited evidence supporting this practice. In a recent meta-analysis of the literature over the past 10 years, 23 studies incorporating 807 patients were combined to analyze the effect of complete resection on 5-year overall and PFS.[23] Complete resection conferred a higher 5-year PFS than incomplete resection based on a random effects model (mean difference in PFS 20.7%; 95% CI 6.57-34.91%). Patients with incomplete resection were 3.83 times more likely to experience a recurrence (95% CI 1.63-9.00) and 5.85 times more likely to die (95% CI 1.40-24.5) at 5 years versus patients with complete resection. In patients with subtotal resection, smaller residual tumor volume in the range of 25-30 cm3 seems to confer a similar rate of local control with adjunctive radiation therapy.[2,38,63] In one study of 42 patients with skull base chordomas, a gross tumor volume ≤25 mL was associated with excellent local control using spot-scanning-based proton radiotherapy.[2]
Achieving aggressive removal of chordomas largely provided the impetus for pioneers in skull base surgery to develop and refine open approaches to the central skull base.[22] These include the extended sub frontal, frontotemporal orbitozygomatic transcavernous, subtemporal-infratemporal, extreme lateral transcondylar, posterior transpetrosal, subtemporal-transzygomatic, and Le Fort I transmaxillary approaches.[71] Depending on the extent of disease, in certain cases staged approaches are necessary to safely maximize surgical resection. In the largest surgical series of cranial base chordomas to date,[21] 95 patients with skull base chordomas were treated from 1988 to 2011; 5-year overall and recurrence-free survival were 74% ± 6% and 56% ± 8%. In 39 patients who were treated from 2000 to 2011, with a 70.5% rate of complete resection. In another large series by Sen et al.,[78] complete resection was achieved in 58% of 71 patients, with a 5-year overall survival rate of 75%. The degree of surgical resection appeared to be most important determinant of survival, based on a multiple Cox proportional hazard model.
Recently a number of centers have published short-term data regarding endoscopic endonasal resection of cranial base chordomas.[19,30,32,36,84,86] In chordomas affecting primarily the midline clivus and without significant lateral extension, an anterior trajectory via the endonasal endoscopic ventral approach is appealing. In a recent systematic review,[41] 639 patients across 26 studies who underwent open resection of skull base chordomas were compared with 16 studies and 127 patients in whom an endoscopic resection was performed. Mean tumor volume was lower in the endoscopic cohort, and there were decreased rates of petrous bone and dural invasion as well. In this carefully selected endoscopic group, the rate of complete resection was 61.0%, with a lower rate of cranial nerve deficits, meningitis, and mortality compared with the open group. Due to the short follow-up in the endoscopic group, no progression-free or overall survival data was compiled. Nevertheless, this early data suggests that for well selected chordomas, particularly those which are smaller and/or affecting the midline clivus, an endoscopic approach can achieve a similar rate of complete resection with low associated morbidity.
Radiation therapy
Despite an apparent benefit to maximal surgical removal of skull base chordomas, recurrence without adjuvant therapy remains high.[92] Radiation therapy is typically used for residual tumor following surgery, as well as for unresectable tumors or recurrent cases. The evidence supporting the use of radiation therapy following complete surgical removal is somewhat more debated,[39] however, is often nevertheless recommended and implemented.
Chordomas are radiosensitive tumors, with a clearly established dose-response relationship and effective dose above 65 Gy.[60] Furthermore, these tumors typically abut critical structures such as the optic apparatus and brainstem, which need to be carefully spared during dosimetric planning. In light of the higher dose required, conformal radiation therapy, in particular proton beam radiation, has been the mainstay of adjuvant therapy following surgery. Proton beam therapy offers several theoretical advantages over photon-based radiation, including an increased linear energy transfer in the Bragg peak region, and rapid dose fall-off.[52] A typical radiotherapy dose is 74 Cobalt Gray equivalents (CGE) to the planning treatment volume, while limiting doses to the brainstem surface and optic apparatus to 60 CGE or less.[17,87] The 5-year recurrence-free survival using proton-based therapy ranges from 59% to 73%.[12,38,52] Similar to proton beam therapy, carbon ions offer similar advantages relative to photon-based treatment; however, carbon ion therapy may have a number of biological differences in vivo, including a higher relative biological effectiveness and reduced oxygen-enhancement ratio in the tumor region.[25] This may be advantageous in chordomas, whose level of proliferation in vitro is enhanced in hypoxic conditions.[59] The 3 year recurrence-free survival for skull base chordomas treated at carbon ion facilities in Germany and Japan ranged from 70.0% to 80.6%.[75,85] There is currently an ongoing monocentric randomized clinical trial of proton versus carbon radiation therapy in newly diagnosed patients with chordoma of the skull base.[54]
Proton and/or carbon ion therapy, however, remains considerably more expensive than photon treatment, particularly in terms of investment cost,[61] and is less available in many parts of the world. Delivery of newer photon-based radiotherapy techniques, especially intensity-modulated radiation therapy (IMRT), has produced encouraging results in skull base chordomas, either alone or in combination with heavy particle therapy.[31,91] For smaller sized tumors, stereotactic radiosurgery may also play a role, and early results have suggested comparable outcomes to other radiation modalities for residual or recurrent cases.[40]
MOLECULAR GENETICS
With advances in molecular biology and sequencing capability, the field of molecularly targeted therapeutics has expanded with the development of drugs inhibiting pathways necessary for uncontrolled cell proliferation [Figure 2]. Due to chordoma's relatively rare occurrence, knowledge of molecular targets specific to chordoma is limited and an area of ongoing research, but current discoveries have provided promising possibilities to be exploited as treatment options.
Figure 2.
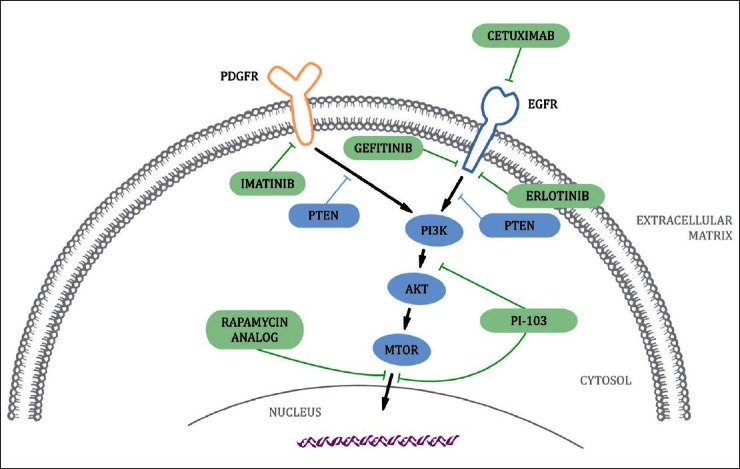
Epidermal growth factor receptor and platelet-derived growth factor receptor signaling pathways in chordoma mediated by tyrosine kinases and downstream effectors. These molecules are potential therapeutic targets of targeted inhibition by cetuximab, erlotinib, gefitinib, imatinib, PI-103, and rapamycin analogs
Brachyury
One such potential therapeutic target is the brachyury (T) gene, which codes for a transcription factor that is uniquely expressed in chordoma cells. Brachyury plays an important role during the development of the notochord in embryos but later remains unexpressed in normal tissue.[81] However, it has been shown that brachyury expression is uncommonly high exclusively in chordoma cells,[96] allowing it to become the differential between chordoma and other neoplasms with similar location and histology.[70,77]
A number of different mechanisms have been suggested to be the cause of brachyury expression in chordoma. It was demonstrated, in a familial chordoma study involving four families, to be due to duplication of a region in 6q27 containing the T gene.[101] In contrast, sporadic chordomas have been evidenced to have a much lower occurrence of copy number gain (CNG) of the T gene, with only 16 of 236 cases positive for T amplification in three separate cohorts,[43,65,80] supporting the view that CNG is not the cause of brachyury expression in the majority of chordomas. Instead, epigenetic factors may be behind the abnormal presence of brachyury in sporadic chordoma,[43] but further research is required to provide a more thorough understanding. Moreover, a common nonsynonymous single nucleotide polymorphism (SNP) (rs2305089) in brachyury was recently found to strongly associated with chordoma development.[62]
In addition to being an identifying marker for chordoma, brachyury also plays an important role in its pathogenesis. It was observed that when the brachyury gene was silenced in chordoma cell line JHC7, cells became more differentiated, displaying senescence and complete growth arrest and were unable to be passaged in vitro, indicating that without brachyury expression, they lose their tumorigenic capabilities. Similarly, brachyury knockdown in chordoma cell line UCH-1 resulted in decreased cell proliferation and senescent appearance.[65] Although the specifics of its involvement remains unexplained, it is evident the expression of brachyury is strongly implicated in the oncogenesis of chordoma.
Since silencing of brachyury results in the interruption of cell growth in vitro, it presents itself as an ideal candidate as a molecular target for therapy. Moreover, targeted silencing of brachyury expression results in the abrogation of transcriptional activation of numerous downstream genes. What makes it an attractive target is that it is only expressed in tumor cells, meaning if it is targeted, the cancer cells will be preferentially affected while leaving the normal cells with little to no adverse side effects. With the lower chance of toxicity to normal cells in combination with the antitumourigenic qualities from its inhibition, brachyury targeting has great potential for the development of a molecular therapy for chordoma patients.
Receptor tyrosine kinases
Receptor tyrosine kinase (RTK) inhibitory drugs for cancer have experienced much advancement in recent years, with the sharp increase in our ability to sequence, and thus detect mutations in, RTK genes in tumor DNA. RTKs are proteins on the cell surface that interact with extracellular ligands to become activated and dimerize, which triggers phosphorylation of a downstream signaling protein and initiates a signaling pathway ultimately leading to a transcriptional change. In cancer cells, the transcriptional change most often gives rise to angiogenesis, cell proliferation or antiapoptosis, which all contribute to tumorigenesis. When an RTK is hyperactivated, its associated pathway also becomes constitutively active as well, leading to loss of control over cell proliferation and cancerous cell growth. The RTK can become hyperactive as a result of autocrine/paracrine loops, ligand or receptor overexpression, or gain of gene mutations.
One commonly targeted RTK in many cancers is the epidermal growth factor receptor (EGFR), which binds a number of ligands including EGF and transforming growth factor alpha (TGF-α). A significant portion of chordomas have EGFR overexpression that causes increased initiation of cell proliferation, leading to higher degree of aggressive behavior of the tumor. This has been evidenced in a study where 69% of cases expressed EGFR and close to 40% of the chordomas tested had amplification of the gene.[79] Likewise, three other studies have seen a cumulative 43 of 54 cases positive for EGFR expression,[29,66,97] indicating that expression is frequently deregulated in chordoma.
Targeting EGFR in chordoma has only just begun, with four studies conducted thus far. The first (Hof et al.[35] ) saw improvement in patients treated with a combination of cetuximab and gefitinib, both of which are EGFR inhibitors. Following the success seen in the first patient case, the same regimen of cetuximab and gefitinib was also administered in a patient with a recurrent chordoma and likewise regression of the tumor as well as a rapid improvement of the patient's neurological function was observed.[44] Two cases of treatment with an alternative EGFR inhibitor, erlotinib, also showed significant response.[42,81] The successes seen with these EGFR inhibitors suggest that targeting EGFR in chordoma is an effective method of treatment that should be incorporated into the arsenal of therapies available for chordoma patients.
Another promising target therapy involves the platelet-derived growth factor receptor (PDGFR). Similar to EGFR, this RTK can display oncogenic properties when hyperactivated, initiating cell proliferation and growth via the PI3K/AKT, RAS/ERK, or STAT pathways. Of the two different types of PDGFR, α and β, overexpression of the β variant is particularly common in chordomas[10,29,57,86] implicating it as an important driver for tumorigenesis. In particular, preferential PDGFR overexpression in the stromal component of tumor tissue with only diffuse expression in neoplastic cells was observed,[57] demonstrating that PDGFR signaling is most significant in the supporting structure of the tumor. Thus, PDGFR targeting therapies may lead to different responses depending on the stromal content in each chordoma case.
Trials of imatinib treatment, a PDGFR inhibitor, on chordoma patients first began in 2004 with a compassionate trial for six patients, of whom four were positive for PDGFR expression.[10] When response to the treatment was observed, the trial was increased to 18 patients and preliminary results were reported.[86] Patients experienced nondimensional responses including decreased cellular density and less contrast enhancement on computed tomography (CT) scan. A minority of the patients also showed dimensional responses with decreased tumor size in magnetic resonance imaging (MRI) and positron emission tomography (PET) scan. Overall, tumors were observed to be responsive to imatinib for nearly 1 year, which is optimistic considering all cases were well advanced. When imatinib was tested in a Phase II trial of 56 chordoma cases, results were similar in that few dimensional responses were observed, with only 9 patients (20%) experiencing minor dimensional changes, but with an overall clinical benefit rate of 64%, and 70% of patients exhibiting stable disease.[83] A complication in an especially bulky tumor was found where imatinib treatment caused the tumor to undergo liquefaction, which led to septic complications and ultimately patient death.[11]
From the results observed in the imatinib trials, patients’ disease progression halted for a period of approximately 9 months, accompanied with symptomatic improvement even in cases with no volumetric response. Particularly in the very advanced chordoma cases, where survival was predicted to be less than 1 year, the benefit of imatinib is evident. However, the limitations of this treatment are also exhibited clearly. Only a minority of cases showed decrease in tumor size, indicating that it does little in terms of dimensional response, reflecting on its inability to decrease cell numbers. This may be due to the preferential overexpression of PDGFR in stromal cells, thereby reducing the variety of tumor cells that are affected by the treatment. Nevertheless, benefits have been observed in patient cases thus far, demonstrating the effectiveness of imatinib overall as an antitumor therapy. Further study of imatinib in vivo will help in determining the extent of its efficacy in chordoma.
Downstream pathways
While RTK inhibitors have proven to be useful target therapies, the immense diversity of receptors in a cell makes it difficult to control cell growth through the inhibition of a single RTK. Many cancers possess multiple oncogenic mutated RTKs. If a particular RTK is inhibited, its function can be replaced by the increased activity of a different RTK able to activate the same downstream signaling pathway, thus leading to the onset of resistance to the RTK inhibitor. This drug resistance has been observed in various cancers and likewise in chordoma. Targeting signaling structures downstream in the pathway can overcome the problem of one RTK making up for the inhibition of another due to the fact that many of the RTK activated pathways converge into a common one downstream.
One such pathway that receives signals from a variety of different RTKs is the phosphoinositide-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway. This pathway is often found to be constitutively activated in cancers because it is a mechanism that cells use to control proliferation, differentiation, and apoptosis. Ungoverned activation of the pathway is caused by an amalgamation of various mutations, including down regulation of normally present suppressor proteins, hyperactivity of its associated RTKs, or an inability to deactivate constituents via the proper feedback loop. As a result of deregulation of this pathway, cells undergo unrestrained growth, leading to cancer and tumor formation.
In human chordoma, activation of this pathway has been observed. The majority of a 13 chordoma sample study had unusually high levels of activated AKT and mTOR,[76] and of another 50 chordoma cases, 92% and 27% were positive for presence of the two, respectively.[64] In addition, Han et al. observed that not only were AKT and mTOR constantly activated, but also that the regulator Phosphatase and tensin homolog ( PTEN), which normally represses the activation of PI3K, was not detected, or was significantly reduced, in the chordoma samples.[33] It is evident from its ubiquity that the continual activation of the PI3K/AKT/mTOR pathway plays an important role in the pathogenesis of chordoma.
The PI3K/AKT/mTOR pathway has been targeted mainly through the inhibition of mTOR with rapamycin and its analogs. mTOR is targeted by rapamycin, as is apparent in its name, causing growth arrest in the early G1 phase and inhibiting translation initiation.[5,74] As a result, rapamycin and analogs such as CC1-779 have been tested in many different cancers.[27] However, a limitation of targeting activated mTOR with rapamycin became clear when rapamycin and analog treated cells began to display buildup in levels of activated AKT that resulted from the disrupted overall pathway.[56] In order to remedy this unwanted increase in phosphorylated AKT, another rapamycin analog with dual inhibitory effects, PI-103, was developed and tested on gliomas, exhibiting proliferative arrest.[28] PI-103 is an effective inhibitory drug because not only does it target mTOR, but also its upstream partner PI3K. As a result, increased levels of phosphorylated AKT are avoided, and improved therapeutic effectiveness is possible. When PI-103 was tested in chordoma cell line UCH-1, PI3K and mTOR inhibition, growth disruption and consequent apoptosis were observed.[76] Although it is evident that PI-103 fulfills the requirements for a potential molecular therapy, further in vivo studies must be conducted before drug trials on chordoma patients can begin.
CHALLENGES AND FUTURE DIRECTIONS
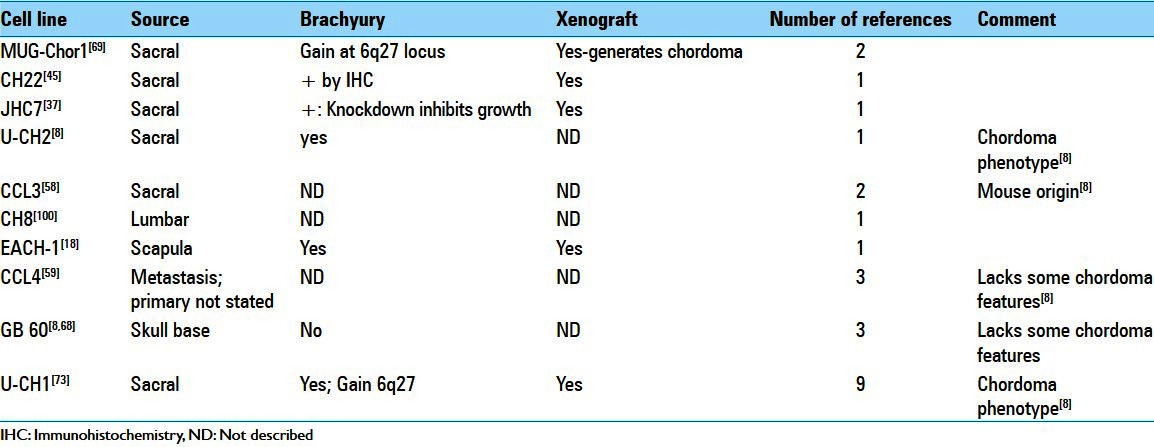
The historical lack of major progress in improving outcomes for chordoma patients stems both from its rarity and difficulties in adapting standard paradigms to its basic scientific investigation. Chordoma is a recognized rare (or orphan) disease,[67] a fact which impacts acquisition of observational data and administration of clinical trials powered to provide meaningful outcomes analysis. The rarity of chordoma also impairs acquisition of research funding as more prevalent cancers take priority. Adding to the roadblocks associated with its orphan status, biological factors also impact basic science investigation and clinical translation. Chordoma cells are slow growing and cell lines have proven extremely difficult to establish. The resulting paucity of available cell lines significantly impedes basic investigation and development of informative animal models required to facilitate preclinical testing of new therapies. The recognition of this shortcoming has stimulated an increase in the number of chordoma cell lines reported in the literature [Table 1][8,18,37,45,58,59,68,69,73,100] including one with cancer stem-like characteristics.[4,46] However, questions raised about the chordoma phenotype of several presumed chordoma cell lines[8] highlights the need for more investigation of phenotypic drift in culture and the generation of a consensus biomarker panel for designating bona fide chordoma cell lines. An additional interesting observation is the fact that the majority of reported chordoma cell lines are of sacral origin. Whether this reflects true differences in biological behavior between sacral and skull base chordoma that affect their adaptation to cell culture or is simply a result of bias in acquisition is a puzzling question requiring additional investigation. As demonstrated in the case of brachyury,[65,102] continued molecular characterization of chordomas is anticipated to reveal additional targets for existing drugs or new therapies.[9,24,53] Finally, driven by these advances, the ongoing development and refinement of animal xenograft models[45,65,82] is expected to provide a platform for basic investigation of chordoma biology and preclinical system to test therapeutic approaches. Currently no genetically engineered mouse models of chordoma exists but would be of great value for understanding the cellular basis and molecular mechanisms driving chordoma formation, malignancy, and progression.
Table 1.
Overview of chordoma cell lines
In response to the needs of orphan diseases such as chordoma, the National Institutes of Health (NIH) created the Office of Rare Diseases Research (ORDR)[55] and Therapeutics for Rare and Neglected Diseases (TRND).[89] However, currently only three extramurally supported NIH research projects of direct relevance to chordoma. In an effort to address ongoing deficiencies and accelerate progress in chordoma therapy, the Chordoma Foundation was cofounded in 2007 by Josh and Simone Sommer.[88] Among its many initiatives, the foundation maintains a central repository of chordoma biospecimens that are then distributed to support research studies abroad. The foundation has held three international research workshops thus far since its inception and supported research enabling the development of in vitro cell lines and animal models, screening of potential drugs to be implemented in clinical trials, genomic sequencing of chordoma cell lines, and seed funding for chordoma-related research grants and publications. By recognizing the roadblocks to progress and galvanizing the research and clinical communities, this "grass roots" organization has laid the groundwork required to realize meaningful improvement in outcomes for chordoma patients. Lastly, AOSpine, the international spine surgery organization, recently began a primary spine tumor biobanking initiative (PST BioNet) to facilitate international collaboration and accumulation of rare surgical specimens that include chordomas.[1] These efforts will certainly accelerate and promote chordoma research. To continue along this positive trajectory, it is imperative for neurosurgeons who treat chordoma patients to actively participate in multi-institutional registries, studies, and clinical trials.
CONCLUSION
As a result of their relative rarity and unfavorable location, the management of cranial base chordomas remains challenging. Refinements in open and endoscopic skull base approaches and improvements in stereotactic radiotherapy and radiosurgery techniques have facilitated local control and survival for new cases as well as at the time of recurrence. Nevertheless, many patients with chordoma ultimately recur and succumb to the disease. Through an increasing knowledge of tumor genetics and molecular biology, advances in laboratory techniques, new in vitro cell lines and xenograft models, preclinical screening for potential therapeutic agents, and collaboration between centers, an improved understanding of chordoma biology that can be translated into treatment paradigms will hopefully occur.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2013/4/1/72/112822
Disclaimer: The authors of this article have no conflicts of interest to disclose, and have adhered to SNI's policies regarding human/animal rights, and informed consent. Advertisers in SNI did not ask for, nor did they receive access to this article prior to publication
Contributor Information
Salvatore Di Maio, Email: sdimaio@jgh.mcgill.ca.
Esther Kong, Email: esthersokong@gmail.com.
Stephen Yip, Email: Stephen.yip@vch.ca.
Robert Rostomily, Email: rosto@u.washington.edu.
REFERENCES
- 1.AOSPine Knowledge Forum. [Last accessed on 2013 Mar 1]. Available from: https://aospine.aofoundation.org/Structure/research/knowledge-forum/Pages/knowledge-forum.aspx .
- 2.Ares C, Hug EB, Lomax AJ, Bolsi A, Timmermann B, Rutz HP, et al. Effectiveness and safety of spot scanning proton radiation therapy for chordomas and chondrosarcomas of the skull base: First long-term report. Int J Radiat Oncol Biol Phys. 2009;75:1111–8. doi: 10.1016/j.ijrobp.2008.12.055. [DOI] [PubMed] [Google Scholar]
- 3.Arnautovic KI, Al-Mefty O. Surgical seeding of chordomas. J Neurosurg. 2001;95:798–803. doi: 10.3171/jns.2001.95.5.0798. [DOI] [PubMed] [Google Scholar]
- 4.Aydemir E, Bayrak OF, Sahin F, Atalay B, Kose GT, Ozen M, et al. Characterization of cancer stem-like cells in chordoma. J Neurosurg. 2012;116:810–20. doi: 10.3171/2011.12.JNS11430. [DOI] [PubMed] [Google Scholar]
- 5.Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell. 1996;7:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borba LA, Al-Mefty O, Mrak RE, Suen J. Cranial chordomas in children and adolescents. J Neurosurg. 1996;84:584–91. doi: 10.3171/jns.1996.84.4.0584. [DOI] [PubMed] [Google Scholar]
- 7.Boriani S, Bandiera S, Biagini R, Bacchini P, Boriani L, Cappuccio M, et al. Chordoma of the mobile spine: Fifty years of experience. Spine (Phila Pa 1976) 2006;31:493–503. doi: 10.1097/01.brs.0000200038.30869.27. [DOI] [PubMed] [Google Scholar]
- 8.Bruderlein S, Sommer JB, Meltzer PS, Li S, Osada T, Ng D, et al. Molecular characterization of putative chordoma cell lines. Sarcoma. 2010;2010:630129. doi: 10.1155/2010/630129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bydon M, Papadimitriou K, Witham T, Wolinsky JP, Bydon A, Sciubba D, et al. Novel therapeutic targets in chordoma. Expert Opin Ther Targets. 2012;16:1139–43. doi: 10.1517/14728222.2012.714772. [DOI] [PubMed] [Google Scholar]
- 10.Casali PG, Messina A, Stacchiotti S, Tamborini E, Crippa F, Gronchi A, et al. Imatinib mesylate in chordoma. Cancer. 2004;101:2086–97. doi: 10.1002/cncr.20618. [DOI] [PubMed] [Google Scholar]
- 11.Casali PG, Stacchiotti S, Sangalli C, Olmi P, Gronchi A. Chordoma. Curr Opin Oncol. 2007;19:367–70. doi: 10.1097/CCO.0b013e3281214448. [DOI] [PubMed] [Google Scholar]
- 12.Castro JR, Linstadt DE, Bahary JP, Petti PL, Daftari I, Collier JM, et al. Experience in charged particle irradiation of tumors of the skull base: 1977-1992. Int J Radiat Oncol Biol Phys. 1994;29:647–55. doi: 10.1016/0360-3016(94)90550-9. [DOI] [PubMed] [Google Scholar]
- 13.CBTRUS. CBTRUS Statistical Report: Primary brain and central nervous system tumors diagnosed in the United States in 2004-2007. Source: Central Brain Tumor Registry of the United States. 2011. [Last accessed on 2013 Mar 1]. Available from: http://www.cbtrus.org .
- 14.Chambers PW, Schwinn CP. Chordoma. A clinicopathologic study of metastasis. Am J Clin Pathol. 1979;72:765–76. doi: 10.1093/ajcp/72.5.765. [DOI] [PubMed] [Google Scholar]
- 15.Chugh R, Tawbi H, Lucas DR, Biermann JS, Schuetze SM, Baker LH. Chordoma: The nonsarcoma primary bone tumor. Oncologist. 2007;12:1344–50. doi: 10.1634/theoncologist.12-11-1344. [DOI] [PubMed] [Google Scholar]
- 16.Cloyd JM, Acosta FL, Jr, Polley MY, Ames CP. En bloc resection for primary and metastatic tumors of the spine: A systematic review of the literature. Neurosurgery. 2010;67:435–44. doi: 10.1227/01.NEU.0000371987.85090.FF. [DOI] [PubMed] [Google Scholar]
- 17.Debus J, Hug EB, Liebsch NJ, O’Farrel D, Finkelstein D, Efird J, et al. Brainstem tolerance to conformal radiotherapy of skull base tumors. Int J Radiat Oncol Biol Phys. 1997;39:967–75. doi: 10.1016/s0360-3016(97)00364-7. [DOI] [PubMed] [Google Scholar]
- 18.DeComas AM, Penfornis P, Harris MR, Meyer MS, Pochampally RR. Derivation and characterization of an extra-axial chordoma cell line (EACH-1) from a scapular tumor. J Bone Joint Surg Am. 2010;92:1231–40. doi: 10.2106/JBJS.I.00594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dehdashti AR, Karabatsou K, Ganna A, Witterick I, Gentili F. Expanded endoscopic endonasal approach for treatment of clival chordomas: Early results in 12 patients. Neurosurgery. 2008;63:299–307. doi: 10.1227/01.NEU.0000316414.20247.32. [DOI] [PubMed] [Google Scholar]
- 20.Deshpande V, Nielsen GP, Rosenthal DI, Rosenberg AE. Intraosseous benign notochord cell tumors (BNCT): Further evidence supporting a relationship to chordoma. Am J Surg Pathol. 2007;31:1573–7. doi: 10.1097/PAS.0b013e31805c9967. [DOI] [PubMed] [Google Scholar]
- 21.Di Maio S, Rostomily R, Sekhar LN. Current surgical outcomes for cranial base chordomas: Cohort study of 95 patients. Neurosurgery. 2012;70:1355–60. doi: 10.1227/NEU.0b013e3182446783. [DOI] [PubMed] [Google Scholar]
- 22.Di Maio S, Sekhar LN. Skull Base Approaches. In: Ellenbogen RG, Abdulrauf SI, editors. Principles of Neurological Surgery: Expert Consult-Online and Print. Elsevier-Health Sciences Division; 2012. [Google Scholar]
- 23.Di Maio S, Temkin N, Ramanathan D, Sekhar LN. Current comprehensive management of cranial base chordomas: 10-year meta-analysis of observational studies. J Neurosurg. 2011;115:1094–105. doi: 10.3171/2011.7.JNS11355. [DOI] [PubMed] [Google Scholar]
- 24.Diaz RJ, Guduk M, Romagnuolo R, Smith CA, Northcott P, Shih D, et al. High-resolution whole-genome analysis of skull base chordomas implicates FHIT loss in chordoma pathogenesis. Neoplasia. 2012;14:788–98. doi: 10.1593/neo.12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durante M, Loeffler JS. Charged particles in radiation oncology. Nat Rev Clin Oncol. 2010;7:37–43. doi: 10.1038/nrclinonc.2009.183. [DOI] [PubMed] [Google Scholar]
- 26.Eriksson B, Gunterberg B, Kindblom LG. Chordoma. A clinicopathologic and prognostic study of a Swedish national series. Acta Orthop Scand. 1981;52:49–58. doi: 10.3109/17453678108991758. [DOI] [PubMed] [Google Scholar]
- 27.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–88. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 28.Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, et al. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007;67:7960–5. doi: 10.1158/0008-5472.CAN-07-2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fasig JH, Dupont WD, LaFleur BJ, Olson SJ, Cates JM. Immunohistochemical analysis of receptor tyrosine kinase signal transduction activity in chordoma. Neuropathol Appl Neurobiol. 2008;34:95–104. doi: 10.1111/j.1365-2990.2007.00873.x. [DOI] [PubMed] [Google Scholar]
- 30.Fatemi N, Dusick JR, Gorgulho AA, Mattozo CA, Moftakhar P, De Salles AA, et al. Endonasal microscopic removal of clival chordomas. Surg Neurol. 2008;69:331–8. doi: 10.1016/j.surneu.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 31.Feuvret L, Noel G, Weber DC, Pommier P, Ferrand R, De Marzi L, et al. A treatment planning comparison of combined photon-proton beams versus proton beams-only for the treatment of skull base tumors. Int J Radiat Oncol Biol Phys. 2007;69:944–54. doi: 10.1016/j.ijrobp.2007.07.2326. [DOI] [PubMed] [Google Scholar]
- 32.Frank G, Sciarretta V, Calbucci F, Farneti G, Mazzatenta D, Pasquini E. The endoscopic transnasal transsphenoidal approach for the treatment of cranial base chordomas and chondrosarcomas. Neurosurgery. 2006;59(1 Suppl 1):ONS50–7. doi: 10.1227/01.NEU.0000219914.17221.55. [DOI] [PubMed] [Google Scholar]
- 33.Han S, Polizzano C, Nielsen GP, Hornicek FJ, Rosenberg AE, Ramesh V. Aberrant hyperactivation of akt and Mammalian target of rapamycin complex1 signaling in sporadic chordomas. Clin Cancer Res. 2009;15:1940–6. doi: 10.1158/1078-0432.CCR-08-2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heffelfinger MJ, Dahlin DC, MacCarty CS, Beabout JW. Chordomas and cartilaginous tumors at the skull base. Cancer. 1973;32:410–20. doi: 10.1002/1097-0142(197308)32:2<410::aid-cncr2820320219>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 35.Hof H, Welzel T, Debus J. Effectiveness of cetuximab/gefitinib in the therapy of a sacral chordoma. Onkologie. 2006;29:572–4. doi: 10.1159/000096283. [DOI] [PubMed] [Google Scholar]
- 36.Hong Jiang W, Ping Zhao S, Hai Xie Z, Zhang H, Zhang J, Yun Xiao J. Endoscopic resection of chordomas in different clival regions. Acta Otolaryngol. 2009;129:71–83. doi: 10.1080/00016480801995404. [DOI] [PubMed] [Google Scholar]
- 37.Hsu W, Mohyeldin A, Shah SR, ap Rhys CM, Johnson LF, Sedora-Roman NI, et al. Generation of chordoma cell line JHC7 and the identification of Brachyury as a novel molecular target. J Neurosurg. 2011;115:760–9. doi: 10.3171/2011.5.JNS11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hug EB, Loredo LN, Slater JD, DeVries A, Grove RI, Schaefer RA, et al. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J Neurosurg. 1999;91:432–9. doi: 10.3171/jns.1999.91.3.0432. [DOI] [PubMed] [Google Scholar]
- 39.Jian BJ, Bloch OG, Yang I, Han SJ, Aranda D, Parsa AT. A comprehensive analysis of intracranial chordoma and survival: A systematic review. Br J Neurosurg. 2011;25:446–53. doi: 10.3109/02688697.2010.546896. [DOI] [PubMed] [Google Scholar]
- 40.Kano H, Iqbal FO, Sheehan J, Mathieu D, Seymour ZA, Niranjan A, et al. Stereotactic radiosurgery for chordoma: A report from the North American Gamma Knife Consortium. Neurosurgery. 2011;68:379–89. doi: 10.1227/NEU.0b013e3181ffa12c. [DOI] [PubMed] [Google Scholar]
- 41.Komotar RJ, Starke RM, Raper DM, Anand VK, Schwartz TH. The endoscope-assisted ventral approach compared with open microscope-assisted surgery for clival chordomas. World Neurosurg. 2011;76:318–27. doi: 10.1016/j.wneu.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 42.Launay SG, Chetaille B, Medina F, Perrot D, Nazarian S, Guiramand J, et al. Efficacy of epidermal growth factor receptor targeting in advanced chordoma: Case report and literature review. BMC cancer. 2011;11:423. doi: 10.1186/1471-2407-11-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le LP, Nielsen GP, Rosenberg AE, Thomas D, Batten JM, Deshpande V, et al. Recurrent chromosomal copy number alterations in sporadic chordomas. PloS one. 2011;6:e18846. doi: 10.1371/journal.pone.0018846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Linden O, Stenberg L, Kjellen E. Regression of cervical spinal cord compression in a patient with chordoma following treatment with cetuximab and gefitinib. Acta Oncol. 2009;48:158–9. doi: 10.1080/02841860802266672. [DOI] [PubMed] [Google Scholar]
- 45.Liu X, Nielsen GP, Rosenberg AE, Waterman PR, Yang W, Choy E, et al. Establishment and characterization of a novel chordoma cell line: CH22. J Orthop Res. 2012;30:1666–73. doi: 10.1002/jor.22113. [DOI] [PubMed] [Google Scholar]
- 46.Lohberger B, Rinner B, Stuendl N, Absenger M, Liegl-Atzwanger B, Walzer SM, et al. Aldehyde dehydrogenase 1, a potential marker for cancer stem cells in human sarcoma. PloS One. 2012;7:e43664. doi: 10.1371/journal.pone.0043664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin MP, Olson S. Intradural drop metastasis of a clival chordoma. J Clin Neurosci. 2009;16:1105–7. doi: 10.1016/j.jocn.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 48.McMaster M. Update on the Epidemiology of chordoma: SEER registry data 1973-2007. Bethesda: Third International Chordoma Research Workshop; 2011. [Google Scholar]
- 49.McMaster ML, Goldstein AM, Bromley CM, Ishibe N, Parry DM. Chordoma: Incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control. 2001;12:1–11. doi: 10.1023/a:1008947301735. [DOI] [PubMed] [Google Scholar]
- 50.Mirra JM, Nelson SD, Della Rocca C, Mertens F. Chordoma. In: Fletcher DM, Unni K, Mertens F, editors. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2002. pp. 316–7. [Google Scholar]
- 51.Müller H. Ueber das Vorkommen von Resten der Chorda dorsalis bei Menschen nach der Geburt und über ihr Verhältniss zu den Gallertgeschwülsten am Clivus. Z Rat Med. 1858;2:202–29. [Google Scholar]
- 52.Munzenrider JE, Liebsch NJ. Proton therapy for tumors of the skull base. Strahlenther Onkol. 1999;175(Suppl 2):57–63. doi: 10.1007/BF03038890. [DOI] [PubMed] [Google Scholar]
- 53.Nelson AC, Pillay N, Henderson S, Presneau N, Tirabosco R, Halai D, et al. An integrated functional genomics approach identifies the regulatory network directed by brachyury (T) in chordoma. J Pathol. 2012;228:274–85. doi: 10.1002/path.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nikoghosyan AV, Karapanagiotou-Schenkel I, Munter MW, Jensen AD, Combs SE, Debus J. Randomised trial of proton vs.carbon ion radiation therapy in patients with chordoma of the skull base, clinical phase III study HIT-1-Study. BMC Cancer. 2010;10:607. doi: 10.1186/1471-2407-10-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Office of Rare Diseases Research. [Last accessed on 2013 Mar 1]. Available from: http://rarediseases.info.nih.gov/
- 56.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orzan F, Terreni MR, Longoni M, Boari N, Mortini P, Doglioni C, et al. Expression study of the target receptor tyrosine kinase of Imatinib mesylate in skull base chordomas. Oncol Rep. 2007;18:249–52. [PubMed] [Google Scholar]
- 58.Ostroumov E, Hunter CJ. Identifying mechanisms for therapeutic intervention in chordoma: c-Met oncoprotein. Spine (Phila Pa 1976) 2008;33:2774–80. doi: 10.1097/BRS.0b013e31817e2d1e. [DOI] [PubMed] [Google Scholar]
- 59.Ostroumov E, Hunter CJ. The role of extracellular factors in human metastatic chordoma cell growth in vitro. Spine (Phila Pa 1976) 2007;32:2957–64. doi: 10.1097/BRS.0b013e31815cde91. [DOI] [PubMed] [Google Scholar]
- 60.Pearlman AW, Friedman M. Radical radiation therapy of chordoma. Am J Roentgenol Radium Ther Nucl Med. 1970;108:332–41. [PubMed] [Google Scholar]
- 61.Peeters A, Grutters JP, Pijls-Johannesma M, Reimoser S, De Ruysscher D, Severens JL, et al. How costly is particle therapy. Cost analysis of external beam radiotherapy with carbon-ions, protons and photons? Radiother Oncol. 2010;95:45–53. doi: 10.1016/j.radonc.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 62.Pillay N, Plagnol V, Tarpey PS, Lobo SB, Presneau N, Szuhai K, et al. A common single-nucleotide variant in T is strongly associated with chordoma. Nat Genet. 2012;44:1185–7. doi: 10.1038/ng.2419. [DOI] [PubMed] [Google Scholar]
- 63.Potluri S, Jefferies SJ, Jena R, Harris F, Burton KE, Prevost AT, et al. Residual postoperative tumour volume predicts outcome after high-dose radiotherapy for chordoma and chondrosarcoma of the skull base and spine. Clin Oncol (R Coll Radiol) 2011;23:199–208. doi: 10.1016/j.clon.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 64.Presneau N, Shalaby A, Idowu B, Gikas P, Cannon SR, Gout I, et al. Potential therapeutic targets for chordoma: PI3K/AKT/TSC1/TSC2/mTOR pathway. Br J Cancer. 2009;100:1406–14. doi: 10.1038/sj.bjc.6605019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Presneau N, Shalaby A, Ye H, Pillay N, Halai D, Idowu B, et al. Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: A genetic and functional-based study. J Pathol. 2011;223:327–35. doi: 10.1002/path.2816. [DOI] [PubMed] [Google Scholar]
- 66.Ptaszynski K, Szumera-Cieckiewicz A, Owczarek J, Mrozkowiak A, Pekul M, Baranska J, et al. Epidermal growth factor receptor (EGFR) status in chordoma. Pol J Pathol. 2009;60:81–7. [PubMed] [Google Scholar]
- 67.RARECARE Cancer List. [Last accessed on 2013 Feb 18]. Available from: http://www.rarecare.eu/rarecancers/rarecancers.asp .
- 68.Ricci-Vitiani L, Pierconti F, Falchetti ML, Petrucci G, Maira G, De Maria R, et al. Establishing tumor cell lines from aggressive telomerase-positive chordomas of the skull base. J Neurosurg. 2006;105:482–4. doi: 10.3171/jns.2006.105.3.482. [DOI] [PubMed] [Google Scholar]
- 69.Rinner B, Froehlich EV, Buerger K, Knausz H, Lohberger B, Scheipl S, et al. Establishment and detailed functional and molecular genetic characterisation of a novel sacral chordoma cell line, MUG-Chor1. Int J Oncol. 2012;40:443–51. doi: 10.3892/ijo.2011.1235. [DOI] [PubMed] [Google Scholar]
- 70.Romeo S, Hogendoorn PC. Brachyury and chordoma: The chondroid-chordoid dilemma resolved? J Pathol. 2006;209:143–6. doi: 10.1002/path.1987. [DOI] [PubMed] [Google Scholar]
- 71.Rostomily RC, Sekhar LN, Elahi F. Chordomas and Chondrosarcomas. In: Sekhar LN, Fessler R, editors. Atlas of Neurosurgical Techniques: Brain. New York: Thieme; 2006. [Google Scholar]
- 72.Salisbury JR, Deverell MH, Cookson MJ, Whimster WF. Three-dimensional reconstruction of human embryonic notochords: Clue to the pathogenesis of chordoma. J Pathol. 1993;171:59–62. doi: 10.1002/path.1711710112. [DOI] [PubMed] [Google Scholar]
- 73.Scheil S, Bruderlein S, Liehr T, Starke H, Herms J, Schulte M, et al. Genome-wide analysis of sixteen chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer. 2001;32:203–11. doi: 10.1002/gcc.1184. [DOI] [PubMed] [Google Scholar]
- 74.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–62. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 75.Schulz-Ertner D, Karger CP, Feuerhake A, Nikoghosyan A, Combs SE, Jakel O, et al. Effectiveness of carbon ion radiotherapy in the treatment of skull-base chordomas. Int J Radiat Oncol Biol Phys. 2007;68:449–57. doi: 10.1016/j.ijrobp.2006.12.059. [DOI] [PubMed] [Google Scholar]
- 76.Schwab J, Antonescu C, Boland P, Healey J, Rosenberg A, Nielsen P, et al. Combination of PI3K/mTOR inhibition demonstrates efficacy in human chordoma. Anticancer Res. 2009;29:1867–71. [PubMed] [Google Scholar]
- 77.Schwab JH, Boland PJ, Agaram NP, Socci ND, Guo T, O’Toole GC, et al. Chordoma and chondrosarcoma gene profile: Implications for immunotherapy. Cancer Immunol Immunother. 2009;58:339–49. doi: 10.1007/s00262-008-0557-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sen C, Triana AI, Berglind N, Godbold J, Shrivastava RK. Clival chordomas: Clinical management, results, and complications in 71 patients. J Neurosurg. 2010 Nov;113:1059–71. doi: 10.3171/2009.9.JNS08596. [DOI] [PubMed] [Google Scholar]
- 79.Shalaby A, Presneau N, Ye H, Halai D, Berisha F, Idowu B, et al. The role of epidermal growth factor receptor in chordoma pathogenesis: A potential therapeutic target. J Pathol. 2011;223:336–46. doi: 10.1002/path.2818. [DOI] [PubMed] [Google Scholar]
- 80.Shalaby AA, Presneau N, Idowu BD, Thompson L, Briggs TR, Tirabosco R, et al. Analysis of the fibroblastic growth factor receptor-RAS/RAF/MEK/ERK-ETS2/brachyury signalling pathway in chordomas. Mod Pathol. 2009;22:996–1005. doi: 10.1038/modpathol.2009.63. [DOI] [PubMed] [Google Scholar]
- 81.Singhal N, Kotasek D, Parnis Fx. Response to erlotinib in a patient with treatment refractory chordoma. Anticancer Drugs. 2009;20:953–5. doi: 10.1097/CAD.0b013e328330c7f0. [DOI] [PubMed] [Google Scholar]
- 82.Siu IM, Salmasi V, Orr BA, Zhao Q, Binder ZA, Tran C, et al. Establishment and characterization of a primary human chordoma xenograft model. J Neurosurg. 2012;116:801–9. doi: 10.3171/2011.12.JNS111123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stacchiotti S, Longhi A, Ferraresi V, Grignani G, Comandone A, Stupp R, et al. Phase II study of imatinib in advanced chordoma. J Clin Oncol. 2012;30:914–20. doi: 10.1200/JCO.2011.35.3656. [DOI] [PubMed] [Google Scholar]
- 84.Stippler M, Gardner PA, Snyderman CH, Carrau RL, Prevedello DM, Kassam AB. Endoscopic endonasal approach for clival chordomas. Neurosurgery. 2009;64:268–77. doi: 10.1227/01.NEU.0000338071.01241.E2. [DOI] [PubMed] [Google Scholar]
- 85.Takahashi S, Kawase T, Yoshida K, Hasegawa A, Mizoe JE. Skull base chordomas: Efficacy of surgery followed by carbon ion radiotherapy. Acta Neurochir (Wien) 2009;151:759–69. doi: 10.1007/s00701-009-0383-5. [DOI] [PubMed] [Google Scholar]
- 86.Tamborini E, Miselli F, Negri T, Lagonigro MS, Staurengo S, Dagrada GP, et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin Cancer Res. 2006;12:6920–8. doi: 10.1158/1078-0432.CCR-06-1584. [DOI] [PubMed] [Google Scholar]
- 87.Tatsuzaki H, Urie MM. Importance of precise positioning for proton beam therapy in the base of skull and cervical spine. Int J Radiat Oncol Biol Phys. 1991;21:757–65. doi: 10.1016/0360-3016(91)90696-2. [DOI] [PubMed] [Google Scholar]
- 88.The Chordoma Foundation. [Last cited in 2012]. Available from: http://www.chordomafoundation.org .
- 89.Therapeutics for Rare and Neglected Diseases. [Last accessed on 2013 Mar 1]. Available from: http://www.ncats.nih.gov/research/rare-diseases/trnd/trnd.html .
- 90.Tomlinson FH, Scheithauer BW, Forsythe PA, Unni KK, Meyer FB. Sarcomatous transformation in cranial chordoma. Neurosurgery. 1992;31:13–8. doi: 10.1227/00006123-199207000-00003. [DOI] [PubMed] [Google Scholar]
- 91.Torres MA, Chang EL, Mahajan A, Lege DG, Riley BA, Zhang X, et al. Optimal treatment planning for skull base chordoma: Photons, protons, or a combination of both? Int J Radiat Oncol Biol Phys. 2009;74:1033–9. doi: 10.1016/j.ijrobp.2008.09.029. [DOI] [PubMed] [Google Scholar]
- 92.Tzortzidis F, Elahi F, Wright D, Natarajan SK, Sekhar LN. Patient outcome at long-term follow-up after aggressive microsurgical resection of cranial base chordomas. Neurosurgery. 2006;59:230–7. doi: 10.1227/01.NEU.0000223441.51012.9D. [DOI] [PubMed] [Google Scholar]
- 93.Uggowitzer MM, Kugler C, Groell R, Lindbichler F, Radner H, Sutter B, et al. Drop metastases in a patient with a chondroid chordoma of the clivus. Neuroradiology. 1999;41:504–7. doi: 10.1007/s002340050792. [DOI] [PubMed] [Google Scholar]
- 94.Vergara G, Belinchon B, Valcarcel F, Veiras M, Zapata I, de la Torre A. Metastatic disease from chordoma. Clin Transl Oncol. 2008;10:517–21. doi: 10.1007/s12094-008-0243-4. [DOI] [PubMed] [Google Scholar]
- 95.Virchow R. Untersuchungen ueber die Entwicklung des Schaedelgrundes im gesunden und krankhaften Zustande und ueber den Einfluss derselben auf Schaedelform, Gesichtsbildung und Gehirnbau. Berlin: Georg Reimer; 1857. [Google Scholar]
- 96.Vujovic S, Henderson S, Presneau N, Odell E, Jacques TS, Tirabosco R, et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209:157–65. doi: 10.1002/path.1969. [DOI] [PubMed] [Google Scholar]
- 97.Weinberger PM, Yu Z, Kowalski D, Joe J, Manger P, Psyrri A, et al. Differential expression of epidermal growth factor receptor, c-Met, and HER2/neu in chordoma compared with 17 other malignancies. Arch Otolaryngol Head Neck Surg. 2005;131:707–11. doi: 10.1001/archotol.131.8.707. [DOI] [PubMed] [Google Scholar]
- 98.Yamaguchi T, Suzuki S, Ishiiwa H, Ueda Y. Intraosseous benign notochordal cell tumours: Overlooked precursors of classic chordomas? Histopathology. 2004;44:597–602. doi: 10.1111/j.1365-2559.2004.01877.x. [DOI] [PubMed] [Google Scholar]
- 99.Yamazaki T, McLoughlin GS, Patel S, Rhines LD, Fourney DR. Feasibility and safety of en bloc resection for primary spine tumors: A systematic review by the Spine Oncology Study Group. Spine (Phila Pa 1976) 2009;34(22 Suppl):S31–8. doi: 10.1097/BRS.0b013e3181b8b796. [DOI] [PubMed] [Google Scholar]
- 100.Yang C, Hornicek FJ, Wood KB, Schwab JH, Choy E, Iafrate J, et al. Characterization and analysis of human chordoma cell lines. Spine (Phila Pa 1976) 2010;35:1257–64. doi: 10.1097/BRS.0b013e3181c2a8b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang C, Schwab JH, Schoenfeld AJ, Hornicek FJ, Wood KB, Nielsen GP, et al. A novel target for treatment of chordoma: Signal transducers and activators of transcription 3. Mol Cancer Ther. 2009;8:2597–605. doi: 10.1158/1535-7163.MCT-09-0504. [DOI] [PubMed] [Google Scholar]
- 102.Yang XR, Ng D, Alcorta DA, Liebsch NJ, Sheridan E, Li S, et al. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat Genet. 2009;41:1176–8. doi: 10.1038/ng.454. [DOI] [PMC free article] [PubMed] [Google Scholar]