

Abstract
Primary hyperparathyroidism (PHP) with pheochromocytoma and neurofibromatosis type 1 is a rare clinical association. We present a case of PHP and pheochromocytoma occurring in a 33-year-old male with familial cutaneous neurofibromatosis.
Keywords: A rare clinical association, neurofibromatosis type 1, pheochromocytoma, primary hyperparathyroidism
INTRODUCTION
Neurofibromatosis is a genetically inherited autosomal disorder affecting the neural crest cells (Schwann cells, melanocyte, and endoneurial fibroblasts). There is excessive proliferation of these cells throughout the body forming tumors formation which results in a variety of skeletal and neurological problems. Malfunctioning melanocytes cause skin pigmentation and café-au- lait spots.[1] A variety of endocrine tumors are known to be associated with NF1, among which Pheochromocytoma is reported to be the most common, occurring in 0.1-5.7% of the patients with NF1.[2] Although pheochromocytoma can be part of the spectrum of disorders associated with neurofibromatosis type 1 (NF1), the occurrences of PHP is a rare but reported entity.[3]
CASE REPORT
A 33-year-old gentleman, diagnosed to have neurofibromatosis type 1 at 14 years of age, presented to us with a history of low back ache for 3 years associated with headache difficulty in walking, urinary urgency, and constipation for 6 months. On examination his height was 149 cm, weight 56 kg, BMI 25.2 kg/m2, head circumference 55.4 cm, pulse 80/minutes, blood pressure 130/90-mm of Hg, and no postural drop in blood pressure. He had multiple neurofibromas with numerous café au lait spots with smooth edges all over the body the largest of which was present over the right leg measuring 15 × 10 cm, axillary freckling was noted bilaterally. He had scoliosis of the spine. His ophthalmic examination revealed lisch nodules with bilateral ptosis. Neurological examination revealed mild spastic quadriperesis with brisk deep tendon reflexes and equivocal plantars. Screening MRI scan of the spine cord was however did not reveal any compressive lesions but it revealed a right suprarenal mass measuring 28 × 20 mm and also a hyperintense lesion measuring 21 × 14 mm, posterior to the left lobe of thyroid. He had biochemical evidence of hypercalcemia (calcium 12 mg/dl; albumin 4.5 g/dl) hyperparathyroidism (PTH 741 pg/unit). Left inferior parathyroid adenoma was confirmed by an ultrasound, parathyroid scintigraphy [Figure 1], and SPECT-CT [Figure 2]. His 24-hour urinary metanephrine level was 356 mcg (normal range <350 mcg/24 hours) and normetanephrine level was 937 mcg (normal range <600 mcg/24 hours). Right adrenal pheochromocytoma was confirmed on the CT scan [Figure 3] and MIBG scan. This patient had features of neurofibromatosis type I (NF-1) with a left inferior parathyroid adenoma and a right adrenal pheochromocytoma. After adequate preparation he was taken up for focused parathyroidectomy with excision of left inferior parathyroid adenoma and right laparoscopic adrenalectomy. The postoperative period was uneventful. Histopathology confirmed pheochromocytoma of the right adrenal gland and a left parathyroid neoplasm with atypical features like nuclear atypia and capsular/vascular invasion, [Figure 4]. Immunohistochemistry of the tumor showed a low MIB-1 index [Figure 5].
Figure 1.
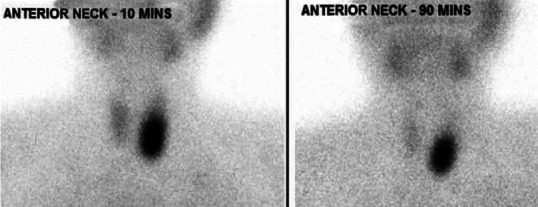
Parathyroid schintigraphy using Tc99 m MIBI showing retaining of tracer in the delayed image at 90 minutes after complete tracer washout from thyroid gland suggestive of left parathyroid adenoma
Figure 2.
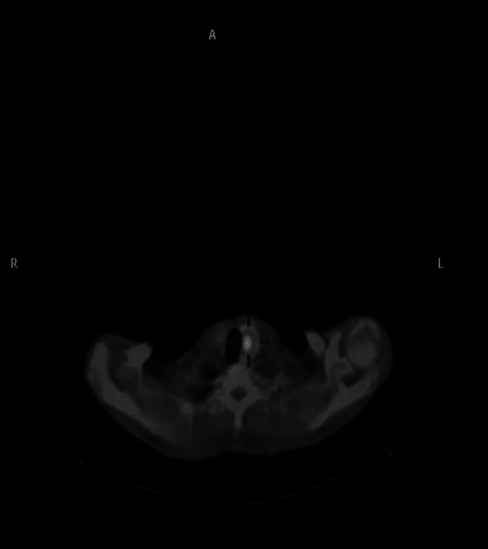
SPECT CT showing left inferior parathyroid adenoma
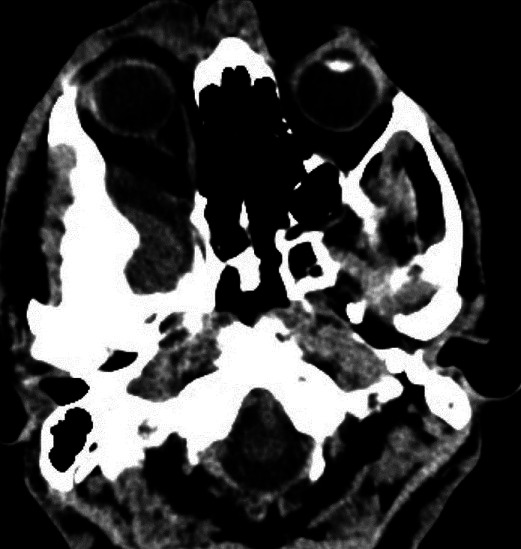
Figure 3.
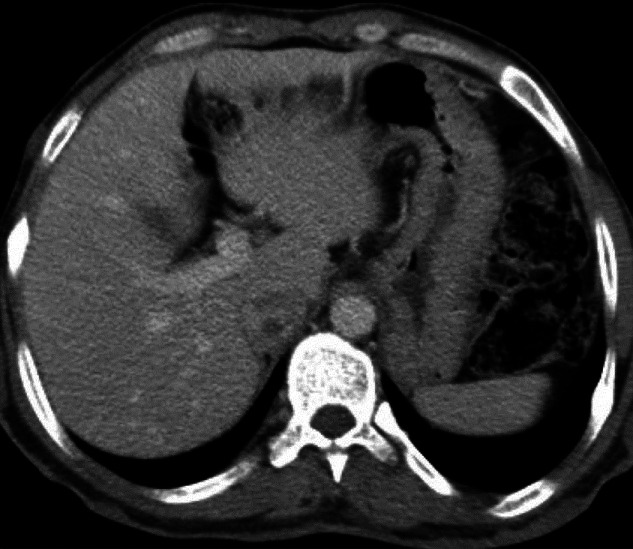
CT abdomen showing right adrenal pheochromacytoma
Figure 4.
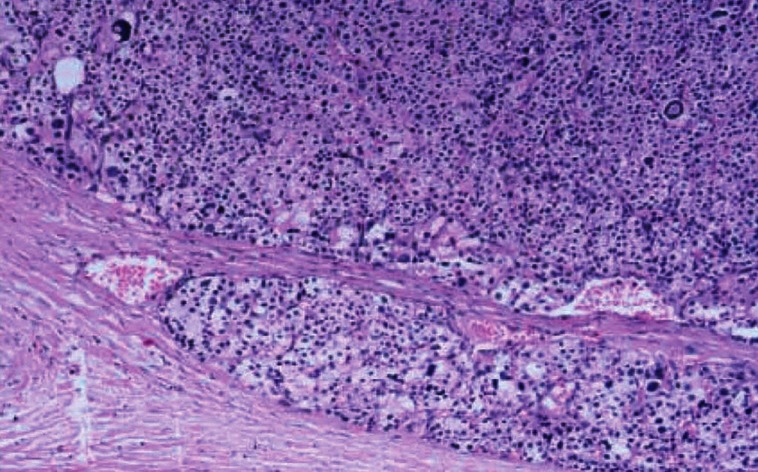
Histopathology showing parathyroid tissue with minimal invasion of capsule
Figure 5.
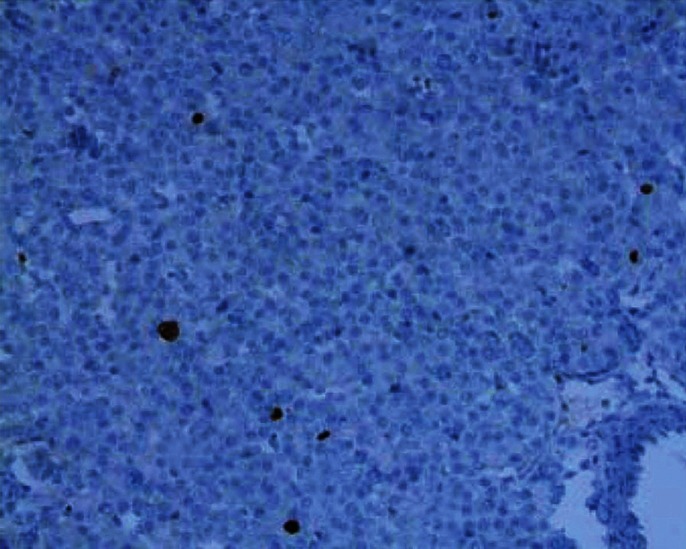
Parathyroid tissue showing low MIB-1 index; arrow mark showing MIB-1 immunostaining
DISCUSSION
Our patient had a combination of two endocrine disorders, pheochromocytoma and hyperparathyroidism, suggesting multiple endocrine neoplasm type IIA (MEN 2A).
In patients with MEN 2A hyperparathyroidism is usually due to hyperplasia of one of more parathyroid glands, but our patient had an atypical parathyroid neoplasm causing hyperparathyroidism. Skeletal lesions are reported in 40-50% of patients with NF1. Our patient had scoliosis and fibrous dysplasia of the right wing of sphenoid [Figure 6]; he had no other features of bone disease.
Figure 6.
Fibrous dysplastic change in right sphenoid bone
Though NF1 has not been included in the syndrome of MEN there are reported cases of hyperparathyroidism occurring in patients with NF, making a genuine association between these two uncommon conditions likely, suggestive of MEN 2A. It would be worthwhile screening all patients with NF1 for hypercalcemia to rule out primary hyperparathyroidism as it is a simple, cost effective and an easily treatable disorder when diagnosed early. It can lead to a high degree of morbidity if is left untreated or if detected late.
Footnotes
Source of Support: Nil
Conflict of Interest: No
REFERENCES
- 1.Rubin R, Strayer DS. 5th ed. Baltimore: Wolters Kluwer Health, Lippincot Williams and Wilkins; 2008. Rubin's Pathology: Clinicopathologic Foundation of Medicine; pp. 201–3. [Google Scholar]
- 2.Zöller ME, Rembeck B, Odén A, Samuelsson M, Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer. 1997;79:2125–31. [PubMed] [Google Scholar]
- 3.Al-Wahhabi B. Parathyroid adenoma and bilateral pheochromocytoma in a patient with neurofibromatosis. Ann Saudi Med. 2005;25:255–7. doi: 10.5144/0256-4947.2005.255. [DOI] [PMC free article] [PubMed] [Google Scholar]