

Abstract
Background
Birth weight (BW) is an economically important trait in beef cattle, and is associated with growth- and stature-related traits and calving difficulty. One region of the cattle genome, located on Bos primigenius taurus chromosome 14 (BTA14), has been previously shown to be associated with stature by multiple independent studies, and contains orthologous genes affecting human height. A genome-wide association study (GWAS) for BW in Brazilian Nellore cattle (Bos primigenius indicus) was performed using estimated breeding values (EBVs) of 654 progeny-tested bulls genotyped for over 777,000 single nucleotide polymorphisms (SNPs).
Results
The most significant SNP (rs133012258, PGC = 1.34 × 10-9), located at BTA14:25376827, explained 4.62% of the variance in BW EBVs. The surrounding 1 Mb region presented high identity with human, pig and mouse autosomes 8, 4 and 4, respectively, and contains the orthologous height genes PLAG1, CHCHD7, MOS, RPS20, LYN, RDHE2 (SDR16C5) and PENK. The region also overlapped 28 quantitative trait loci (QTLs) previously reported in literature by linkage mapping studies in cattle, including QTLs for birth weight, mature height, carcass weight, stature, pre-weaning average daily gain, calving ease, and gestation length.
Conclusions
This study presents the first GWAS applying a high-density SNP panel to identify putative chromosome regions affecting birth weight in Nellore cattle. These results suggest that the QTLs on BTA14 associated with body size in taurine cattle (Bos primigenius taurus) also affect birth weight and size in zebu cattle (Bos primigenius indicus).
Keywords: GWAS, Birth weight, Bos primigenius indicus, Nellore cattle, Stature
Background
Birth weight (BW) is an economically important trait in beef cattle, and is usually the first characteristic measured in a calf. Birth weight is associated with growth-related traits [1], mature size [2] and carcass weight, thus being a valuable production indicator, as well as a selection criterion to improve calving ease [3,4].
Despite the beef industry’s pursuit of animals with rapid growth, yielding heavier carcasses, selection for these objectives needs to be properly balanced against selection for reproductive traits, which have great economic importance in beef cattle production systems [5,6]. While low estimated breeding values (EBVs) for BW are associated with reduced calf viability [4] and lower growth rates [3,7], the use of sires with high EBVs for BW on dams with small pelvic size may result in higher rates of dystocia [7] and increased perinatal mortality [8]. These antagonisms result from the strong association of birth weight with the body size of the calf, i.e., with the stature of the animal [2]. Calving difficulties can result from a mismatch between pelvic opening and calf size [6]. This relationship between BW with reproductive and growth/size traits highlights the importance of understanding the underlying genetic architecture of BW.
Birth weight exhibits sufficient variability and heritability in the Nellore breed (Bos primigenius indicus), with an average of 29.8 ± 2.7 kg [9] and estimated heritability between 0.25 and 0.33 [1,9,10]. Despite the low frequency of dystocia in Nellore cows, BW has been recorded and used to monitor genetic trend. One selection strategy of the breeding programs in Brazil has been to preferentially use sires with higher EBVs for weaning and yearling weights, but with low or close to average EBVs for BW [11]. The identification of major genes and variants affecting multiple weight and carcass traits or influencing BW alone would be of help to balance these conflicting goals, because BW is positively correlated with weaning and yearling weights [1].
In the past two decades, linkage studies attempting to map quantitative trait loci (QTLs) affecting weight, growth, or stature in cattle have been published (e.g. [12-16]). The release of the reference bovine genome [17], the discovery of common single nucleotide polymorphisms (SNPs) across breeds [18,19], and the availability of high-throughput microarrays have enhanced the process of mapping loci that affect complex traits. This has led to several population-based investigations of associations between weight/growth/height phenotypes with genome-wide variants in different cattle breeds [15,20-23].
In particular, two regions of the bovine genome associated with stature and growth have been highlighted recently. The first, located on Bos primigenius taurus (BTA) autosome 6 [20,23], shelters the orthologous genes NCAPG and LCORL, which have been also found to be associated with adult height in humans [24,25]. The second, located on BTA14 [15,21-23], contains the genes PLAG1, CHCHD7, RDHE2, MOS, RPS20, LYN, PENK and TGS1, that were previously found to affect stature in both cattle and humans [22,24,26-28]. Importantly, the majority of the genome-wide association studies (GWAS) reported in literature were conducted in the humpless subspecies of cattle (Bos primigenius taurus, known as taurine cattle), and GWAS in the humped bovine subspecies (Bos primigenius indicus, often referred as indicine or zebu cattle) are only now emerging, especially because the first SNP microarrays were optimized for taurine cattle [19].
In this paper, results from a genome-wide scan for SNPs associated with BW variation in Nellore cattle using EBVs of progeny-tested Brazilian bulls are reported. As EBVs take into account information from performance of the individual, progeny and parents, pedigree relationships, and systematic management and environmental factors, they can be used as composite phenotypes for proceeding with association analyses (see, e.g., [29]). The objective of this study was to identify putative SNP associated with differences in BW and to explore the genomic regions around them to unravel prospective functional relationships among weight, fertility, and growth/size traits.
Results
Genotype informativeness and quality control
From the initial set of 777,961 SNPs, 42,669 (5.5%) were non-autosomal markers. Fifty four autosomal SNPs with redundant genomic coordinates were identified and excluded from further analyses. The identity by state (IBS) check revealed no unexpected sample duplicates. A total of 223,309 (30.4%) markers were excluded due to minor allele frequency (MAF) < 0.02. The number of SNPs excluded due to SNP Call Rate (CRSNP) < 0.98 and Fisher’s exact test P-value for Hardy-Weinberg Equilibrium (HWE) < 1 × 10-5 were 122,611 (16.7%) and 13,194 (1.8%), respectively. Five individuals were removed due to low Call Rate (CRIND). The final dataset included data for 649 individuals and 434,020 SNPs.
Descriptive statistics of dependent variables
Based on the results for the Shapiro-Wilk test, there was no evidence that EBVs deviated from normality (P = 0.415), and no outliers were observed. Average accuracy was 0.87 ± 0.11, with minimum, median and maximum accuracies of 0.51, 0.91 and 0.99, respectively. After fitting the regression in formula (2), there was no evidence against the hypotheses of normally distributed (P = 0.57) and homoskedastic residuals (studentized Breusch-Pagan test, P = 0.11). These findings suggest that the dependent variables used were reliable and did not violate possible assumptions of the statistical analyses used hereafter.
Population substructure
The Principal Coordinates Analysis (PCoA) revealed genetic stratification among the Nellore samples (Figure 1). After using k-means clustering to assign individuals to two different groups according to their coordinates in the PCoA, a highly significant association (P = 5.41 × 10-34) between the k-means assignments (ncluster1 = 531, ncluster2 = 118) and breeding program subgroups (nsubgroup1 = 352, nsubgroup2 = 297) was found. For the k-means groups, BW average was 0.434 ± 1.306 in cluster 1 and -0.216 ± 1.295 in cluster 2. For the breeding program subgroups, the trait averages in subgroup 1 and subgroup 2 were 0.683 ± 1.276 and -0.119 ± 1.255, respectively. When considering either k-means clusters or breeding program subgroups as subpopulation labels, the EBVs showed homogeneity of variance (P > 0.05), but the trait mean was significantly different between groups (P = 1.21 × 10-6 and P = 4.37 × 10-15 for k-means and breeding program, respectively). Thus, population substructure was a potential confounder in the genotype-EBV association analysis, which justified the inclusion of eigenvectors from the PCoA as fixed effects in the linear model.
Figure 1.

Principal Coordinates Analysis based on the genomic kinship coefficient. Percentages inside brackets correspond to the variance explained by each respective eigenvector. Each ‘+’ represents an individual and ovals are 95% inertia ellipses. A) Subjects colored according to breeding program subgroups. B) Subjects colored according to k-means clustering results.
Association analysis
A total of 32 eigenvectors from the PCoA were significantly correlated with the phenotypes, which together explained 15.23% of the genotypic variability. Residuals from the weighted regression on these significant eigenvectors were used as the dependent variable for the SNP association analysis. The quantile-quantile (Q-Q) plot (Figure 2) showed that the deviation of the observed test statistics from the theoretical quantiles was mild and acceptable (λ = 1.002831), and the values were adjusted for the inflation factor via Genomic Control (GC). The T2 values deviating from the expected values were interpreted as SNPs departing from the null hypothesis. The genome-wide deflated P-values are shown in Figure 3.
Figure 2.

Quantile-quantile plot for the test statistics (χ2) used in the association analysis.
Figure 3.
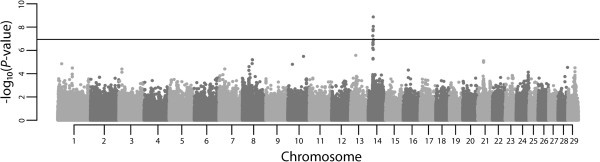
Manhattan plot of genome-wide -log10(P-values) for birth weight estimated breeding values in Nellore cattle. The horizontal line represents the Bonferroni significance threshold (α = 1.15 × 10-7).
A peak crossing the boundary for Bonferroni significance (α = 1.15 × 10-7) was detected on BTA14, comprising 5 SNPs (Table 1) which were highly linked (mean r2 = 0.728 ± 0.12). The most significant SNP (rs133012258, PGC = 1.34 × 10-9), located at BTA14:25376827, had an estimated allele substitution effect of 0.452 kg (i.e., for each extra A allele, the BW breeding value is expected to increase 0.452 kg), with lower and upper limits for the 95% confidence interval (CI) of 0.306 kg and 0.598 kg, respectively, and the percentage of the variance in sires EBVs explained by the SNP was 4.62% (with a 95% CI of 2.12-8.09%). The overall rs133012258 A allele frequency was 0.274, whereas the breeding program subgroups 1 and 2 had frequencies of 0.351 and 0.184, respectively. Figure 4 shows the distribution of the EBVs (in standard deviations) for the three genotype classes of rs133012258. In both Illumina TOP and Forward allele notation, the AB correspondence for rs133012258 was A = A and B = G.
Table 1.
Summary of parameters and statistics estimated for the identified significant SNPs
| Ensembl variant ID | Illumina probe ID | BTA14 Position (bp) | n | Effect allele | Allele frequency | CR a (%) | HWE P-value | βb (kg) | SE | T2c | GC-T2d | P-value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs133012258 |
BovineHD1400007343 |
25376827 |
649 |
A |
0.274 |
100.00 |
0.920 |
0.452 |
0.074 |
36.858 |
36.753 |
1.34×10-9 |
| rs41627948 |
BovineHD1400007374 |
25504073 |
648 |
B |
0.181 |
99.85 |
0.110 |
0.522 |
0.090 |
33.202 |
33.108 |
8.72×10-9 |
| rs42646720 |
BovineHD1400007144 |
24590812 |
649 |
B |
0.243 |
100.00 |
0.390 |
0.457 |
0.081 |
31.999 |
31.909 |
1.62×10-8 |
| rs136764901 |
BovineHD1400007159 |
24651537 |
641 |
B |
0.244 |
98.77 |
0.200 |
0.459 |
0.082 |
31.546 |
31.457 |
2.04×10-8 |
| rs136287861 | BovineHD1400006765 | 23313228 | 645 | A | 0.272 | 99.38 | 0.766 | 0.408 | 0.075 | 29.580 | 29.497 | 5.60×10-8 |
aCR Call rate.
bβ Estimated allele substitution effect.
cT2 Chi-squared statistics for β: .
dGC-T2 Corrected Chi-squared statistics for β. Genomic control correction was performed by dividing the χ2 statistics by the distribution inflation/deflation factor estimate (λ = 1.002831).
Figure 4.

Box plots for the birth weight estimated breeding values according to rs133012258 genotypes. Values in the y axis are expressed in terms of standard units.
One of the 5 significant SNPs, rs42646720 (BTA14:24590812, PGC = 1.62 × 10-8), is located within intron 2 of the gene Kell blood group complex subunit-related family, member 4 (XKR4 or KIAA1889, ENSBTAG00000044050). Thirteen genes were found within 500 kb of the most significant SNP (Figure 5), including the human height-associated orthologous genes PLAG1, CHCHD7, MOS, RPS20, LYN, RDHE2 (SDR16C5) and PENK (Table 2). The bovine reference genome sequence of this region was found to have high identity with human (Homo sapiens - HSA), pig (Sus scrofa - SSC) and mouse (Mus musculus - MMU) autosomes 8 (HSA8), 4 (SSC4) and 4 (MMU4), respectively (Figure 6), and the majority of the genes had homology across species (Table 2). The most significant SNP also overlapped 28 QTLs previously reported in the literature by linkage mapping studies using different cattle breeds (Table 3), including QTLs for birth weight, mature height, carcass weight, stature, pre-weaning average daily gain, calving ease and gestation length.
Figure 5.

Regional association plot for birth weight in the 1 Mb window around rs133012258. Upper box: each dot represents a SNP, and its color heat the degree of linkage disequilibrium with rs133012258 (black diamond). The horizontal dashed line represents the Bonferroni significance threshold (α = 1.15 × 10-7). Lower box: genes (green arrows; right-handed = positive strand, left-handed = negative strand) within the region in the UMD v3.1 assembly.
Table 2.
List of genes within the 1 Mb region surrounding the most significant SNP (rs133012258)
| Gene | Ensembl ID | BTA14 coordinates | Distance from SNP (kb) | Strand | HSA8 homology | SSA4 homology | MMA4 homology | Description |
|---|---|---|---|---|---|---|---|---|
| U6 |
ENSBTAG00000043923 |
25492090:25492184 |
115.4 |
+ |
No homologues |
No homologues |
No homologues |
U6 spliceosomal RNA |
| PENK |
ENSBTAG00000004924 |
25218586:25222991 |
153.8 |
- |
ENSG00000181195 |
ENSSSCG00000006243 |
ENSMUSG00000045573 |
Proenkephalin-A |
| IMPAD1 |
ENSBTAG00000015637 |
25544907:25560879 |
168.1 |
- |
ENSG00000104331 |
ENSSSCG00000006242 |
ENSMUSG00000066324 |
Inositolmonophosphatase 3 |
| SDR16C6 |
ENSBTAG00000040321 |
25153583:25179651 |
197.2 |
- |
No homologues |
No homologues |
ENSMUSG00000071019 |
Short-chain dehydrogenase/reductase family 16C member 6 |
| SDR16C5 (RDHE2) |
ENSBTAG00000018570 |
25105062:25117554 |
259.3 |
- |
ENSG00000170786 |
ENSSSCG00000006245 |
ENSMUSG00000028236 |
Epidermal retinol dehydrogenase 2 |
| Unknown |
ENSBTAG00000039031 |
25067486:25067823 |
309.0 |
- |
No homologues |
No homologues |
No homologues |
Uncharacterizedprotein |
| CHCHD7 |
ENSBTAG00000033284 |
25052885:25058779 |
318.0 |
+ |
ENSG00000170791 |
ENSSSCG00000006246 |
ENSMUSG00000042198 |
Coiled-coil-helix-coiled-coil-helix domain-containing protein 7 |
| PLAG1 |
ENSBTAG00000004022 |
25007291:25009296 |
367.5 |
- |
ENSG00000181690 |
ENSSSCG00000006247 |
ENSMUSG00000003282 |
Pleiomorphic adenoma gene 1 |
| MOS |
ENSBTAG00000019145 |
24975950:24976948 |
399.9 |
- |
ENSG00000172680 |
ENSSSCG00000006248 |
ENSMUSG00000078365 |
V-mosMoloneymurine sarcoma viral oncogenehomolog |
| U1 |
ENSBTAG00000028889 |
24970516:24970679 |
406.1 |
+ |
No homologues |
No homologues |
No homologues |
U1 spliceosomal RNA |
| RPS20 |
ENSBTAG00000019147 |
24955079:24956324 |
420.5 |
- |
ENSG00000008988 |
ENSSSCG00000006249 |
ENSMUSG00000028234 |
40S ribosomalprotein S20 |
| snoU54 |
ENSBTAG00000045097 |
24955769:24955835 |
421.0 |
- |
No homologues |
No homologues |
No homologues |
Small nucleolar RNA U54 |
| LYN | ENSBTAG00000020034 | 24847257:24920713 | 456.1 | + | ENSG00000254087 | ENSSSCG00000006250 | ENSMUSG00000042228 | Tyrosine-proteinkinaseLyn |
Figure 6.

Ensembl alignments of UMD v3.1 sequence for the 1 Mb region surrounding rs133012258. The bovine reference genome sequence was aligned against (from top to bottom) the human (GRCh37 assembly), pig (Sscrofa10.2 assembly) and mouse (GRCm38 assembly) genome builds. Gene colors: yellow - merged Ensembl/Havana, red - protein coding, blue - processed transcript, grey - pseudogene, purple - RNA gene. Triangles: black - breakpoint between different chromosomes, blue - inversion in chromosome, brown - breakpoint on chromosome, red - gap between two underlying slices.
Table 3.
QTLdb hits within the 1 Mb region surrounding the most significant SNP (rs133012258)
| Trait | BTA14 coordinates | QTLdb ID | PubMed ID |
|---|---|---|---|
| Body weight (birth) |
6311565:71762521 |
5375 |
19016677 |
| Height (mature) |
19204282:42398519 |
10962 |
20477797 |
| Carcass weight |
25224396:30870876 |
1375 |
16151698 |
| |
10808022:28658498 |
10960 |
20477797 |
| Stature |
25219037:65017465 |
4613 |
10575619 |
| Pre-weaning average daily gain |
25224396:35530275 |
2630 |
15537758 |
| Calving ease (maternal) |
19204282:28658498 |
10959 |
20477797 |
| Gestation length |
6311565:30372479 |
5374 |
19016677 |
| |
17512260:48646289 |
5385 |
19016677 |
| Rump angle |
25224396:29928419 |
1592 |
16230715 |
| Longissimus muscle area |
19204282:42398519 |
10964 |
20477797 |
| Fat thickness at the 12th rib |
19204282:28658498 |
10961 |
20477797 |
| Marbling score |
16670076:73064076 |
1334 |
14677852 |
| Abnormal flavor intensity |
5565085:28658498 |
4833 |
18254735 |
| Tick resistance |
5545944:77366190 |
9917 |
17894560 |
| Milk yield |
25372161:25525285 |
6209 |
18650300 |
| |
16243029:28716621 |
3608 |
12729552 |
| Milk fat yield (or percentage) |
13401044:35992517 |
2733 |
9691050 |
| |
1641277:25448723 |
3408 |
12605852 |
| |
25224396:29928419 |
2676 |
14762090 |
| |
13401044:35992517 |
2732 |
9691050 |
| Milk protein yield (or percentage) |
1641277:56300551 |
3413 |
12605852 |
| |
9479897:44651695 |
2604 |
12778594 |
| |
1641277:81189386 |
10099/10100/10101 |
18298934 |
| Somatic cell score |
13401044:35992517 |
2734 |
9691050 |
| |
13401044:35992517 |
2776 |
14556700 |
| |
20567087:44651695 |
4884 |
17954769 |
| Clinical mastitis | 9479131:44651695 | 3177 | 14762087 |
Discussion
Five SNPs on BTA14 were identified as associated with BW in Nellore cattle (P < 1.15 × 10-7), whose surrounding region has been shown to contain many QTLs, genes and variants affecting stature-related traits in cattle by several independent studies [12-15,21-23]. More particularly, the genes PLAG1, CHCHD7, RDHE2, MOS, RPS20, LYN and PENK have been found to influence both human and cattle height [22-28].
The BTA14 region pointed out by the present study has also been shown to be associated with reproductive traits. Cole et al. [16] reported a QTL on BTA14 associated with stillbirth, which also has been associated with body size in dairy cattle [8,30], but found no effect on stature or other conformation traits on that chromosome. The region also associates with many fertility and growth-related traits in the indicine breed Brahman, for example scrotal circumference [31,32], age at the first corpus luteum[31,33], blood levels of insulin-like growth factor 1 (IGF1) [32,33] and hip height [33].
A significant SNP was found within intron 2 of the XKR4 gene in the present study. Lindholm-Perry et al. [34] identified five SNPs near XKR4 associated with feed intake and gain in crossbred steers. Bolormaa et al. [35] found five SNPs in a narrow region of BTA14 encompassing XKR4 associated with rump fat thickness measured at the P8 position (CHILLP8) in seven breeds of cattle, including taurine, indicine and composite breeds. The authors found that four of these SNPs were also associated with CHILLP8 in a confirmatory sample of 1,338 animals, including Angus, Hereford and Brahman cattle. Furthermore, Porto Neto et al. [36] performed a replication study using samples of Belmont Red, Santa Gertrudis and Brahman animals genotyped for SNPs within XKR4 and found that although the SNP effect may vary depending on the breed, the variant rs42646708 (BTA14:24573257) explain around 1.3% of CHILLP8 variance in cattle. This SNP is also located within intron 2 of XKR4, only 17.6 kb apart from the intronic SNP detected in the present study, which strongly suggests XKR4 as a candidate gene for being further explored in future studies of weight and carcass traits in Nellore cattle.
The most significant SNP (rs133012258, PGC = 1.34 × 10-9) was found to explain 4.62% of the variance in sires EBVs, with a 95% CI of 2.12-8.09%. One hundred and eighty loci associated with human adult height explain only 10% of the phenotypic variance together, while individual loci account for 0.4% or less [37]. SNPs analyzed by [22] within a nearby BTA14 region explain from 0.29 to 2.53% of the bovine stature variability, and the quantitative trait nucleotides (QTN) spanning MOS, CHCHD7 and PLAG1 described by [27] explain from 1.10 to 3.50% of height in Jersey and Holstein breeds. Furthermore, the genome-wide survey performed by [21] provided strong evidence for two QTL on BTA14 and BTA21 that together explain at least 10% of the variation of EBVs for calving ease in the German Fleckvieh.
Considering that multiple stature-related traits are governed by variants with small effects, and that the genomic region identified in this study has been previously found to be associated with several of these traits, the putative SNP detected in the present analysis can be considered as a marker in linkage disequilibrium (LD) with major untyped (i.e., not probed by the SNP assay used) causative variants affecting BW and other height-associated traits in Nellore cattle, and further studies would be needed to determine if the QTNs reported by [27] are also segregating in the Nellore population. Also, future investigations are needed to better characterize the effect of nearby SNPs on other weight and carcass traits in Nellore cattle, as it is not clear yet how the putative pleiotropic effect of these variants would be used towards balancing conflicting selection goals for birth, weaning and yearling weights. Although we cannot confirm that the allele substitution effects of these SNPs work in the same direction for all three traits, because only birth weight was analyzed here, these findings suggest that the SNPs identified would be key polymorphisms to be monitored over time. In a scenario where the SNP effects have the same direction in all three traits, one strategy could be avoiding strong positive selection or drifting of the allele that contributes to higher BW EBVs, and identify and promote positive selection of other variants that have effects on weaning and yearling weights only.
The high identity found in the alignment of this BTA14 region against other mammalian species genomes suggests that these orthologous genes are located in a conserved syntenic block which may have arisen and been maintained after speciation from a common ancestor of the mammal clade. Moreover, the evidence for variants associated with growth and stature within this BTA14 region in both taurine and zebu cattle raises two hypotheses: 1) these variants have been introgressed into Nellore via historical admixture with taurine Creole cattle in the maternal line, and was maintained in the breed in spite of several generations of backcrossing; 2) these are ancient polymorphisms, probably already segregating in the founder population of wild Aurochs (Bos primigenius) before subspecies formation.
Regarding functional meaning, the set of genes reported participate in diverse growth and tumor development mechanisms. Among these genes, PLAG1 is the most appealing functional candidate. It is an oncogene that encodes a transcription factor broadly expressed during fetal development, but is down-regulated at birth [27]. It interacts with several growth factors controlling body size, including IGF2[38]. In addition, PLAG1 knock-out mice have been shown to have marked growth retardation and reduced fertility [39]. In a replication study, [28] confirmed the findings reported by [27], demonstrating association of growth rate and early life and peripubertal body weight with PLAG1 polymorphisms, supporting its status as a key regulator of mammalian growth.
The lack of significant association between BW and SNPs within other previously described weight- and height-related chromosome regions in the present study should not be interpreted as a lack of existence of true association, but rather it might be due to limitations specific to this study. Firstly, because complex trait mapping requires large sample sizes and only 649 bulls were analyzed here. Secondly, the significance level adopted was highly stringent, which may have caused inflation of type II errors. In spite of these limitations, it was possible to demonstrate that a well-characterized chromosome region affecting human and taurine cattle stature also associates with BW in a zebu breed. The release of a Bos primigenius indicus reference genome assembly, as well as the application of re-sequencing and replication studies would help improve resolution to narrow down the genomic region as close as possible to the true causative variants.
Conclusions
This study is believed to be the first genome-wide association study applying a high-density SNP panel to identify putative chromosome regions affecting birth weight in zebu cattle. The findings presented, which are strongly supported by the literature, point to orthologous genes already known to affect growth- and stature-related traits in both humans and cattle, which may shelter ancient polymorphisms responsible for variation in those traits since before cattle subspecies divergence.
Methods
Estimated breeding values
Estimated breeding values for BW were obtained from routine genetic evaluations using performance and pedigree data from the Aliança database [11], containing data from different commercial Nellore breeding programs, including more than 250 farms distributed across Brazil and Paraguay. The genetic evaluation for BW was calculated using a subset of that data that included 542,918 animals, born from 1985 to 2011, and distributed in approximately 5,000 distinct contemporary groups. These data were collected in 243 grazing-based herds in Brazil. Estimated breeding values were obtained using an animal model that included fixed effects for the age of dam at calving and contemporary group (defined as animals from the same herd, born in the same year and season, and belonging to the same management group at birth, and sex), as well as random effects that include direct additive genetic, maternal additive genetic, maternal permanent environmental and residual error effects. The variance ratios required to solve the mixed model equations were computed based on restricted maximum likelihood (REML) estimates of the variance components from previous studies in this population. Only EBVs of progeny-tested bulls whose accuracy (i.e., square root of reliability, calculated based on prediction error variance estimates) was ≥ 0.50 were used for sample collection and genotyping (described later). The majority of the bulls were used under artificial insemination service.
Genotyping, informativeness, and quality assurance
A total of 654 progeny-tested Nellore bulls were genotyped with the Illumina® BovineHD Genotyping BeadChip assay, according to the manufacturer’s protocol. Genotype calls (i.e. successfully determined genotypes) were defined as genotypes with GenCall Scores greater than 0.70, using the validated standard cluster file provided by the manufacturer. As chromosomes X, Y and mtDNA present different mode of inheritance from the rest of the genome, only autosomal markers with unique genomic coordinates were included into the analyses. After this initial screening, potential duplicated samples were determined by calculating the proportion of alleles identical by state (IBS) shared between all pairs of individuals. Any pair of samples with IBS ≥ 0.95 for 2,000 randomly sampled markers was considered unexpected duplicates, and resulted in the exclusion of both members of the pair. Individual SNPs were removed from the dataset if they did not exhibit: 1) minor allele frequency (MAF) greater than or equal to 0.02, 2) Fisher’s exact test P-value for Hardy-Weinberg Equilibrium (HWE) greater than or equal to 1 × 10-5 (i.e. extremely deviating from HWE, suggesting potential genotyping error) or 3) Call rate (CRSNP) of at least 98%. After the SNP pruning, individuals exhibiting call rate (CRIND) below 90% were also removed. These procedures and many others described later were performed in the R v2.15.0 environment [40], using combinations of functions from the R base, locally developed scripts, and the GenABEL v1.7-2 package [41].
Assessment of population substructure
Sires genotyped in this study were known to belong to one of two major breeding program subgroups in the Aliança database [11] that have different selection objectives. One group emphasizes selection for weaning and yearling weight (subgroup 1) and the other emphasizes selection for fertility and carcass traits (subgroup 2). Thus, genetic stratification was expected and therefore population substructure was evaluated by performing a Principal Coordinates Analysis (PCoA). Pair-wise genomic kinship coefficients for all subjects under study were calculated first, following [42,43]:
| (1) |
Where is the estimated genomic kinship between individuals i and j, L is the total number of loci used for the calculation, pl is the reference allele frequency for locus l, and gl,i and gl,j are the locus l genotypes for individuals i and j, respectively (coded as 0, 1 or 2 reference alleles). Calculations were based on 10,000 randomly sampled markers using GenABEL [41]. The calculated genomic kinship coefficients within the yielded n x n symmetric matrix (where n is the total number of samples) were then transformed to squared Euclidean distances, and the dissimilarities between the subjects within the matrix were captured in n-1 dimensional spaces of n observations (eigenvectors), via classical multidimensional scaling [44].
A clustering analysis was applied to the two eigenvectors that explained the largest proportion of the data variance using the k-means algorithm [45] implemented in R[40]. Individuals were clustered into 2 groups, and the association between the prior information on breeding program and the k-means clustering results was tested using Pearson’s χ2 with Yates’ continuity correction in order to see if the algorithm could reproduce the known breeding programs subgroups. Additionally, an F-test for homogeneity of variance between subpopulations and a t-test for difference between subpopulation means, defining subpopulations either as k-means assignments or breeding program of origin, was performed to determine if there was confounding due to stratification.
Association analysis
In order to reduce computation time, the ideas of [46] were abstracted and a three-step association analysis was performed. In the first step, a linear regression using the weighted least squares method with weights equal to the squared accuracy (i.e., reliability) of the EBVs was applied. By weighting the EBVs by their respective accuracies, the uncertainty around the estimates was taken into account when estimating the regression parameters. The following model was fitted:
| (2) |
Where yi is the EBV of sire i, μ is the overall mean, Xij is value i (corresponding to sire i) in the eigenvector j calculated in the PCoA, βj is the estimated effect of eigenvector j, and ϵi is the residual effect for animal j. Only eigenvectors significantly (P < 0.05) correlated with the dependent variable, as assessed by Pearson correlations, were included in the model. Next, the residuals were obtained from the fitted model in (2):
| (3) |
and had their homoskedasticity and normality tested by using the studentized Breusch-Pagan test and the Shapiro-Wilk test, respectively. Then, these residuals were used as the new dependent variable for a single-marker linear regression:
| (4) |
Where βg is the marker regression coefficient (i.e., the allele substitution effect of the SNP) and gi is the genotype (0, 1 or 2) of the sire i. For each SNP, βg and its respective standard error (SEg) were estimated using ordinary least squares. The association between the SNP and the trait was assessed via a test statistic, calculated as:
| (5) |
The test statistics are assumed to asymptotically follow a χ2 distribution with one degree of freedom under the null hypothesis. To assess the validity of this assumption, the deviation of the distribution of the test statistics from the expected theoretical quantiles was examined via 1) a quantile-quantile (Q-Q) plot, and 2) calculation of the inflation/deflation factor:
| (6) |
If λ < 1.1, the inflation was considered acceptable, and the Genomic Control (GC) correction was applied to adjust for that inflation [47]. Then, P-values were derived from the χ2 cumulative distribution function for the corrected test statistics. Finally, markers within the smallest 0.1% P-value percentile (i.e., most significant) were considered for re-analysis with the full model:
| (7) |
The EBVs were again weighted by their respective accuracies. The conservative Bonferroni adjustment for multiple testing (α=0.05/N, where N is the number of tests, i.e., number of SNPs) was used to reject the null hypothesis (βg = 0, i.e. there is no association between the SNP and the EBVs), which resulted in an adjusted significance of α = 1.15 × 10-7.
Exploratory view of significant SNPs
For any peak crossing the Bonferroni significance threshold, the estimated regression parameters were reported. For the most significant SNP, a 95% confidence interval (CI) for the estimated allele substitution effect size () was calculated, and the percentage of the EBV variance explained was calculated as:
| (8) |
Where p and q are the allele frequencies and S2 is the sample EBVs variance. The upper and lower limits of the estimated 95% CI for were used to derive a 95% CI for % .
The genomic region containing the most significant SNP of a peak was explored by inspecting a 1 Mb window around the location of this SNP using the BioMart tool and the Ensembl genes 69 database [48] to interrogate 500 kb to each side of the marker using the UMD v3.1 assembly. The cattle QTLdb database [49] was also examined to find out if the significant SNP mapped against any previously described bovine QTL. Additionally, the alignments of the UMD v3.1 assembly sequence of the 1 Mb window against the human (Homo sapiens, GRCh37 assembly), pig (Sus scrofa, Sscrofa10.2 assembly) and mouse (Mus musculus, GRCm38 assembly) genome builds were inspected in the Ensembl Comparative genomics alignments and Comparative genomics synteny tools in order to determine if any homologous genes were present in the putative region.
Abbreviations
BW: Birth weight; EBV: Estimated breeding value; SNP: Single nucleotide polymorphism; BTA: Bos primigenius taurus; GWAS: Genome-wide association study; IBS: Identity by state; MAF: Minor allele frequency; HWE: Hardy-weinberg equilibrium; PCoA: Principal coordinates analysis; Q-Q plot: quantile-quantile plot; GC: Genomic control; CI: Confidence interval; HSA: Homo sapiens; SSC: Sus scrofa; MMU: Mus musculus; QTL: Quantitative trait locus; CHILLP8: Rump fat thickness measured at the P8 position; QTN: Quantitative trait nucleotide; LD: Linkage disequilibrium
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JFG conceived and led the coordination of the study. JS, JM, JBC, CPVT, FSS, MVGBS, LRPN and TSS contributed to the study design and coordination. ASC directed the genotyping work and contributed to the data analysis. RC and HHRN provided EBVs and assisted the data analysis. YTU led the data analysis and the manuscript preparation. MCM, LBZ and AMPO contributed in the data preparation and analysis. JBC, FSS, LRPN, TSS, CPVT, JFG, RC, HHRN, ASC and YTU interpreted the results and contributed to edit the manuscript. All authors read and approved the final manuscript.
Contributor Information
Yuri T Utsunomiya, Email: ytutsunomiya@gmail.com.
Adriana S do Carmo, Email: adrianasantanacarmo@gmail.com.
Roberto Carvalheiro, Email: r.carvalheiro@gensys.com.br.
Haroldo HR Neves, Email: haroldozoo@hotmail.com.
Márcia C Matos, Email: marciacmattos@yahoo.com.br.
Ludmilla B Zavarez, Email: luzavarez@hotmail.com.
Ana M Pérez O’Brien, Email: anita_op@hotmail.com.
Johann Sölkner, Email: johann.soelkner@boku.ac.at.
John C McEwan, Email: john.mcewan@agresearch.co.nz.
John B Cole, Email: John.Cole@ars.usda.gov.
Curtis P Van Tassell, Email: curt.vantassell@ars.usda.gov.
Flávio S Schenkel, Email: schenkel@uoguelph.ca.
Marcos VGB da Silva, Email: marcos@cnpgl.embrapa.br.
Laercio R Porto Neto, Email: laercioporto@gmail.com.
Tad S Sonstegard, Email: Tad.Sonstegard@ars.usda.gov.
José F Garcia, Email: jfgarcia@fmva.unesp.br.
Acknowledgements
We thank Guilherme Penteado Coelho Filho and Daniel Biluca for technical assistance in sample acquisition. This research was supported by: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) - process 560922/2010-8 and 483590/2010-0; Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) - process 2011/16643-2 and 2010/52030-2; Next-Generation BioGreen 21 Program (No. PJ008196), Rural Development Administration, Republic of Korea.
Mention of trade name proprietary product or specified equipment in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the authors or their respective institutions.
References
- Boligon AA, Albuquerque LG, Mercadante MEZ, Lôbo RB. Herdabilidades e correlações entre pesos do nascimento à idade adulta em rebanhos da raça Nelore. Rev. Bras. Zootec. 2009;38(12):2320–2326. doi: 10.1590/S1516-35982009001200005. [DOI] [Google Scholar]
- Meyer K. Estimates of genetic parameters for mature weight of Australian beef cows and its relationship to early growth and skeletal measures. Livest Prod Sci. 1995;44:125–137. doi: 10.1016/0301-6226(95)00067-4. [DOI] [Google Scholar]
- Bourdon RM, Brinks JS. Genetic, environmental and phenotypic relationships among gestation length, birth weight, growth traits and age at first calving in beef cattle. J Anim Sci. 1982;55(3):543–553. doi: 10.2527/jas1982.553543x. [DOI] [PubMed] [Google Scholar]
- Eriksson S, Näsholm A, Johansson K, Philipsson J. Genetic parameters for calving difficulty, stillbirth, and birth weight for Hereford and Charolais at first and later parities. J Anim Sci. 2004;82:375–383. doi: 10.2527/2004.822375x. [DOI] [PubMed] [Google Scholar]
- Phocas F, Bloch C, Chapelle P, Bécherel F, Renand G, Ménissier F. Developing a breeding objective for a French purebred beef cattle selection programme. Livest Prod Sci. 1998;57:49–65. doi: 10.1016/S0301-6226(98)00157-2. [DOI] [Google Scholar]
- Gutiérrez JP, Goyache F, Fernández I, Alvarez I, Royo LJ. Genetic relationships among calving ease, calving interval, birth weight, and weaning weight in the Asturiana de los Valles beef cattle breed. J Anim Sci. 2007;85:69–75. doi: 10.2527/jas.2006-168. [DOI] [PubMed] [Google Scholar]
- Cook BR, Tess MW, Kress DD. Effects of selection strategies using heifer pelvic area and sire birth weight expected progeny difference on dystocia in first-calf heifers. J Anim Sci. 1993;71:602–607. doi: 10.2527/1993.713602x. [DOI] [PubMed] [Google Scholar]
- Johanson JM, Berger PJ. Birth weight as a predictor of calving ease and perinatal mortality in Holstein cattle. J Dairy Sci. 2003;86(11):3745–3755. doi: 10.3168/jds.S0022-0302(03)73981-2. [DOI] [PubMed] [Google Scholar]
- Nobre PR, Misztal I, Tsuruta S, Bertrand JK, Silva LO, Lopes OS. Analyses of growth curves of Nellore cattle by multiple-trait and random regression models. J Anim Sci. 2003;81:918–926. doi: 10.2527/2003.814918x. [DOI] [PubMed] [Google Scholar]
- Albuqueque LG, Meyer K. Estimates of direct and maternal genetic effects for weights from birth to 600 days of age in Nelore cattle. J Anim Breed Genet. 2001;118:83–92. doi: 10.1046/j.1439-0388.2001.00279.x. [DOI] [Google Scholar]
- Aliança. Sumário de touros 2011 Aliança Nelore. Porto Alegre, Brazil: GenSys Consultores Associados; 2011. [Google Scholar]
- Spelman RJ, Huisman AE, Singireddy SR, Coppieters W, Arranz J, Georges M, Garrick DJ. Short communication: quantitative trait loci analysis on 17 nonproduction traits in the New Zealand dairy population. J Dairy Sci. 1999;82(11):2514–2516. doi: 10.3168/jds.S0022-0302(99)75503-7. [DOI] [PubMed] [Google Scholar]
- Kneeland J, Li C, Basarab J, Snelling WM, Benkel B, Murdoch B, Hansen C, Moore SS. Identification and fine mapping of quantitative trait loci for growth traits on bovine chromosomes 2, 6, 14, 19, 21, and 23 within one commercial line of Bos taurus. J Anim Sci. 2004;82(12):3405–3414. doi: 10.2527/2004.82123405x. [DOI] [PubMed] [Google Scholar]
- Maltecca C, Weigel KA, Khatib H, Cowan M, Bagnato A. Whole-genome scan for quantitative trait loci associated with birth weight, gestation length and passive immune transfer in a Holstein x Jersey crossbred population. Anim Genet. 2009;40(1):27–34. doi: 10.1111/j.1365-2052.2008.01793.x. [DOI] [PubMed] [Google Scholar]
- McClure MC, Morsci NS, Schnabel RD, Kim JW, Yao P, Rolf MM, McKay SD, Gregg SJ, Chapple RH, Northcutt SL, Taylor JF. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet. 2010;41(6):597–607. doi: 10.1111/j.1365-2052.2010.02063.x. [DOI] [PubMed] [Google Scholar]
- Cole JB, Wiggans GR, Ma L, Sonstegard TS, Lawlor TJ, Crooker BA, Van Tassell CP, Yang J, Wang S, Matukumalli LK, Da Y. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics. 2011;12:408. doi: 10.1186/1471-2164-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsik CG, Tellam RL, Worley KC, Gibbs RA, Muzny DM, Weinstock GM, Adelson DL, Eichler EE, Elnitski L, Guigó R, Hamernik DL, Kappes SM, Lewin HA, Lynn DJ, Nicholas FW, Reymond A, Rijnkels M, Skow LC, Zdobnov EM, Schook L, Womack J, Alioto T, Antonarakis SE, Astashyn A, Chapple CE, Chen HC, Chrast J, Câmara F, Ermolaeva O. Bovine Genome Sequencing and Analysis Consortium et al. The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science. 2009;324(5926):522–528. doi: 10.1126/science.1169588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Taylor JF, Van Tassell CP, Barendse W, Eversole KA, Gill CA, Green RD, Hamernik DL, Kappes SM, Lien S, Matukumalli LK, McEwan JC, Nazareth LV, Schnabel RD, Weinstock GM, Wheeler DA, Ajmone-Marsan P, Boettcher PJ, Caetano AR, Garcia JF, Hanotte O, Mariani P, Skow LC, Sonstegard TS, Williams JL, Diallo B, Hailemariam L, Martinez ML, Morris CA, Bovine HapMap Consortium Science. 2009. pp. 528–532. [DOI] [PMC free article] [PubMed]
- Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, O’Connell J, Moore SS, Smith TP, Sonstegard TS, Van Tassell CP. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009;4(4):e5350. doi: 10.1371/journal.pone.0005350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snelling WM, Allan MF, Keele JW, Kuehn LA, McDaneld T, Smith TP, Sonstegard TS, Thallman RM, Bennett GL. Genome-wide association study of growth in crossbred beef cattle. J Anim Sci. 2010;88(3):837–848. doi: 10.2527/jas.2009-2257. [DOI] [PubMed] [Google Scholar]
- Pausch H, Flisikowski K, Jung S, Emmerling R, Edel C, Götz KU, Fries R. Genome-wide association study identifies two major loci affecting calving ease and growth-related traits in cattle. Genetics. 2011;187:289–297. doi: 10.1534/genetics.110.124057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryce JE, Hayes BJ, Bolormaa S, Goddard ME. Polymorphic regions affecting human height also control stature in cattle. Genetics. 2011;187:981–984. doi: 10.1534/genetics.110.123943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura S, Watanabe T, Mizoshita K, Tatsuda K, Fujita T, Watanabe N, Sugimoto Y, Takasuga A. Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 2012;13:40. doi: 10.1186/1471-2156-13-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson DF, Walters GB, Thorleifsson G, Stefansson H, Halldorsson BV, Zusmanovich P, Sulem P, Thorlacius S, Gylfason A, Steinberg S, Helgadottir A, Ingason A, Steinthorsdottir V, Olafsdottir EJ, Olafsdottir GH, Jonsson T, Borch-Johnsen K, Hansen T, Andersen G, Jorgensen T, Pedersen O, Aben KK, Witjes JA, Swinkels DW, den Heijer M, Franke B, Verbeek AL, Becker DM, Yanek LR, Becker LC. et al. Many sequence variants affecting diversity of adult human height. Nat Genet. 2008;40:609–615. doi: 10.1038/ng.122. [DOI] [PubMed] [Google Scholar]
- Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, Freathy RM, Perry JR, Stevens S, Hall AS, Samani NJ, Shields B, Prokopenko I, Farrall M, Dominiczak A, Johnson T, Bergmann S, Beckmann JS, Vollenweider P, Waterworth DM, Mooser V, Palmer CN, Morris AD, Ouwehand WH, Zhao JH, Li S, Loos RJ. Diabetes Genetics Initiative; Wellcome Trust CaseControl Consortium et al. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, Sanna S, Eyheramendy S, Voight BF, Butler JL, Guiducci C, Illig T, Hackett R, Heid IM, Jacobs KB, Lyssenko V, Uda M, Boehnke M, Chanock SJ, Groop LC, Hu FB, Isomaa B, Kraft P, Peltonen L, Salomaa V. Diabetes Genetics Initiative; FUSION; KORA; Prostate, Lung Colorectal and Ovarian Cancer Screening Trial; Nurses’ HealthStudy; SardiNIA et al. Identification of ten loci associated withheight highlights new biological pathways in human growth. Nat Genet. 2008;40:584–591. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim L, Takeda H, Lin L, Druet T, Arias JA, Baurain D, Cambisano N, Davis SR, Farnir F, Grisart B, Harris BL, Keehan MD, Littlejohn MD, Spelman RJ, Georges M, Coppieters W. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet. 2011;43:405–413. doi: 10.1038/ng.814. [DOI] [PubMed] [Google Scholar]
- Littlejohn M, Grala T, Sanders K, Walker C, Waghorn G, Macdonald K, Coppieters W, Georges M, Spelman R, Hillerton E, Davisand S, Snell R. Genetic variation in PLAG1 associates with early life body weight and peripubertal weight and growth in Bos taurus. Anim Genet. 2012;43(5):591–594. doi: 10.1111/j.1365-2052.2011.02293.x. [DOI] [PubMed] [Google Scholar]
- Garrick DJ, Taylor JF, Fernando RL. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet Sel Evol. 2009;41:55. doi: 10.1186/1297-9686-41-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JB, VanRaden PM, O’Connell JR, Van Tassell CP, Sonstegard TS, Schnabel RD, Taylor JF, Wiggans GR. Distribution and location of genetic effects for dairy traits. J Dairy Sci. 2009;92:2931–2946. doi: 10.3168/jds.2008-1762. [DOI] [PubMed] [Google Scholar]
- Fortes MRS, Lehnert SA, Bolormaa S, Reich C, Fordyce G, Corbet NJ, Whan V, Hawken RJ, Reverter A. Finding genes for economically important traits: Brahman cattle puberty. Animal Production Science. 2012;52:143–150. doi: 10.1071/AN11165. [DOI] [Google Scholar]
- Fortes MR, Reverter A, Hawken RJ, Bolormaa S, Lehnert SA. Candidate genes associated with testicular development, sperm quality, and hormone levels of inhibin, luteinizing hormone, and insulin-like growth factor 1 in Brahman bulls. BiolReprod. 2012;87(3):58. doi: 10.1095/biolreprod.112.101089. [DOI] [PubMed] [Google Scholar]
- Hawken RJ, Zhang YD, Fortes MRS, Collis E, Barris WC, Corbet NJ, Williams PJ, Fordyce G, Holroyd RG, Walkley JRW, Barendse W, Johnston DJ, Prayaga KC, Tier B, Reverter A, Lehnert SA. Genome-wide association studies of female reproduction in tropically adapted beef cattle. J Anim Sci. 2012;90:1398–1410. doi: 10.2527/jas.2011-4410. [DOI] [PubMed] [Google Scholar]
- Lindholm-Perry AK, Kuehn LA, Smith TP, Ferrell CL, Jenkins TG, Freetly HC, Snelling WM. A region on BTA14 that includes the positional candidate genes LYPLA1, XKR4 and TMEM68 is associated with feed intake and growth phenotypes in cattle. Anim Genet. 2012;43(2):216–219. doi: 10.1111/j.1365-2052.2011.02232.x. [DOI] [PubMed] [Google Scholar]
- Bolormaa S, Porto Neto LR, Zhang YD, Bunch RJ, Harrison BE, Goddard ME, Barendse W. A genome-wide association study of meat and carcass traits in Australian cattle. J Anim Sci. 2011;89(2):297–309. doi: 10.2527/jas.2010-3138. [DOI] [PubMed] [Google Scholar]
- Porto Neto LR, Bunch RJ, Harrison BE, Barendse W. Variation in the XKR4 gene was significantly associated with subcutaneous rump fat thickness in indicine and composite cattle. Anim Genet. 2012;43(6):785–789. doi: 10.1111/j.1365-2052.2012.02330.x. [DOI] [PubMed] [Google Scholar]
- Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S, Ferreira T, Wood AR, Weyant RJ, Segrè AV, Speliotes EK, Wheeler E, Soranzo N, Park JH, Yang J, Gudbjartsson D, Heard-Costa NL, Randall JC, Qi L, Vernon Smith A, Mägi R, Pastinen T, Liang L, Heid IM, Luan J, Thorleifsson G. et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyck F, Declercq J, Braem CV, Van de Ven WJ. PLAG1, the prototype of the PLAG gene family: versatility in tumour development. Int J Oncol. 2007;30:765–774. [PubMed] [Google Scholar]
- Hensen K, Braem C, Declercq J, Van Dyck F, Dewerchin M, Fiette L, Denef C, Van de Ven WJM. Targeted disruption of the murine PLAG1 proto-oncogene causes growth retardation and reduced fertility. Dev Growth Differ. 2004;46:459–470. doi: 10.1111/j.1440-169x.2004.00762.x. [DOI] [PubMed] [Google Scholar]
- R Development Core Team: R. A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. http://www.R-project.org. [Google Scholar]
- Aulchenko YS, Ripke S, Isaacs A, Van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23(10):1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- Amin N, Van Duijn CM, Aulchenko YS. A genomic background based method for association analysis in related individuals. PLoS One. 2007;2:e1274. doi: 10.1371/journal.pone.0001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astle W, Balding DJ. Population structure and cryptic relatedness in genetic association studies. Stat Sci. 2009;24(4):451–471. doi: 10.1214/09-STS307. [DOI] [Google Scholar]
- Mardia KV. Some properties of classical multi-dimensional scaling. Communications on Statistics-Theory and Methods. 1978;A7(13):1233–1241. [Google Scholar]
- Hartigan JA, Wong MA. A K-means clustering algorithm. Applied Statistics. 1979;28:100–108. doi: 10.2307/2346830. [DOI] [Google Scholar]
- Aulchenko YS, de Koning D-J, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics. 2007;177:577–585. doi: 10.1534/genetics.107.075614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341X.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A, Kersey P, Flicek P. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011;2011:Bar030. doi: 10.1093/database/bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZL, Park CA, Wu XL, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2012. in press. [DOI] [PMC free article] [PubMed]