

Abstract
Background
Detailed study of genetic variation at the population level in humans and other species is now possible due to the availability of large sets of single nucleotide polymorphism data. Alleles at two or more loci are said to be in linkage disequilibrium (LD) when they are correlated or statistically dependent. Current efforts to understand the genetic basis of complex phenotypes are based on the existence of such associations, making study of the extent and distribution of linkage disequilibrium central to this endeavour. The objective of this paper is to develop methods to study fine-scale patterns of allelic association using probabilistic graphical models.
Results
An efficient, linear-time forward-backward algorithm is developed to estimate chromosome-wide LD models by optimizing a penalized likelihood criterion, and a convenient way to display these models is described. To illustrate the methods they are applied to data obtained by genotyping 8341 pigs. It is found that roughly 20% of the porcine genome exhibits complex LD patterns, forming islands of relatively high genetic diversity.
Conclusions
The proposed algorithm is efficient and makes it feasible to estimate and visualize chromosome-wide LD models on a routine basis.
Background
Alleles at two loci are said to be in linkage disequilibrium (LD) when they are correlated or statistically dependent. The term refers to the idea that in a large homogeneous population subject to random mating, recombination between two loci will cause any initial association between them to vanish over time. In observed data, however, non-zero allelic associations are pervasive, particularly at short distances, but also at long distances and even between chromosomes. These associations arise in a complex interplay between processes such as mutation, selection, genetic drift and population admixture, and are broken down by recombination. The patterns of association are of interest, partly because they underpin the relation of genotype to phenotype at the population level, and partly because they reflect population history.
Patterns of LD may be represented in different ways. A common method is to display pairwise measures of LD as triangular heatmaps [1,2]: in these displays, LD blocks (genomic intervals within which all loci are in high LD) stand out clearly. Early work in the HapMap project led researchers to hypothesize that the human genome consists of a series of disjoint blocks, within which there is high LD, low haplotype diversity and little recombination, and that are punctuated by short regions with high recombination (recombination hotspots) [3-6]. Subsequently various authors [7,8] reported that genetic variation follows more complex patterns, for which richer models are required.
Discrete graphical models [9] (also known as discrete Markov networks) provide a rich family of statistical models to describe the distribution of multivariate discrete data. They may be represented as undirected graphs in which the nodes represent variables (here, SNPs) and absent edges represent conditional independence relations, in the sense that two variables that are not connected by an edge are conditionally independent given some other variables. To motivate this focus on conditional rather than marginal associations, consider three loci , and suppose that initially s2 is polymorphic and s1 and s2 monomorphic, so that two haplotypes (1,1,1) and (1,2,1) are initially present. Suppose further that a mutation subsequently occurs at s1 in the haplotype (1,1,1), and another at s3 in the haplotype (1,2,1), so that the population now contains the four haplotypes (1,1,1), (1,2,1), (2,1,1) and (1,2,2). Observe that in general s1 and s3 are marginally associated (are in LD), but in the subpopulations corresponding to s2=1 and s2=2 they are unassociated: in other words, they are conditionally independent given s2. More complex mutation histories give rise to more complex patterns of conditional independences that can be represented as graphical models [8].
Other authors have used graphical models for the joint distributions of allele frequencies. Usually, in high-dimensional applications, attention is restricted to a tractable subclass, the decomposable graphical models [10]. In the first use of decomposable models in this context [11], models were selected using a greedy algorithm based on significance tests. In [8,12] methods and programs for selecting decomposable graphical models using Monte Carlo Markov Chain (MCMC) sampling were described. These methods are computationally feasible for modest numbers of markers (say, several hundreds), but not for modern SNP arrays with hundreds of thousands of SNPs per chromosome. To improve efficiency, the search space may be restricted to graphical models whose dependence graphs are interval graphs[13,14]. These are graphs for which each vertex may be associated with an interval of the real line such that two vertices are connected by an edge if and only if their intervals overlap. In this context the ordering of SNPs along the real line is their physical ordering along the chromosome. MCMC sampling from this model class may be performed more efficiently [13,14]. This work was extended in [15] to a more general subclass of decomposable models, namely those in which distant marker pairs (i.e., with more than a given number of intervening markers) are conditionally independent given the intervening markers.
In an alternative approach [16-18] latent mixtures of forests have been applied, in order to accommodate short-, medium- and long-range LD patterns. Also directed graphs (Bayesian networks) have been applied, selecting edges and their directions using causal discovery algorithms [19]. There are close links between decomposable models and Bayesian networks ([10], Sect. 4.5.1).
A rather different approach to modelling the joint distribution of allele frequencies [20,21] is implemented in the software package BEAGLE [22], which is widely used to process data from SNP arrays. The approach is based on a class of models arising in the machine learning literature called acyclic probabilistic finite automata (APFA) [23]. These are related to time-variant variable length Markov chains. For phase estimation and imputation BEAGLE uses an iterative scheme analogous to the EM algorithm, alternating between sampling from a haplotype-level model given the observed genotype data (the E-step) and selecting a haplotype-level model given the samples (the M-step). A similar computational scheme for decomposable graphical models has been described and implemented in the FitGMLD program [15].
Characterization of genetic variation at the population level is of fundamental importance to understanding how phenotypes relate to genotypes. Some specific uses to which joint models for allele frequencies have been put include
1. Insight into the population history of different genomic intervals. Under simplifying assumptions, the ancestral history of a short genomic interval can be reconstructed from a decomposable graphical model for the SNPs in the interval [8].
2. Quality control of genome assembly, in that some motifs may suggest errors in SNP positioning.
3. Phase estimation and imputation as described above [15,21].
4. Derivation of more informative covariates involving multiple loci to plug into genomic prediction models [20].
5. Use in pedigree simulation to model LD in founders, for example in connection with gene drop simulation [14] and assessment of SNP streak statistics [24].
In this paper decomposable graphical models are used to model fine-scale, local LD patterns. By local is meant that the proposed methods are designed to capture short-range associations between loci, but not long range ones. The same is true of other approaches [13-15] mentioned above. Here, an efficient, linear-time algorithm is developed to select a model using a penalized likelihood criterion, and it is shown how such a model may conveniently be displayed, allowing fine-scale LD structure to be visualized.
Methods
Graphs and graphical models
The following notation and terminology is mainly based on [9]. A graph is defined as a pair , where V is a set of vertices or nodes and E is a set of edges. Each edge is associated with a pair of nodes, its endpoints. Here only undirected graphs are considered, that is, with graphs undirected edges only. Two vertices α and β are said to be adjacent, written α∼β, if there is an edge between them. The neighbours of a vertex is the set of nodes that are adjacent to it. A subset A⊆V is complete if all vertex pairs in A are connected by an edge. A clique is a maximal complete subset, that is to say, a complete subset that is not contained in a larger complete subset.
A path (of length n) between vertices α and β in an undirected graph is a set of vertices where αi−1∼αi for . If a path has α=β then the path is said to be a cycle of length n. If a cycle has adjacent elements αi∼αj with j∉{i−1,i+1} then it is said to have a chord. If it has no chords it is said to be chordless. A graph with no chordless cycles of length ≥4 is called triangulated or chordal.
A subset D⊂V in an undirected graph is said to separateA⊂V from B⊂V if every path between a vertex in A and a vertex in B contains a vertex from D. The graph is said to be a subgraph of if V0⊆V and E0⊆E. For A⊆V, let EA denote the set of edges in E between vertices in A. Then is the subgraph induced byA.
The boundary bd(A) of a vertex set A⊆V is the set of vertices adjacent to a vertex in A but not in A, that is ,.
Let be an undirected graph with cliques . Consider a joint density f() of the variables in V. If this admits a factorization of the form
for some functions where gj() depends on x only through then f() is said to factorize according to . If all the densities in a model factorize according to , then the model is said to be -Markov. When this is true encodes the conditional independence structure of the model, through the following result (the global Markov property): whenever sets A and B are separated by a set C in , A and B are conditionally independent given C under the model. This is written as A⊥ ⊥B | C. A decomposable graphical model is one whose dependence graph is triangulated.
Selecting graphical models for chromosome-wide LD
Suppose that N observations of p SNPs from the same chromosome are sampled from some population. The variable set is written , and it is assumed that these are ordered by physical position on the chromosome. The variables may be either observed genotypes or inferred haplotypes, if these have been imputed: the former are trichotomous and the latter binary. Here an algorithm to use these data to select a graphical model for the distribution of V is described. It is based on a penalized likelihood criterion
| (1) |
where is the maximized log-likelihood under , is the number of free parameters under , and α is a penalization constant. For the AIC, α=2 and for the BIC, α= log(N). The latter penalizes complex models more heavily and so selects simpler models. Under suitable regularity conditions, the BIC is consistent in the sense that for large N it will select the simplest model consistent with the data ([25], Sect. 2.6).
A technical but nonetheless important point is that calculation of the correct model dimension is not straightforward for high-dimensional models, since not all parameters may be estimable. A simple expression exists however for the difference in model dimension between two decomposable models that differ by a single edge ([10], pp. 37-40). This is useful when the search space is restricted to decomposable models and the search algorithm only involves comparison of models differing by single edges (as here).
A forward-backward approach to estimate the graphical model for V is used. The first (forward) step is based on a greedy forward search algorithm. To take account of the physical ordering of the SNPs, the algorithm starts from a model where E0 is the set of edges between physically adjacent SNPs: this model is called the skeleton. To seek the minimum BIC (or AIC) model, the algorithm repeatedly adds the edge associated with the greatest reduction in BIC (AIC): only edges whose inclusion results in a decomposable model are considered. The process continues until no more edges improve the criterion. The search space in this step is the class of decomposable models that include the skeleton. Note that this algorithm – as with almost all greedy algorithms — is not guaranteed to find the global optimum.
There are several advantages to initially constraining the search space to include the skeleton. An unconstrained search starting off from the null model (with no edges) would not take the physical ordering into account. Since the graphs considered are consistent with this ordering, they are conveniently displayed as LD maps, as illustrated below. Because decomposable models contain no chordless cycles of length ≥4, two distal loci cannot be linked by an edge unless there are sufficiently many intermediate (chordal) edges to prevent the occurrence of such cycles. Thus the algorithm proceeds by filling out around the skeleton by incrementally adding edges. The effect of this is that only local LD is modelled. Since both association and (implicitly) proximity are required, the edges included are more likely to be for real (that is, less likely to be due to chance), so in this sense the model selection process is more reliable. In addition, restricting the search space in this way improves computational efficiency. The linear-time algorithm described in the following section also builds on the physical vertex ordering.
In the second (backward) step the graph found in the first step is pruned, again using a greedy algorithm that seeks to minimize the BIC (AIC) criterion by removing edges, without requiring that the skeletal edges are retained. Keeping these in the model would be appropriate if adjacent SNPs are in high LD, but this is not always the case. For example, there may be recombination hotspots, or genome assembly errors may have led to errors in positioning the SNPs. The graphs may be used to examine whether this has occurred.
To display the resulting graph, a suitable graph layout is needed. After some experimentation, one such was obtained by modifying the horizontal dot layout in Rgraphviz [26], by exponentially smoothing the y-coordinates and replacing the x-coordinates with the SNP-number. In this way the y-coordinates are chosen so as to clarify the graphical structure: a consequence of this is that nodes with large absolute y-coordinates tend to signal genomic regions of high structural complexity.
For some purposes it is helpful to use more objective measures of complexity, and two such measures are used here. Consider a chromosome with p SNPs. For each , the heighthi of the interval between SNPs i and i+1 is defined to be the number of edges of the form (j,k), where j≤i and i+1≤k, and the widthwi is defined to be the maximum value of |k−j| of such edges. Note that by the global Markov property, when hi=0 or equivalently wi=0, the SNPs V≤i={vj:j≤i} are independent of V>i={vj:j>i}. Similarly, when hi=1 or equivalently wi=1, V<i⊥⊥V>i|vi.
As a measure of haplotype diversity, the entropy[27] is used here. Suppose that in some genomic interval there are k distinct haplotypes with relative frequencies . Then the entropy is defined as . It is large when there are many distinct haplotypes that are equally frequent.
A useful way to display marker data over short genomic intervals is the sample tree[23]. This summarizes a set of discrete longitudinal data of the form for . A rooted tree is constructed in which the non-root nodes represent partial outcomes of the form for k≤q present in the sample data. Edges may be coloured with the marker value and labelled with the corresponding sample counts, or drawn with width proportional to the sample counts.
A fast selection algorithm
For high-dimensional models, the first step of the algorithm described above can be computationally demanding. To address this a much faster algorithm for the same task is now described. This involves combining a series of overlapping marginal models of lower dimension. Call the block length (model dimension) L and the overlap K. Thus the first block is , the second is and so on. In the applications described here L=100 and K=20 are used.
Suppose that the true model for is , and that is the estimate of obtained by performing the first step of the algorithm described in the previous section. The goal is to construct an approximation to by combining models obtained by applying that same algorithm to blocks Vi for .
A way to construct a model for V1∪V2 by combining and is now described. Let . Then is defined as , where and .
The rationale for this is that marginal models may include spurious associations on the boundaries. For example, let and be the subgraphs of induced by V1 and V2, respectively. Then the marginal distribution of V2 will not in general be -Markov, but it will be -Markov for a graph derived from by completing the boundary of each connected component of in [28]. So will tend to contain edges not in . To estimate the boundary of V1∖V2 in , is used: the boundary is estimated to be contained in the set . Hence by only including edges with w>m∗ edges in this boundary are omitted. Similarly, the boundary of V2∖V1 in may contain spurious associations, so may contain unnecessary edges. To avoid this the overlap length K is chosen to be sufficiently large so that m∗< min{v:∃ (v,w)∈E2 with w≥L+1}. If the maximum width was known in advance this inequality could be ensured by setting K to be twice the maximum width, but so large a value appears to be unnecessary in practice.
The algorithm proceeds in the obvious fashion combining with and so on. Since the construction may result in chordless cycles it is necessary to triangulate the resulting graph . This can be done using the maximum cardinality search algorithm [29] which can be implemented in linear time.
Assuming that the time for deriving the estimates for fixed L is bounded, the algorithm described here is by construction linear in p. But the proportionality constant can be expected to be depend on various factors, such as the density of the minimum AIC/BIC model.
Implementation
The methods are implemented in a set of R functions which are provided as Additional files 1 and 2 to the paper. The selection algorithms build on the forward search algorithm implemented in the stepw function in the gRapHD package [30]. Currently this function requires that there are no missing data. The backward search algorithm was developed by the author in the course of preparing this paper, and has subsequently been implemented in the gRapHD package. The functions to perform the selection algorithms, and others to work with and display the graphs, build on the following packages: igraph [31], graph [32], Rgraphviz [26] and gRapHD. The triangular heatmaps were produced using the LDheatmap package [33]. Simulation from a fitted decomposable model was carried out using the gRain package [34]. The package rJPSGCS provides an interface between R and the FitGMLD program which was used to perform the algorithm of [15].
Results
To illustrate the methods described above, they were applied to SNP data obtained from three commercial pig breeds. In all 4239, 1979 and 2123 pigs of the Duroc, Landrace and Yorkshire breeds were genotyped using the Illumina Porcine SNP60 BeadChip [35]. After preprocessing on the basis of call rate, minimal allele frequency and other quality criteria, missing values were imputed using BEAGLE [22]. Using the methods described above, decomposable graphical models were selected for each chromosome and breed, using data at the genotype level. The BIC penalizing constant α= log(N) was used throughout.
Figure 1 compares the running times of the two algorithms, and confirms that the fast algorithm is approximately linear in the number of SNPs. Table 1 gives more detailed information. It is seen that the estimate is indeed a good approximation to , and that the algorithm is considerably faster than the standard greedy algorithm.
Figure 1.
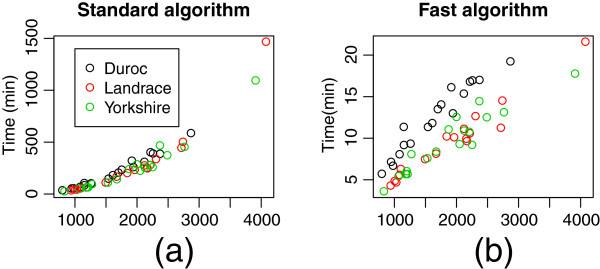
Running times of the two algorithms. The running times (in minutes) of the forward step of the standard and fast algorithms for the different chromosomes and breeds are shown in (a) and (b), respectively.
Table 1.
The performance of the two algorithms
| Breed | chr | N | p | |Es| | under | over | |Esp| | under | over | ts | tf | tsp | tfp | width |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| duroc |
1 |
4249 |
2863 |
4762 |
0 |
0 |
4340 |
0 |
0 |
35084 |
1153 |
37 |
37 |
15 |
| duroc |
2 |
4249 |
2113 |
3676 |
0 |
3 |
3339 |
2 |
2 |
18450 |
920 |
26 |
27 |
10 |
| duroc |
3 |
4249 |
1535 |
2873 |
1 |
13 |
2643 |
4 |
15 |
8695 |
679 |
20 |
20 |
8 |
| duroc |
4 |
4249 |
2211 |
4392 |
0 |
0 |
4012 |
0 |
0 |
24005 |
1007 |
35 |
36 |
11 |
| duroc |
5 |
4249 |
1255 |
2357 |
7 |
2 |
2166 |
8 |
2 |
6030 |
559 |
16 |
16 |
10 |
| duroc |
6 |
4249 |
1937 |
3358 |
0 |
0 |
3069 |
0 |
0 |
15396 |
778 |
24 |
24 |
12 |
| duroc |
7 |
4249 |
1911 |
3978 |
0 |
3 |
3621 |
0 |
2 |
19112 |
966 |
36 |
36 |
12 |
| duroc |
8 |
4249 |
1694 |
3136 |
3 |
3 |
2871 |
3 |
3 |
12193 |
808 |
24 |
24 |
12 |
| duroc |
9 |
4249 |
1749 |
3399 |
3 |
14 |
3087 |
5 |
17 |
13904 |
841 |
26 |
27 |
14 |
| duroc |
10 |
4249 |
1078 |
2178 |
0 |
0 |
1980 |
0 |
0 |
3991 |
482 |
15 |
15 |
12 |
| duroc |
11 |
4249 |
1145 |
2493 |
0 |
2 |
2294 |
0 |
3 |
6206 |
680 |
19 |
19 |
16 |
| duroc |
12 |
4249 |
973 |
1857 |
0 |
0 |
1689 |
0 |
0 |
3138 |
399 |
13 |
13 |
10 |
| duroc |
13 |
4249 |
2365 |
3998 |
0 |
7 |
3634 |
0 |
2 |
23112 |
1019 |
29 |
29 |
9 |
| duroc |
14 |
4249 |
2252 |
4037 |
0 |
3 |
3731 |
0 |
2 |
23291 |
1016 |
29 |
29 |
14 |
| duroc |
15 |
4249 |
1605 |
2977 |
0 |
0 |
2717 |
0 |
0 |
10757 |
707 |
22 |
22 |
11 |
| duroc |
16 |
4249 |
1148 |
2249 |
0 |
0 |
2032 |
0 |
0 |
4936 |
549 |
16 |
16 |
10 |
| duroc |
17 |
4249 |
941 |
1856 |
1 |
3 |
1703 |
1 |
3 |
3034 |
427 |
12 |
12 |
10 |
| duroc |
18 |
4249 |
792 |
1402 |
0 |
0 |
1262 |
0 |
0 |
2032 |
341 |
9 |
10 |
9 |
| landrace |
1 |
1979 |
4071 |
7679 |
0 |
5 |
7020 |
0 |
4 |
87941 |
1295 |
63 |
64 |
12 |
| landrace |
2 |
1979 |
2153 |
4058 |
0 |
0 |
3690 |
0 |
0 |
14868 |
595 |
28 |
29 |
12 |
| landrace |
3 |
1979 |
1957 |
4036 |
1 |
5 |
3664 |
4 |
3 |
13841 |
605 |
30 |
31 |
17 |
| landrace |
4 |
1979 |
2210 |
4498 |
1 |
1 |
4111 |
3 |
2 |
16629 |
634 |
33 |
33 |
12 |
| landrace |
5 |
1979 |
1489 |
3368 |
0 |
7 |
3090 |
1 |
4 |
6511 |
447 |
24 |
25 |
16 |
| landrace |
6 |
1979 |
2163 |
4113 |
0 |
0 |
3753 |
0 |
0 |
14664 |
577 |
28 |
28 |
12 |
| landrace |
7 |
1979 |
2116 |
4634 |
0 |
0 |
4238 |
0 |
0 |
16385 |
666 |
36 |
37 |
13 |
| landrace |
8 |
1979 |
1836 |
3989 |
2 |
4 |
3682 |
4 |
4 |
11989 |
611 |
28 |
29 |
14 |
| landrace |
9 |
1979 |
2301 |
4897 |
0 |
4 |
4415 |
0 |
5 |
20077 |
758 |
40 |
40 |
13 |
| landrace |
10 |
1979 |
1077 |
2285 |
3 |
11 |
2074 |
9 |
9 |
2997 |
334 |
16 |
16 |
14 |
| landrace |
11 |
1979 |
999 |
2150 |
0 |
0 |
1954 |
0 |
0 |
2492 |
289 |
15 |
15 |
11 |
| landrace |
12 |
1979 |
1098 |
2573 |
0 |
0 |
2332 |
0 |
0 |
3420 |
376 |
21 |
21 |
11 |
| landrace |
13 |
1979 |
2734 |
5295 |
0 |
7 |
4868 |
3 |
6 |
30046 |
870 |
39 |
40 |
12 |
| landrace |
14 |
1979 |
2709 |
4819 |
1 |
4 |
4454 |
5 |
3 |
26602 |
673 |
33 |
33 |
17 |
| landrace |
15 |
1979 |
1666 |
3507 |
8 |
13 |
3202 |
10 |
12 |
9888 |
486 |
25 |
25 |
15 |
| landrace |
16 |
1979 |
1181 |
2403 |
0 |
0 |
2179 |
0 |
0 |
3747 |
339 |
15 |
16 |
10 |
| landrace |
17 |
1979 |
1025 |
2227 |
1 |
3 |
2017 |
3 |
3 |
2390 |
279 |
15 |
15 |
11 |
| landrace |
18 |
1979 |
934 |
1890 |
0 |
2 |
1721 |
3 |
3 |
1981 |
256 |
12 |
12 |
9 |
| yorkshire |
1 |
2123 |
3904 |
6700 |
1 |
3 |
6112 |
1 |
1 |
65538 |
1065 |
51 |
52 |
11 |
| yorkshire |
2 |
2123 |
2202 |
4311 |
1 |
8 |
3968 |
3 |
9 |
16548 |
644 |
32 |
33 |
11 |
| yorkshire |
3 |
2123 |
1994 |
4440 |
0 |
9 |
4016 |
1 |
10 |
17020 |
751 |
39 |
39 |
13 |
| yorkshire |
4 |
2123 |
2246 |
4359 |
3 |
3 |
3979 |
4 |
3 |
15429 |
550 |
32 |
32 |
12 |
| yorkshire |
5 |
2123 |
1525 |
3145 |
0 |
0 |
2864 |
0 |
0 |
6471 |
454 |
22 |
22 |
16 |
| yorkshire |
6 |
2123 |
2045 |
3876 |
2 |
2 |
3513 |
2 |
2 |
13313 |
556 |
27 |
27 |
11 |
| yorkshire |
7 |
2123 |
2363 |
5342 |
0 |
11 |
4894 |
4 |
10 |
27929 |
866 |
44 |
46 |
14 |
| yorkshire |
8 |
2123 |
1868 |
3887 |
1 |
5 |
3593 |
1 |
3 |
14254 |
662 |
27 |
28 |
11 |
| yorkshire |
9 |
2123 |
2119 |
4174 |
0 |
3 |
3757 |
1 |
5 |
15108 |
653 |
31 |
31 |
11 |
| yorkshire |
10 |
2123 |
1193 |
2518 |
0 |
0 |
2268 |
0 |
0 |
3757 |
360 |
19 |
19 |
12 |
| yorkshire |
11 |
2123 |
1269 |
2851 |
0 |
0 |
2633 |
0 |
0 |
5501 |
483 |
20 |
20 |
12 |
| yorkshire |
12 |
2123 |
1061 |
2287 |
0 |
0 |
2068 |
0 |
0 |
2885 |
324 |
17 |
17 |
10 |
| yorkshire |
13 |
2123 |
2759 |
4935 |
0 |
0 |
4544 |
0 |
0 |
27100 |
785 |
35 |
35 |
13 |
| yorkshire |
14 |
2123 |
2482 |
4732 |
0 |
6 |
4353 |
1 |
4 |
22286 |
749 |
34 |
33 |
11 |
| yorkshire |
15 |
2123 |
1669 |
3321 |
0 |
4 |
3030 |
2 |
5 |
8385 |
501 |
24 |
24 |
12 |
| yorkshire |
16 |
2123 |
1207 |
2575 |
1 |
5 |
2345 |
2 |
5 |
3734 |
339 |
17 |
17 |
10 |
| yorkshire |
17 |
2123 |
1176 |
2446 |
2 |
7 |
2223 |
3 |
8 |
3621 |
339 |
16 |
17 |
14 |
| yorkshire | 18 | 2123 | 825 | 1618 | 2 | 6 | 1445 | 6 | 6 | 1434 | 215 | 10 | 11 | 9 |
The table shows the performance of the algorithms applied to the pig data. N and p denote the numbers of observations in the data and number of SNPs available after filtering. The edge sets found in steps 1 and 2 of the standard algorithm are denoted Es and Esp, respectively: for the fast algorithm they are denoted Ef and Efp. The numbers of undershoots and overshoots, i.e. |Es∖Ef| and |Ef∖Es| for step 1, and |Esp∖Efp| and |Efp∖Esp| for step 2, are also shown. The corresponding running times in seconds are denoted ts, tf, tsp and tfp, respectively. The backward step is much faster than the forward step in both cases (tsp and tfp). Overall, the fast algorithm is 23.4 times faster than the standard algorithm, and its inaccuracy, assessed as , is 0.0012. The last column shows the maximum width of . The computations were run under Redhat Fedora 10 Linux on a Intel i7 four-core 2.93GHz machine with 48 GB RAM.
Figure 2 shows the LD graph for one chromosome (Duroc chromosome 1): the same graph is obtained with both algorithms. It is a long thin graph with 2863 nodes and 4340 edges. It has 32 connected components, of which 20 are isolated vertices — suggesting SNP positioning errors — and four are long intervals with 997, 569, 786 and 452 SNPs. Curiously, the subgraph induced by the last 35 SNPs contains 7 connected components, which suggests positioning errors in this region. The most striking feature of the graph is that for about 80% of the chromosome, a simple serial or near-serial association structure is found, but for the remaining 20% more complex patterns of LD are observed. Similar results are found for all chromosomes and breeds.
Figure 2.

The LD graph for Duroc chromosome 1. The estimated LD graph for chromosome, constructed using the unphased genotypes, is shown.
Genomic intervals with simple association structure tend to be associated with low haplotype diversity, and intervals with complex structure with high diversity (Figure 3). To further compare the low and high complexity regions, two representative intervals of the same length were selected: SNPs numbered 1800-1825 (low complexity), and SNPs numbered 2470-2495 (high complexity). These have sample entropies of 0.79 and 4.05, respectively. Their subgraphs are shown in Figure 4, and their sample trees in Figure 5. These show the low diversity of the low complexity interval, and the relatively high diversity of the high complexity interval. Corresponding triangular heatmaps are shown in Figures 6 and 7. The low complexity interval has high LD whereas the high complexity interval shows a more mosaic structure.
Figure 3.
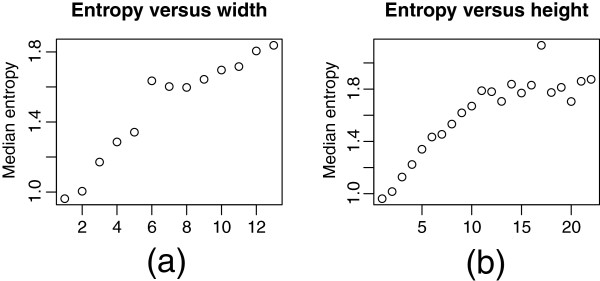
Entropy versus height and width for Duroc chromosome 1. In (a) a plot of median entropy, a measure of haplotype diversity, versus width for Duroc chromosome 1 is shown. In (b), a corresponding plot of median entropy versus height is shown. The entropy was calculated for each interval of 7 adjacent SNPs using the phased data.
Figure 4.

The subgraphs of typical low and high complexity intervals. The subgraphs of a typical low complexity interval (SNPs 1800-1825) and high complexity interval (SNPs numbered 2470-2495) are shown in (a) and (b), respectively.
Figure 5.

The sample tree for the low and high complexity intervals. Sample trees for the low and high complexity intervals are shown in (a) and (b), respectively. The trees are based on the phased haplotype data. The root node is drawn on the left. Edge widths are proportional to the numbers of observations passing through the edges. The colour of the edges signifies the allele: red is 1 and blue 2. It is seen that the interval shown in (a) has low haplotype diversity, dominated by a single common haplotype, whereas that shown in (b) has high haplotype diversity, with one or two relatively common haplotypes.
Figure 6.

A triangular heatmap for the low complexity interval. A heatmap of the pairwise LD values in the low complexity interval is shown. All associations are high.
Figure 7.

A triangular heatmap for the high complexity interval. A heatmap of the pairwise LD values in the high complexity interval is shown. The LD values form a mosaic of mostly low associations.
Thus low complexity regions tend to consist of series of haplotype blocks with high LD and low haplotype complexity, and may be dominated by a few common haplotypes. In contrast, regions of high complexity tend to have high haplotype diversity and little or no haplotype block structure. It must be stressed that the SNPs in such regions are not generally in linkage equilibrium: on the contrary, complex patterns of association, not marginal independences, are observed.
Figure 2 and Table 1 show the results of applying the algorithms to unphased, genotype data, but as mentioned above they may also be applied to phased haplotype-level data. This implicitly regards the inferred haplotypes as a random sample of size 2N from an underlying population of haplotypes. Analyses based on inferred haplotypes may be subject to a loss of efficiency [36]. For the current data phase imputation was carried out using the BEAGLE software [22]: applying the fast algorithm to the phased data for Duroc chromosome 1 resulted in the graph shown in Figure 8. This is slightly denser than the graph in Figure 2, with 375 more edges. This may be ascribed to the reduced model dimension due to the use of binary rather than trichotomous variables, which leads to a weaker penalization of complex models in (1). Haplotype- and genotype-level graphical models are related but in general distinct: it has been shown that they are identical only when the haplotype-level graph is acyclic [37].
Figure 8.

The LD graph for Duroc chromosome 1 based on phased data. The estimated LD graph for Duroc chromosome 1, constructed using the phased haplotypes, is shown.
To assess the accuracy of the algorithm, twenty simulated data sets each with the same number of observations as the data sets analyzed above (N=4239) were constructed. The first ten were generated by taking N random samples from the skeleton, that is, the graphical model with edges between physically adjacent SNPs only, fitted to the Duroc chromosome 1 genotype data. The second ten were generated by taking N random samples from the model shown in Figure 2, fitted to the same data. The results of applying the fast algorithm to each data set are summarized in Table 2. For the skeleton, the edgewise false negative rate was 0.029 and the edgewise false inclusion rate was 0.00024. For the model in Figure 2 the corresponding rates were 0.031 and 0.019. To visually represent the latter results, Figure 9 shows a graph , in which the edge colours represent the frequency that the edge was found in the 10 models. The graph suggests that model uncertainty is primarily restricted to the genomic intervals with complex dependence structure.
Table 2.
Results from simulations
|
Skeleton |
LD graph |
||
|---|---|---|---|
| Undershoot | Overshoot | Undershoot | Overshoot |
| 81 |
0 |
117 |
73 |
| 84 |
0 |
146 |
82 |
| 82 |
0 |
138 |
81 |
| 84 |
0 |
122 |
94 |
| 83 |
0 |
161 |
84 |
| 76 |
1 |
141 |
77 |
| 88 |
2 |
127 |
70 |
| 81 |
0 |
133 |
92 |
| 79 |
3 |
138 |
79 |
| 85 | 1 | 133 | 90 |
The table summarizes the inaccuracy of the fast selection algorithm applied to data sets simulated under two models for Duroc chromosome 1: the skeleton, and the more complex LD graph shown in Figure 2. The columns labelled undershoot and overshoot show |E∖Ej| and |Ej∖E|, where E and Ej are the edge sets of the true model and the model found by the algorithm, respectively. For the data sets simulated under the skeleton, the edgewise false negative rate is 0.029 and the false positive rate is 0.00024. Under the more complex LD graph, the corresponding rates are 0.031 and 0.019.
Figure 9.

The union of LD graphs for Duroc chromosome 1 derived from ten simulated data sets. The figure represents the results of applying the fast algorithm to ten data sets simulated under the LD graph shown in Figure 1. The union of the ten selected graphs is shown. Edges present in all ten graphs are black, and the colour of the others is interpolated between light blue (those found once) and blue (those found nine times).
Finally, for comparison purposes the algorithm of [15] was applied to the Duroc chromosome 1 genotype data. This algorithm automatically imputes phase and missing marker data, cycling between imputation and model selection as sketched in the background section. The selected graph is shown in Figure 10. Like the graphs shown in Figures 2 and 8, it is long and stringy, but rather more dense. This greater density may be explained by the use in [15] of a Metropolis acceptance rule that is based on the penalized likelihood (1), but with a smaller penalizing constant α=(1/8) log(N). A detailed comparison of the two algorithms would be valuable but is not attempted here.
Figure 10.

Haplotype-level graph found using the algorithm of [[15]]. The figure shows the result of applying the algorithm of [15] to the same data set as used with Figure 2. The algorithm estimates a haplotype-level graph for the data.
Discussion
This paper has introduced an efficient, linear-time forward-backward algorithm to estimate chromosome-wide probabilistic graphical models of fine-scale linkage disequilibrium, and has described a convenient way to display these models. In illustration, the methods have been applied to data obtained from three commercial breeds of pigs using the Illumina Porcine SNP60 BeadChip.
The forward part of the algorithm proceeds by combining a series of overlapping marginal decomposable models of dimension L and overlap length K. This implicitly assumes that the maximum width of the true graph is at most K. The resulting model is then triangulated and backward selection performed. The search space closely resembles that of [15], which samples from decomposable models with a given maximum width, using sliding windows. The difference in approaches lies primarily in the search method: here greedy search to optimize a penalized likelihood criterion is used, whereas in [15] MCMC sampling methods are applied.
The R function used for greedy forward search currently requires that the input data contain no missing values, so it was necessary to impute missing values prior to performing the algorithm, and the BEAGLE software [22] was used for this. This raises the possibility of circularity, or more precisely, that the model selection is influenced by constraints or assumptions implicit in the models used by BEAGLE. But given BEAGLE’s high imputation accuracy with such data it seems unlikely that this plays an important role here.
The algorithm was found to have high accuracy when applied to simulated data sets of the dimensions considered here (N∼4000;p∼3000), with edgewise false positive and negative rates of around 3%. That it is good at reconstructing the model generating the data is reassuring. Needless to say, the algorithm does not necessarily identify the “true” model, which may not be in the search space. As noted previously, the approach captures short range associations, but not long range ones. Moreover, higher edgewise false negative rates may occur when observed data are used if there are infinitesimal departures from a model that are not detectable at the given sample size, as these are filtered out when data are simulated under a selected model.
A comparison of triangular heatmaps with LD graphs is instructive. The former are compact graphical representations of all pairwise marginal associations for a set of SNPs. They are particularly well-suited to identify LD blocks, which stand out as highlighted triangles. LD graphs supplement heatmaps by showing patterns of association but not their strength. Since they are parametric models they can be put to a number of quantitative uses as described in the background section. At one level, heatmaps and LD graphs convey similar information, since intervals with simple dependence structures tend to appear as series of LD blocks in the heatmap, whereas those with complex structures tend to occur in the inter-block regions and have a more mosaic appearance. But the graphs provide a more incisive characterisation of genetic variation, building on the concept of conditional rather than marginal dependence. In this regard, it may be helpful to regard conditional independence statements as expressing the notion of irrelevance, in the sense that A⊥ ⊥B | C implies that if we know C, information about A is irrelevant for knowledge of B. Thus the graphs say something about connections between specific SNPs, for example about which SNPs are required to predict a specific SNP.
The graphs may also be useful in fine mapping. Genome-wide association studies seek to find the genetic basis of complex traits, typically using single locus methods - that is, by identifying SNPs with strong marginal association with the complex trait. Due to LD, many SNPs in a genomic region may exhibit strong association with the trait, making it hard to identify the causal loci. A way to address this is to assess the effect of a putative causal SNP on the complex trait in a linear model that also includes terms for the two flanking SNPs, in order to adjust for confounding with nearby effects due to LD. This implicitly assumes a simple serial dependence structure between the SNPs, and when the dependence structure is more complex such adjustment might be insufficient, leading to false positives. This may be prevented by including terms for the neighbours of the putative causal SNP, not just for the flanking SNPs. Any SNP is separated from the remaining SNPs by its neighbours, so by the global Markov property it is independent of the remaining SNPs given its neighbours. A similar method has been proposed based on Bayesian networks [38].
Like graphical models, the APFA models that underlie BEAGLE may be represented as graphs that encode a set of conditional independence relations, but in a very different way. In the present context, for example, nodes in APFA graphs represent haplotype clusters rather than SNPs. Where the model classes intersect, APFA graphs are much more complex than the corresponding dependence graphs, and so less amenable to visualization.
A striking feature of the LD graphs for the pig data was that for roughly 80% of the genome, simple serial or near-serial LD patterns were found, but for the remaining 20%, more complex patterns were observed. The regions with the simple serial structure tend to have low haplotype diversity, which is to be expected in livestock breeds with small effective population sizes [39]. Perhaps more unexpected is that roughly 20% of the porcine genome exhibits complex LD patterns, forming islands of relatively high genetic diversity. This information may be useful in an animal breeding context, to identify regions with high genetic variation. It will also be interesting to compare graphs obtained using different SNP densities in a given breed or species to examine whether and how their topologies change with varying marker densities.
Conclusions
The proposed algorithm is efficient and makes it feasible to estimate and visualize chromosome-wide LD models on a routine basis.
Competing interests
The author declares he has no competing interests.
Supplementary Material
LDgraph. The R functions implementing the methods of the paper.
exampleScript. R code illustrating how the R functions may be used.
Acknowledgements
The data were kindly provided by the Danish Pig Research Centre and in particular Tage Ostersen. The work was performed in a project funded through the Green Development and Demonstration Programme (grant no. 3405-11-0279) by the Danish Ministry of Food, Agriculture and Fisheries, the Pig Research Centre and Aarhus University. Ole F. Christensen, Peter Sørensen, Luc Janss and Lei Wang are acknowledged for helpful discussions.
References
- Barrett J, Fry B, Maller J, Daly M. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Abad-Grau M, Montes R, Sebastiani P. Building chromosome-wide LD maps. Bioinformatics. 2006;22(16):1933–1934. doi: 10.1093/bioinformatics/btl288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardon L, Abecasis G. et al. Using haplotype blocks to map human complex trait loci. Trends Genet. 2003;19(3):135–140. doi: 10.1016/S0168-9525(03)00022-2. [DOI] [PubMed] [Google Scholar]
- Daly M, Rioux J, Schaffner S, Hudson T, Lander E. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29(2):229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Schaffner S, Nguyen H, Moore J, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M. et al. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Stumpf M. Haplotype diversity and the block structure of linkage disequilibrium. Trends Genet. 2002;18(5):226–228. doi: 10.1016/S0168-9525(02)02641-0. [DOI] [PubMed] [Google Scholar]
- Crawford D, Carlson C, Rieder M, Carrington D, Yi Q, Smith J, Eberle M, Kruglyak L, Nickerson D. Haplotype diversity across 100 candidate genes for inflammation, lipid metabolism, and blood pressure regulation in two populations. Am J Hum Genet. 2004;74(4):610. doi: 10.1086/382227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Camp N. Graphical modeling of the joint distribution of alleles at associated loci. Am J Hum Genet. 2004;74(6):1088–1101. doi: 10.1086/421249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen SL. Graphical Models. Oxford: Oxford University Press; 1996. [Google Scholar]
- Højsgaard S, Edwards D, Lauritzen S. Graphical Models with. R. New York, Dordrecht, Heidelberg, London: Springer; 2012. [Google Scholar]
- Edwards D. Linkage analysis using loglinear models. Comput Stat Data Anal. 1992;13(3):281–290. doi: 10.1016/0167-9473(92)90136-4. [DOI] [Google Scholar]
- Thomas A. Characterizing allelic associations from unphased diploid data by graphical modeling. Genet Epidemiol. 2005;29:23–35. doi: 10.1002/gepi.20076. [DOI] [PubMed] [Google Scholar]
- Thomas A. Estimation of graphical models whose conditional independence graphs are interval graphs and its application to modelling linkage disequilibrium. Comput Stat Data Anal. 2009;53(5):1818–1828. doi: 10.1016/j.csda.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A. A method and program for estimating graphical models for linkage disequilibrium that scale linearly with the number of loci, and their application to gene drop simulation. Bioinformatics. 2009;25(10):1287–1292. doi: 10.1093/bioinformatics/btp146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel H, Thomas A. Accuracy and computational efficiency of a graphical modeling approach to linkage disequilibrium estimation. Stat Appl Genet Mol Biol. 2011;10:1–15. doi: 10.2202/1544-6115.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourad R, Sinoquet C, Leray P, MODGRAPH 2009 Probabilistic Graphical Models for Integration of Complex Data and Discovery of Causal Models in Biology, Satellite meeting of JOBIM 2009. : ; 2009. A Bayesian network approach to model local dependencies among SNPs. [Google Scholar]
- Mourad R, Sinoquet C, Dina C, Leray P. Visualization of Pairwise and Multilocus Linkage Disequilibrium Structure Using Latent Forests. PloS one. 2011;6(12):e27320. doi: 10.1371/journal.pone.0027320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourad R, Sinoquet C, Leray P. Probabilistic graphical models for genetic association studies. Brief Bioinform. 2012;13:20–33. doi: 10.1093/bib/bbr015. [DOI] [PubMed] [Google Scholar]
- Morota G, Valente B, Rosa G, Weigel K, Gianola D. An assessment of linkage disequilibrium in Holstein cattle using a Bayesian network. J Anim Breed Genet. 2012;129(6):474–487. doi: 10.1111/jbg.12002. [ http://dx.doi.org/10.1111/jbg.12002] [DOI] [PubMed] [Google Scholar]
- Browning S. Multilocus association mapping using variable-length Markov chains. Am J Hum Genet. 2006;78(6):903–913. doi: 10.1086/503876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning B, Browning S. Efficient multilocus association testing for whole genome association studies using localized haplotype clustering. Genet Epidemiol. 2007;31(5):365–375. doi: 10.1002/gepi.20216. [DOI] [PubMed] [Google Scholar]
- Browning S, Browning B. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Gen. 2007;81(5):1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Singer Y, Tishby N. On the Learnability and Usage of Acyclic Probabilistic Finite Automata. J Comput Sys Sci. 1998;56:133–152. doi: 10.1006/jcss.1997.1555. [DOI] [Google Scholar]
- Thomas A. Assessment of SNP streak statistics using gene drop simulation with linkage disequilibrium. Genet Epidemiol. 2010;34(2):119–124. doi: 10.1002/gepi.20440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripley BD. Pattern Recognition and Neural Networks. Cambridge, United Kingdom: Cambridge University Press; 1996. [Google Scholar]
- Gentry J, Long L, Gentleman R, Falcon S, Hahne F, Sarkar D, Hansen K. Rgraphviz: Provides plotting capabilities for R graph objects. [R package version 2.2.1]
- Su SC, Kuo CC, Chen T. Inference of missing SNPs and information quantity measurements for haplotype blocks. Bioinformatics. 2005;21(9):2001–2007. doi: 10.1093/bioinformatics/bti261. [DOI] [PubMed] [Google Scholar]
- Asmussen S, Edwards D. Collapsibility and response variables in contingency tables. Biometrika. 1983;70(3):567–578. doi: 10.1093/biomet/70.3.567. [DOI] [Google Scholar]
- Tarjan R, Yannakakis M. Simple linear-time algorithms to test chordality of graphs, test acyclicity of hypergraphs, and selectively reduce acyclic hypergraphs. SIAM J Comput. 1984;13:566–579. doi: 10.1137/0213035. [DOI] [Google Scholar]
- Abreu G, Edwards D, Labouriau R. High-Dimensional Graphical Model Search with the gRapHD R Package. J Stat Softw. 2010;37:1–18. [ http://www.jstatsoft.org/v37/i01/] [Google Scholar]
- Gabor C, Tamas N. The igraph software package for complex network research. InterJournal 2006. p. 1695. [ http://igraph.sf.net]
- Gentleman R, Whalen E, Huber W, Falcon S. graph: A package to handle graph data structures. [R package version 1.36.0]
- Shin JH, Blay S, McNeney B, Graham J. LDheatmap: An R Function for Graphical Display of Pairwise Linkage Disequilibria Between Single Nucleotide Polymorphisms. J Stat Softw. 2006;16:Code Snippet 3. [ http://jstatsoft.org/v16/c03] [Google Scholar]
- Højsgaard S Graphical Independence Networks with the gRain Package for R J Stat Softw 201246101–26.[http://www.jstatsoft.org/v46/i10/]22837731 [Google Scholar]
- Ramos AM, Crooijmans R, Affara N, Amaral A, Archibald A, Beever J, Bendixen C, Churcher C, Clark R, Dehais P. et al. Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS One. 2009;4(8):e6524. doi: 10.1371/journal.pone.0006524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris A, Whittaker J, Balding D. Little loss of information due to unknown phase for fine-scale linkage-disequilibrium mapping with single-nucleotide–polymorphism genotype data. Am J Hum Genet. 2004;74(5):945. doi: 10.1086/420773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A. The conditional independences between variables derived from two independent identically distributed Markov random fields when pairwise order is ignored. Math Med Bio. 2010;27(3):283–288. doi: 10.1093/imammb/dqp022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Park M, Chen Xw. A Markov blanket-based method for detecting causal SNPs in GWAS. BMC Bioinformatics. 2010;11(Suppl 3):S5. doi: 10.1186/1471-2105-11-S3-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uimari P, Tapio M. Extent of linkage disequilibrium and effective population size in finnish landrace and finnish yorkshire pig breeds. J An Sci. 2011;89(3):609–614. doi: 10.2527/jas.2010-3249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
LDgraph. The R functions implementing the methods of the paper.
exampleScript. R code illustrating how the R functions may be used.