

Abstract
Arginase from parasitic protozoa belonging to the genus Leishmania is a potential drug target for the treatment of leishmaniasis because this binuclear manganese metalloenzyme catalyzes the first committed step in the biosynthesis of polyamines that enable cell growth and survival. The high resolution X-ray crystal structures of the unliganded form of Leishmania mexicana arginase (LmARG) and four inhibitor complexes are now reported. These complexes include the reactive substrate analogue 2(S)-amino-6-boronohexanoic acid (ABH) and the hydroxylated substrate analogue nor-Nω-hydroxy-L-arginine (nor-NOHA), which are the most potent arginase inhibitors known to date. Comparisons of the LmARG structure with that of the archetypal arginase, human arginase I, reveal that all residues important for substrate binding and catalysis are strictly conserved. However, three regions of tertiary structure differ between the parasitic enzyme and the human enzyme corresponding to the G62 – S71, L161 – C172, and I219 – V230 segments of LmARG. Additionally, variations are observed in salt link interactions that stabilize trimer assembly in LmARG. We also report biological studies in which we demonstrate that localization of LmARG to the glycosome, a unique subcellular organelle peculiar to Leishmania and related parasites, is essential for robust pathogenesis.
Keywords: Metalloenzyme, arginase, leishmaniasis, X-ray crystallography
Introduction
Leishmaniasis is a neglected tropical disease caused by parasitic protozoa belonging to the genus Leishmania. Transmitted by the bite of the female sand fly, the overall prevalence of leishmaniasis is estimated to be 12 million cases with approximately 2 million new cases reported per year in nearly one hundred countries worldwide [1–4]. Visceral leishmaniasis, also known as black fever or kala-azar, is lethal if left untreated due to infection of liver, spleen, and bone marrow; cutaneous leishmaniasis, also known as Baghdad boil or Khandahar sore, is characterized by benign but disfiguring skin lesions [5]. With the growing occurrence of leishmaniasis infection in western countries due to population migration and the return of military troops from endemic regions [6], and with cutaneous leishmaniasis outbreaks in Texas caused by L. mexicana [7] projected to expand northward toward the Canadian border [8], this neglected tropical disease represents a significant international health problem and an increasingly prominent threat to the U.S. public health.
Therapeutic strategies for the treatment of leishmaniasis depend on the form of the disease, the species of parasite causing the disease, and the availability of cost-effective health care to infected individuals [1–4]. For example, some forms of cutaneous leishmaniasis will resolve without medical intervention, while others require treatment with pentavalent antimony-based drugs. Other more expensive drugs, such as the antifungal drug amphotericin B, the antiprotozoal drug miltefosine, or the broad spectrum antibiotic paromomycin, are also available for the treatment of cutaneous and visceral leishmaniasis. However, these drugs are neither universally effective nor universally available. Accordingly, the search for new targets for the treatment of leishmaniasis remains an urgent area of investigation.
Enzymes of polyamine biosynthesis have been increasingly studied as drug targets for the treatment of parasitic diseases such as leishmaniasis, since polyamines are essential for parasite growth and survival [9, 10]. For example, ornithine decarboxylase is a critical enzyme in polyamine biosynthesis that catalyzes the decarboxylation of L-ornithine to yield putrescine and carbon dioxide (Figure 1). Ornithine decarboxylase is irreversibly inhibited by D,L-α-difluoromethylornithine (DFMO)5, which is used to treat patients with Trypanosoma brucei gambiense infections (African sleeping sickness) [11, 12]. DFMO is also cytotoxic to L. donovani [13] and L. infantum promastigotes [14]. However, when tested in mice or hamsters infected with L. donovani or L. infantum, DFMO failed to cure the animals although infectivity was reduced [15–17]. Surprisingly, however, an ornithine decarboxylase-deficient null mutant of L. donovani was dramatically compromised in its ability to establish an infection in the mouse model [18], while a genetic lesion in spermidine synthase also negatively impacted infectivity in mice [19]. Thus, the polyamine pathway of Leishmania offers several potential targets for chemotherapeutic intervention in the treatment of leishmaniasis.
Figure 1.

L-Arginine metabolism in polyamine and nitric oxide biosynthesis (SAMPA = S-adenosylmethylthiopropylamine).
The binuclear manganese metalloenzyme arginase precedes ornithine decarboxylase in the polyamine biosynthetic pathway and may represent an alternative drug target for the treatment of leishmaniasis (Figure 1). Arginase catalyzes the hydrolysis of L-arginine to yield L-ornithine and urea [20, 21] and thereby regulates the flux of L-ornithine for polyamine biosynthesis. By depleting local L-arginine concentrations at sites of infection, arginase also suppresses the immune response in part by reducing the generation of L-arginine-derived nitric oxide by activated macrophages. Thus, arginase plays a critical role not only in polyamine biosynthesis, which is essential for parasite growth and survival, but also in the evasion of the host immune response against the invading parasite [22, 23]. Studies of arginase knockout mutants in L. mexicana [24], L. amazonensis [25], and L. major [26, 27] specifically confirm the role of arginase in polyamine biosynthesis and parasite survival [24]. Furthermore, the arginase inhibitors Nω-hydroxy-L-arginine (NOHA) and nor-Nω-hydroxy-L-arginine (nor-NOHA) [28] reduce the growth of L. infantum and L. major in macrophages and mice, thereby validating arginase as a drug target for the treatment of leishmaniasis [29–31].
The genomes of Leishmania species encode for a single arginase enzyme [32]. Arginase genes have been cloned and expressed, and the recombinant enzymes have been kinetically characterized [33, 34]. Arginase from L. mexicana (LmARG) is the best characterized with regard to in vitro inhibition, and it is strongly inhibited by known inhibitors of human arginase I (Table 1) [34]. Of particular interest is the inhibitor 2(S)-amino-6-boronohexanoic acid (ABH) [35], which is the best known inhibitor of human arginase I reported to date [36]. The essentially isosteric inhibitor S-(2-boronoethyl)-L-cysteine (BEC) [37] also binds tightly to human arginase I [36]. Boronic acid inhibitors such as ABH and BEC undergo nucleophilic attack in the active site of arginase to form a tetrahedral boronate anion that mimics the tetrahedral intermediate and its flanking transition states in catalysis [21]. Also notable is the N-hydroxyguanidinium inhibitor nor-NOHA [28], which binds with exceptionally high affinity for an inhibitor that is not a transition state analogue [38]. The overall amino acid sequence identity, as determined by both pairwise protein alignment and structure-based sequence alignment, is only 40% between LmARG and human arginase I (Figure 2), but residues important for substrate and inhibitor binding are strictly conserved between these two enzymes.
Table 1.
Arginase Inhibitors.
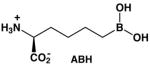
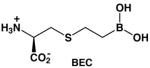
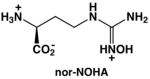
Figure 2.

Structure-based sequence alignment of LmARG and human arginase I (PDB entry 2AEB) [36], as determined with MacPyMOL [49] and STRIDE [64, 65]. The structural alignment was created with the program ESPript (version 2.2) [66]. The overall sequence identity is 40% based on this alignment. The symbols α, η, and β indicate α-helices, 310-helices, and β-strands, respectively. Identical residues are boxed and indicated by white lettering on red background; similar residues are boxed and indicated by red lettering on white background.
Here, we report the X-ray crystal structures of unliganded LmARG, its complex with the catalytic product L-ornithine, and its complexes with the inhibitors ABH, BEC, and nor-NOHA. These structures reveal the molecular basis of inhibitor affinity and may provide clues toward the design of inhibitors that may selectively inhibit the parasitic enzyme. While L. mexicana arginase may be a validated target for antiparasitic drugs that would block polyamine biosynthesis and thereby compromise parasite viability, we also show that the arginase inhibitors NOHA and nor-NOHA exhibit blunted effects in vivo, perhaps due to inefficient uptake by the parasite.
Materials and Methods
Materials
Manganese(II) chloride tetrahydrate (≥99%) was purchased from Sigma-Aldrich. 2(S)-Amino-6-boronohexanoic acid (ABH) ammonium salt was purchased from Enzo Life Sciences (Farmingdale, NY). A prepared solution of 12% (w/v) polyethylene glycol 3350, 100 mM HEPES (pH 7.5) was purchased from Hampton Research (Aliso Viejo, CA). All other chemicals were purchased from Fisher Scientific unless otherwise specified.
Preparation of L. mexicana Arginase cDNA
The pETLmARG plasmid containing the full length LmARG gene (GenBank accession number AY386701) was previously cloned into the kanamycin-resistant pET200/D-TOPO Escherichia coli expression vector [34]. This construct also encodes for an N-terminal hexahistidine tag and a 26-residue linker segment preceding the actual N-terminal residue of LmARG. However, we were not successful in crystallizing the full-length N-terminally-tagged protein. Analysis of the amino acid sequence using the program DISOPRED2 [39] suggested that the hexahistidine tag, the linker segment, and a short C-terminal segment might be disordered and thereby hinder crystallization. Accordingly, the construct was modified by two rounds of deletion mutagenesis to substitute the 13-residue segment MRGSHHHHHHGMA for the N-terminal hexahistidine tag, the 26-residue linker segment, and residues M1-E12 of LmARG (the C-terminal segment was left intact). This new construct was designated Δ12-LmARG.
Oligonucleotide primers (Integrated DNA Technologies) used in this mutagenesis were: (first-round) 5′-AGC ATG ACT GGT GGA CAG CAA ATG GAG CAC GTG CAG CAG TAC AAG-3′ (sense), 5′-CTT GTA CTG CTG CAC GTG CTC CAT TTG CTG TCC ACC AGT CAT GCT-3′ (antisense); and (second-round) 5′-CAT CAT CAT CAT CAT CAT GGT ATG GCT AAG AAG ATG AGC ATT GTG CTT GCC C-3′ (sense), 5′-GGG CAA GCA CAA TGC TCA TCT TCT TAG CCA TAC CAT GAT GAT GAT GAT GAT G-3′ (antisense). All polymerase chain reaction protocols used the following thermal cycling settings: first step (95 °C for 1 min), one cycle; second step [melt (95 °C for 30 s), anneal (57 °C for 1 min), and extension (72 °C for 2 min)], 10 cycles; third step (72 °C for 5 min), one cycle. The sequence of the resulting gene was verified by DNA sequencing, which was performed at the University of Pennsylvania DNA Sequencing Facility.
Expression and Purification of L. mexicana Arginase
The plasmid encoding Δ12-LmARG (pETΔ12-LmARG) was transformed into E. coli strain BL21(DE3) (Stratagene) by the heat shock method and grown on Luria-Bertani (LB) agar plates with 50 μg/mL of kanamycin. Culture tubes containing 5 mL of LB media to which 50 μg/mL kanamycin was added, were each inoculated with a single colony of E. coli from the transformation plate and allowed to grow at 37 °C and 250 rpm for 8 hours. Culture flasks (2 L) containing 1 L of LB media and 50 μg/mL of kanamycin were each inoculated with a single 5 mL starter culture and were allowed to incubate at 37 °C and 220 rpm until the OD600 reached 0.80, at which point isopropyl β-D-thiogalactopyranoside (IPTG) (Carbosynth) was added to a final concentration of 1 mM, the incubating temperature was lowered to 22 °C, and the cultures were allowed to continue incubating at 220 rpm overnight. The E. coli were centrifuged and the 27 g cell pellet was stored at −80 °C overnight. The cell pellet was thawed to room temperature and resuspended in lysis buffer [50 mM potassium phosphate (pH 7.0), 300 mM NaCl, 10% (v/v) glycerol]. Lysozyme (lyophilized, MP Biomedicals) was added to the E. coli and stirred at 4 °C for 1 hour to lyse the cells followed by sonication in a water bath type sonicator containing ice for 30 minutes. DNAse (Sigma) and RNAse (Sigma) at final concentrations of 8 μg/mL and 13 μg/mL, respectively, were added and the cell lysate was stirred at 4 °C for 1 hour, followed by overnight freezing at −80 °C. The cell lysate was thawed to room temperature and centrifuged at 15,000 rpm for 30 minutes. The supernatant containing Δ12-LmARG enzyme was loaded onto a cobalt-nitrilotriacetic acid (Co-NTA, BD Biosciences) immobilized-metal affinity chromatography column [1.5 cm (internal diameter) × 3 cm (bed height)] that was pre-equilibrated with lysis buffer. This same buffer was used as a mobile phase to elute impurities from the column. Once the A280 reached baseline on the UV detector, the Δ12-LmARG enzyme was eluted from an isocratic step of 60% mobile phase B, where 100% mobile phase B was 50 mM potassium phosphate (pH 7.0), 300 mM NaCl, 150 mM imidazole, 10% (v/v) glycerol.
Fractions of Δ12-LmARG enzyme were combined and concentrated to 25 mL using Amicon concentrators (Millipore) equipped with the YM-10 membrane (10 kDa molecular mass cutoff). The Δ12-LmARG enzyme was dialyzed against 25 mM potassium phosphate (pH 7.5), 2 mM β-mercaptoethanol (BME), 100 μM MnCl2, 10% (v/v) glycerol followed by centrifugation at 15,000 rpm for 30 minutes to pellet down any precipitate. The supernatant was diluted with the same buffer to a volume of 5 mL and then loaded onto a 5 mL Q-HP anion-exchange column (GE Life Sciences). Mobile phase A [10 mM potassium phosphate (pH 7.5), 2 mM BME, 100 μM MnCl2, 10% (v/v) glycerol] was used to elute impurities off the column while maintaining the strong retention of Δ12-LmARG. A gradient elution was subsequently performed with mobile phase B [50 mM potassium phosphate (pH 7.5), 2 mM BME, 100 μM MnCl2, 10% (v/v) glycerol]. The Δ12-LmARG protein remained on the column at this point; after the absorbance at 280 nm reached baseline on the UV detector, a second gradient elution with mobile phase C [100 mM potassium phosphate (pH 7.5), 300 mM NaCl, 2 mM BME, 100 μM MnCl2, 10% (v/v) glycerol] was performed to elute Δ12-LmARG. The estimated purity of the protein sample was >99% based on SDS-PAGE. The Δ12-LmARG protein was concentrated to 2.5 mL using Amicon concentrators (Millipore) equipped with the YM-10 membrane (10 kDa molecular mass cutoff), followed by a buffer exchange into 50 mM bicine (pH 8.5), 100 μM MnCl2, 2 mM BME, 5% (v/v) glycerol using a PD-10 column (GE Healthcare). The Δ12-LmARG protein was further concentrated to 7.8 mg/mL (ε280 = 26,570 M−1 cm−1).
Crystallization of L. mexicana Arginase
Unliganded Δ12-LmARG crystals were prepared by the hanging-drop vapor diffusion method by combining a 3 μL drop of protein solution [7.0 mg/mL Δ12-LmARG, 45 mM bicine (pH 8.5), 1.4 mM thymine, 90 μM MnCl2, 4.5% (v/v) glycerol, and 1.8 mM BME] and a 3 μL drop of precipitant solution [10% (w/v) polyethylene glycol 10,000, 5% (v/v) (+/−)-2-methyl-2,4-pentanediol (MPD), and 0.1 M N-(2-hydroxyethyl)piperazine-N′-(2-ethansulfonic acid) (HEPES, pH 7.3)] on a siliconized cover slide and equilibrated against 500 μL of precipitant solution at room temperature. While thymine is not observed to bind anywhere in the protein structure, it is an additive that improves the physical quality of unliganded human arginase I crystals, so we incorporated it into the crystallization conditions for unliganded Δ12-LmARG. Crystals first appeared after 12 hours, and a crystal was transferred into a cryoprotectant solution [precipitant solution supplemented with 15% (v/v) glycerol] after 4 days. The Δ12-LmARG-ABH complex was crystallized by the hanging drop vapor diffusion method. Typically, a 5 μL drop of protein solution [7.0 mg/mL Δ12-LmARG, 45 mM bicine (pH 8.5), 10 mM ABH, 90 μM MnCl2, 4.5% (v/v) glycerol, and 1.8 mM BME] and a 5 μL drop of precipitant solution [12% (w/v) polyethylene glycol 3,350 and 0.1 M HEPES (pH 7.5)] were combined on a siliconized cover slide and equilibrated against 300 μL of precipitant solution. Crystals appeared after 4 days at room temperature and were immediately transferred into a cryoprotectant solution [precipitant solution supplemented with 15% (v/v) glycerol]. The Δ12-LmARG-nor-NOHA complex was crystallized by the sitting-drop vapor diffusion method by combining a 1.0 μL drop of protein solution [7.0 mg/mL Δ12-LmARG, 45 mM bicine (pH 8.5), 10 mM nor-NOHA, 90 μM MnCl2, 4.5 % (v/v) glycerol, and 1.8 mM BME] with a 1.0 μL drop of precipitant solution [0.1 M 2-(N-morpholino)ethanesulfonic acid (MES, pH 6.0) and 20% (w/v) polyethylene glycol monomethyl ether 2,000] using the Nanodrop NS-2 Stage crystallization robot on a 96-well sitting-drop plate (Innovadyne) and was equilibrated against 100 μL of precipitant solution at room temperature. Crystals appeared after 2 hours but were allowed to grow for 1 day, and a crystal was transferred into a cryoprotectant solution [precipitant solution supplemented with 15% (v/v) glycerol]. The Δ12-LmARG-BEC complex was crystallized by combining a 3 μL drop of protein solution [7.0 mg/mL Δ12-LmARG, 45 mM bicine (pH 8.5), 10 mM BEC, 90 μM MnCl2, 4.5 % (v/v) glycerol, and 1.8 mM BME] with a 3 μL drop of precipitant solution [0.1 M HEPES (pH 7.5) and 12% (w/v) polyethylene glycol 3,350] on the pedestal of a 24-well sitting-drop plate equilibrated against 500 μL of precipitant solution. Crystals appeared after 3 days and were transferred into a cryoprotectant solution [precipitant solution supplemented with 15% (v/v) glycerol]. A crystal of the Δ12-LmARG-L-ornithine complex was prepared by soaking a crystal of unliganded Δ12-LmARG in 10 μL of an L-ornithine soak solution [50 mM L-ornithine, 0.1 M N-(2-hydroxyethyl)piperazine-N′-(3-propanesulfonic acid) (HEPPS, pH 8.5), and 15% (w/v) polyethylene glycol 3,350] for 23 hours using a 24-well sitting-drop crystallization plate. This crystal was transferred into a cryoprotectant solution [L-ornithine soak solution supplemented with 15% (v/v) glycerol]. All crystals were flash-cooled in liquid nitrogen for data collection after they were immersed for at least 10 seconds in their corresponding cryoprotectant solutions.
X-ray Crystal Structure Determination
X-ray diffraction data from crystals of unliganded Δ12-LmARG and its inhibitor complexes were collected on beamline X29 (λ = 1.075 Å) of the National Synchrotron Light Source at Brookhaven National Laboratory (Upton, NY). The HKL-2000 suite of programs was used for indexing, integrating, and scaling diffraction data [40]. Diffraction data were initially processed using the H3 space group (the hexagonal setting of the R3 space group) with unit cell dimensions a = 89.5 Å, b = 89.5 Å, c = 115.1 Å (one molecule in the asymmetric unit). Deviations from ideal Wilson statistics were observed, with the second moment of intensity, 〈I2〉/〈I〉2, ranging between 1.5–2.0 but closer to 1.5 for most resolution bins as calculated with the CNS script detect_twinning.inp [41, 42]. For example, for the Δ12-LmARG-ABH complex, 〈I2〉/〈I〉2 = 1.75. The twinning fraction for each data set was subsequently calculated with the CNS script twin_fraction.inp [41, 42] and is recorded in Table 2.
Table 2.
Data Collection and Refinement Statistics.
| LmARG | LmARG-ABH | LmARG-nor-NOHA | LmARG-BEC | LmARG- L - | |
|---|---|---|---|---|---|
| Data Collection | |||||
| resolution limits (Å) | 50.0 – 1.80 | 50.0 – 1.77 | 50.0 – 1.95 | 50.0 – 1.80 | 50.0 – 1.95 |
| total/unique reflections measured | 149263/31710 | 156434/33141 | 117046/24840 | 144883/30860 | 116890/24867 |
| space group symmetry | H3 | H3 | H3 | H3 | H3 |
| unit cell dimensions a, b, c (Å) | 89.5, 89.5, 115.1 | 89.4, 89.4, 114.4 | 89.6, 89.6, 114.6 | 88.5, 88.5, 114.0 | 89.6, 89.6, 114.4 |
| R mergea,b | 0.087 (0.525) | 0.067 (0.699) | 0.053 (0.526) | 0.099 (0.525) | 0.077 (0.624) |
| I/σ (I)a | 16.91 (2.77) | 21.90 (2.37) | 28.96 (2.92) | 16.52 (3.45) | 20.20 (2.71) |
| completeness (%)a | 99.8 (100) | 100 (100) | 99.4 (98.1) | 100 (99.9) | 99.9 (100) |
| Refinement | |||||
| reflections used in refinement/test set | 30734/2382 | 31693/1559 | 24016/1795 | 29594/2260 | 23754/2181 |
| twinning fraction | 0.37 | 0.38 | 0.37 | 0.24 | 0.30 |
| R twin c | 0.1440 | 0.1663 | 0.1418 | 0.1614 | 0.1643 |
| R twin/freed | 0.1903 | 0.1995 | 0.2008 | 0.1932 | 0.2064 |
| protein chainse | 1 | 1 | 1 | 1 | 1 |
| protein residuese | 310 | 310 | 310 | 310 | 310 |
| protein atomse | 2362 | 2362 | 2362 | 2362 | 2362 |
| solvent moleculese | 107 | 81 | 137 | 96 | 68 |
| ligand moleculese,h | 1 | 2 | 2 | 3 | 2 |
| metal ionse | 2 | 2 | 2 | 2 | 2 |
| Root Mean Square Deviation f | |||||
| bonds (Å) | 0.007 | 0.007 | 0.007 | 0.007 | 0.008 |
| angles (deg) | 1.4 | 1.4 | 1.4 | 1.4 | 1.4 |
| Average B-factors g (Å 2) | |||||
| main chain | 34 | 27 | 40 | 27 | 52 |
| side chain | 37 | 30 | 42 | 30 | 54 |
| solvent | 39 | 32 | 44 | 33 | 49 |
| ligandh | 37 | 29 | 49 | 38 | 51 |
| metal ions | 23 | 16 | 26 | 16 | 36 |
| Ramachandran Plot f (%) | |||||
| allowed | 89.8 | 89.4 | 87.9 | 90.9 | 83.7 |
| additionally allowed | 10.2 | 10.6 | 11.7 | 9.1 | 15.2 |
| generously allowed | 0.0 | 0.0 | 0.4 | 0.0 | 0.8 |
| disallowed | 0.0 | 0.0 | 0.0 | 0.0 | 0.4 |
| PDB accession code | 4ITY | 4IU0 | 4IU1 | 4IU4 | 4IU5 |
Values in parentheses are for the outer 0.06 Å shell of data.
Rmerge = Σ |I − 〈I〉|/ Σ I, where I is the observed intensity, 〈I〉 is the average intensity calculated from replicate data.
Rtwin = Σ ||Fo|−|Fc||/ Σ |Fo| for reflections contained in the working set.
Rtwin/free = Σ ||Fo|−|Fc||/ Σ |Fo| for 5% of reflections contained in the test set held aside during refinement. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
Per asymmetric unit.
Calculated using PROCHECK [47].
Calculated using MOLEMAN [48].
Values pertain to all ligands and/or inhibitors observed in the asymmetric unit (e.g., ABH and glycerol).
The program Phaser [43] in the CCP4 suite [44] was used for molecular replacement calculations using the A-chain (residues R6 – N319) of the ligand-free and metal-free structure of human arginase I (PDB entry 3TF3) [45] as a search probe for rotation and translation function calculations. Refinement was performed with CNS (version 1.2) [42] in space group R3 (instead of H3) using hemihedral twinning operators h, -h-k, and -l. Model building was performed using Coot (version 0.6.1) [46]. Refinement against twinned intensity data was performed with CNS. Water molecules were included in the later stages of refinement. Crystals of LmARG complexes with ABH, BEC, nor-NOHA, and L-ornithine were generally isomorphous with those of unliganded LmARG; unit cell parameters are listed in Table 2. Gradient omit maps showed clear electron density for the corresponding ligand bound in the active site of each monomer in the asymmetric unit. Atomic coordinates of bound ligands were included in later stages of refinement, and all ligand atoms were refined with 100% occupancy. Thermal B factors for ligands were generally consistent with the average B factor calculated for the whole protein (Table 2). Disordered segments at the N-terminus (the hexahistidine tag and linker sequence MRGSHHHHHHGMA) and at the C-terminus (P323-L329) were excluded from all final models. The programs PROCHECK [47], MOLEMAN [48], and MacPyMOL [49] were used for structural analysis. Data collection and refinement statistics for all structure determinations are recorded in Table 2.
Electrostatic Surface Potential Calculations
The electrostatic potential of the protein surface was calculated using the Adaptive Poisson-Boltzmann Solver (version 1.3) available as a PyMOL plug-in [49–52]. Atomic coordinates of the LmARG-ABH complex and the human arginase I-ABH complex (PDB entry 2AEB, chain A) were used as input files. However, these coordinate files required some editing: the ABH ligand, anisotropic temperature factors, and hydrogen atoms were deleted from each PDB file if present. The following parameters were implemented in the calculations: T = 298.15 K, pH = 8.5, the AMBER forcefield was used, a linearized Poisson-Boltzmann equation was used, and the charge and ionic radius of Mn2+ ions were set to +2 and 0.8 Å, respectively. During file conversion using the web-based server PDB2PQR (version 1.8) [51], the Mn2+ ions were removed from the file and were hand-edited back into the PQR file prior to calculation of the electrostatic surface potential of the protein.
Treatment of Leishmania Promastigotes with Arginase Inhibitors
The L. mexicana wild-type strain (MNYC/BZ/62/M379) and mutant cell lines derived from wild-type parasites, Δarg, Δarg[pGFP-ARG], and Δarg[pGFP-argΔskl], have been described previously [24, 53]. Parasites were routinely incubated in Dulbecco’s modified Eagle’s medium–Leishmania containing 10% heat inactivated chicken serum, with the exception of Δarg parasites, which are polyamine auxotrophs [10] that require media supplementation with putrescine, which was routinely added to a concentration of 200 μM. To evaluate proliferation, parasites were seeded at 3 × 104/100 μL in 96-well plates to which serial dilutions of 2 mM NOHA or 2 mM nor-NOHA were added to media in the absence or presence of 200 μM putrescine in a volume of 100 μL. After 5 days, 10 μL of 520 μM resazurin was added to each well, and plates were incubated for an additional 24 hours. Reduction of resazurin to resorufin was evaluated on a BioTek Synergy plate reader by monitoring absorbance at 570 nm, using 600 nm as a reference wavelength. Reduction observed in wells containing parasites without drug was taken as maximal proliferation. The effective concentration of drug that inhibited growth by 50% (EC50 value) was determined using Prism Graphpad. For long-term inhibition studies, 5 μL of stationary phase parasites were seeded into multiple 96-well plates at the same drug concentrations and with and without putrescine as for the 5 day experiment described above. Parasites were then passed every 5–6 days into 96-well plates containing fresh media and identical drug concentrations for up to 4 weeks. Parasite densities were measured every 5–6 days using the resazurin reduction method.
Western Blot Analysis
Parasite lysates prepared from exponentially growing L. mexicana promastigotes were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and blotted onto either nitrocellulose or Nytran membranes (Schleicher and Schuell, Keene, NH), and Western blot analysis was performed according to standardized procedures. The membranes were probed with polyclonal rabbit antibodies raised to the L. mexicana arginase and commercially available anti-tubulin mouse monoclonal antibody (DM1A) (Calbiochem, La Jolla, CA).
Mouse Infectivity Studies
Infectivity studies in BALB/c mice have been described previously [53]. Briefly, five groups of 5 female mice (4–6 weeks old) were inoculated i.d. on day zero in the right hind footpad with a single injection of 1 × 106 stationary phase wild-type L. mexicana promastigotes: either wild-type, one of two Δarg clones (labeled “2” and “5”), Δarg [pGFP-Arg] complemented with wild-type ARG on an extrachromosomal plasmid (labeled “ARG”), or Δarg [pGFP-argΔskl] complemented with a mutant ARG that homes incorrectly within the promastigote. Footpad thickness was measured weekly with a Mitutoyo digital caliper as described [53].
Results and Discussion
Unliganded LmARG
The overall fold of Δ12-LmARG (henceforth designated simply “LmARG”) is generally similar to that of unliganded human arginase I (PDB entry 2ZAV) [54], and the monomers of each enzyme superimpose with a root-mean-square (r.m.s.) deviation of 0.80 A for 264 Cα atoms as calculated with the program MacPyMOL [49]. Each Mn2+ ion is coordinated in octahedral or distorted octahedral fashion by 2 histidine residues, 4 aspartate residues, and 2 non protein ligands. The metal-bridging nonprotein ligand is expected to be a hydroxide ion in the catalytically-active form of the enzyme, and the Mn2+A-bound nonprotein ligand is interpreted as a water molecule (Figure 3a). Metal coordination distances and geometries in the binuclear manganese cluster are essentially identical to those observed in unliganded human arginase I (Figure 3b) [54].
Figure 3.

(a) Simulated annealing omit map (green) of the Mn2+-bound solvent molecules in unliganded LmARG, contoured at 5.3σ. Atoms and intermolecular interactions are color-coded as follows: yellow for C, blue for N, red for O, pink spheres for Mn2+ ions, and red spheres for solvent. Metal coordination and hydrogen bond interactions are represented as blue and green dashed lines, respectively. (b) Superposition of unliganded LmARG (color-coded as in panel (a)) and chain A of unliganded human arginase I (PDB entry 2ZAV [54], all atoms and intermolecular interactions colored light blue). LmARG and human arginase I residue labels are black and red, respectively.
Despite the overall structural similarity between LmARG and human arginase I, some local tertiary structural differences are observed (Figure 4) and appear to be rooted in primary structure differences; none of these differences appear to result from crystal packing interactions. First, a new α-helix (G62 – S71) is found in the F60 – D75 segment of LmARG (L53 – S62 segment in human arginase I), which corresponds to a loop region in human arginase I. Second, both arginases contain a large loop that exhibits a different conformation in each enzyme: the corresponding segments are L161 – C172 in LmARG and L148 – G161 in human arginase I, which is 2 residues longer in the latter enzyme due to a dipeptide insertion in the amino acid sequence. Finally, LmARG has a 12-residue α-helix and human arginase I has a corresponding 14-residue α-helix (α-helix F2) that when superimposed become gradually more offset from each other, leading to a sideways shift of 2.2 Å at the C-terminal end of the α-helix. This α-helix is the I219 – V230 segment in LmARG (Figure 4), and in human arginase I, it is the I208 – G221 segment. One key structural feature appears to enable this helix shift, namely, R225 in human arginase I, which appears as E236 in LmARG. The side chain of R225 forms an intermonomer salt link with D173 in human arginase I; R225 also donates two hydrogen bonds to the backbone carbonyl of L219, which is part of α-helix F2 (Figure 5). Accordingly, intermolecular and intramolecular interactions of R225 prevent α-helix F2 from a closer approach to D173. In LmARG, a salt link pair corresponding to R225-D173 of human arginase I is absent. Instead, a hydrogen bond is donated by the side chain of K184 (D173 in human arginase I) to the backbone carbonyl of V230 (L219 in human arginase I).
Figure 4.
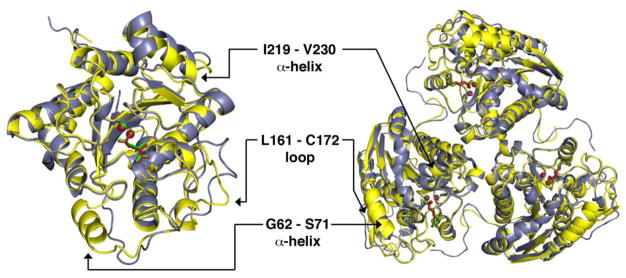
Superposition of monomers (left) and trimers (right) of the LmARG-ABH complex and the human arginase I-ABH complex (PDB entry 2AEB) [36]. Atoms are color-coded as follows: LmARG-ABH complex, yellow for protein and green for ABH and Mn2+ ions; human arginase I-ABH complex, blue-grey for protein and dark red for ABH and Mn2+ ions.
Figure 5.
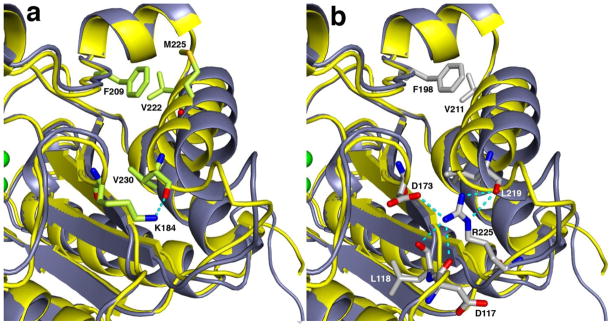
Close-up view of intermonomer interactions involving α-helix F2 and β-sheet 5 in LmARG (yellow backbone) and human arginase I (blue-grey backbone). (a) Superposition of the LmARG-ABH complex with the human arginase I-ABH complex showing key interactions of residues in LmARG. (b) The same superposition as in (a) showing key interactions of residues in human arginase I. Selected side chain atoms and intermolecular interactions are color-coded as follows: green for carbon (LmARG), grey for carbon (human arginase I), blue for nitrogen, and red for oxygen.
Like human arginase I [36], LmARG is a trimer (Figure 4) stabilized by intermolecular ionic interactions (e.g., salt bridges) and hydrogen bonds. Indeed, the subunit-subunit contact surface area of 877 Å2 is predominantly polar in nature (57%), as determined by the web-based Protein Interfaces, Surfaces, and Assemblies (PISA) server [55]. Each trimer is stabilized by the conserved salt link cluster designated SLC-α: D204A/D215A, E256A/E267A, and R255B/R266B (human arginase I/LmARG numbering; subscripts A and B represent adjacent monomers) (Figure 6). A second salt link cluster, SLC-β, adjoins SLC-α in human arginase I and involves R308A, E262B, and D204B. However, SLC-β is not conserved as such in LmARG due to the lack of an arginine residue corresponding to R308 in human arginase I. Consequently, new hydrogen bonds and salt links are formed in LmARG involving H212A, D215A, R266B, and E273B (Figure 6a). LmARG contains L319 in place of R308 in human arginase I, which facilitates the binding of a glycerol molecule that hydrogen bonds with E273 and H215 (from an adjacent monomer) (Figure 6a). Finally, a new intermolecular salt link found in LmARG is designated salt link-γ (SL-γ) and is formed by E277A and R216B (Figure 6b). This salt link is absent in human arginase I. Another intermolecular interaction unique to LmARG is the hydrogen bond donated by the side chain of K198B to the backbone carbonyl of T318A.
Figure 6.
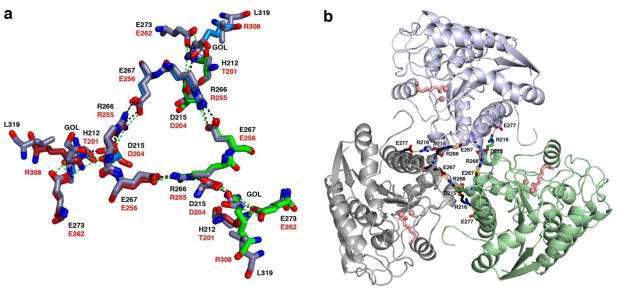
(a) Superposition of human arginase I with LmARG at the trimer interface illustrates salt link networks SLC-α and SLC-β. Atoms in human arginase I are color-coded as red, blue, and green for subunit carbon atoms, blue for nitrogen, and red for oxygen. Atoms are color-coded similarly in LmARG except that subunit carbons are colored blue-grey. Intermolecular interactions are represented as dashed green lines in human arginase I and as dashed black lines in LmARG. Residue labels are black for LmARG and red for human arginase I. (b) The trimeric LmARG-ABH complex, in which salt link networks SLC-α and SL-γ are represented as dashed orange lines. Monomers are grey, light blue, and green; Mn2+ ions and ABH are pink in each monomer.
LmARG-ABH Complex
The overall structure of the LmARG-ABH complex is essentially identical to that of unliganded LmARG with an r.m.s. deviation of 0.28 A for 310 Cα atoms. Accordingly, no significant structural changes are triggered by inhibitor binding. Salt link networks at each subunit-subunit interface are identical to those observed in the unliganded enzyme. The structure of the LmARG-ABH complex is generally similar to that of the human arginase I-ABH complex structure (PDB entry 2AEB) [36], in which the monomers superimpose with an r.m.s. deviation of 0.82 A for 266 Cα atoms.
A simulated annealing omit map of ABH bound in the LmARG active site (Figure 7a) shows that the boronic acid group of ABH undergoes nucleophilic attack by the metal-bridging hydroxide ion of the native enzyme to form the tetrahedral boronate anion. A similar binding mode for ABH is observed in its complexes with rat arginase I [56], human arginase I [36], and arginase from Plasmodium falciparum [57]. The tetrahedral boronate anion mimics the tetrahedral intermediate and its flanking transition states in the arginase reaction. The α-amino and α-carboxylate groups of ABH also make conserved hydrogen bond interactions with active site residues.
Figure 7.

Simulated annealing omit maps of the boronic acid inhibitors (a) ABH (orange map, contoured at 3.1σ) and (b) BEC (magenta map, contoured at 4.0σ) bound in the active site of LmARG. Atoms are color-coded as follows: yellow (protein) or grey (inhibitor) for C, blue for N, red for O, green for B, pink spheres for Mn2+ ions, and red spheres for solvent. Metal coordination and hydrogen bond interactions are represented by blue and green dashed lines, respectively.
Three amino acid substitutions are found in the active site of LmARG compared with human arginase I; however, these differences do not directly impact the binding mode of ABH. First, the T246 side chain in LmARG is located near two metal binding residues, D243 and D245, and the hydroxyl group of T246 donates a hydrogen bond to the backbone carbonyl of metal binding residue D243. In human arginase I, the corresponding residue is G235, so T246 may additionally stabilize the metal binding site in LmARG. Second, the hydroxyl group of T127 in human arginase I donates a hydrogen bond to E186, which in turn hydrogen bonds to a water molecule that accepts a hydrogen bond from the α-amino group of the substrate. LmARG has an alanine residue (A140) in place of T127 in human arginase I, but the glutamate side chain that engages the substrate α-amino group with a water-mediated hydrogen bond (E197 in LmARG) is in the same position as found for E186 in human arginase I. Finally, the backbone carbonyl of D181 in human arginase I accepts a water-mediated hydrogen bond from the α-amino group of the substrate. Although this residue corresponds to A192 in LmARG, the backbone carbonyl of A192 maintains this interaction in the LmARG-ABH complex.
LmARG-BEC Complex
The structure of the LmARG-BEC complex is essentially identical to that of the unliganded LmARG structure in terms of its overall fold (r.m.s. deviation = 0.27 A for 310 Cα atoms). As also observed for ABH, the boronic acid moiety of BEC undergoes nucleophilic attack by the metal-bridging hydroxide ion to form a tetrahedral boronate anion that mimics the tetrahedral intermediate and its flanking transition states in catalysis. Intermolecular interactions within the active site of the LmARG-BEC complex are very similar to those observed in the human arginase I-BEC complex (PDB entry 1WVA) [36]. However, some differences in side chain torsion angles are observed as the binding conformations of ABH and BEC are compared. These differences presumably arise from the longer C-S bond lengths and the compressed C-S-C bond angle introduced by the substitution of the Sγ atom of BEC for the corresponding CH2 group of ABH. A simulated annealing omit map of the LmARG-BEC complex is shown in Figure 7b.
LmARG-nor-NOHA Complex
The overall fold of LmARG in the LmARG-nor-NOHA complex is essentially identical to that of unliganded LmARG, with an r.m.s. deviation of 0.32 Å for 310 Cα atoms. The intermolecular interactions within the active site of the LmARG-nor-NOHA complex are similar to those observed in the human arginase I-nor-NOHA complex (PDB entry 3KV2) [38]. A simulated annealing omit map is shown for the LmARG-nor-NOHA complex in Figure 8a. The hydroxyl group of nor-NOHA displaces the metal-bridging hydroxide ion observed in the unliganded enzyme and coordinates to the Mn2+A ion with a Mn2+A-O separation of 2.1 Å; the Mn2+B-O separation of 2.7 Å is too long to be considered an inner-sphere coordination interaction. The Mn2+A-bound water molecule observed in the unliganded enzyme is also absent. The metal coordination mode of nor-NOHA is very different from that of ABH or BEC, and nor-NOHA exhibits approximately 40-fold weaker inhibitory potency compared with ABH (Table 1) [34]. Nevertheless, it is surprising that micromolar potency is achieved for an inhibitor that does not resemble the transition state in catalysis.
Figure 8.

Simulated annealing omit maps of inhibitors (a) nor-NOHA (blue map, contoured at 4.0σ) and (b) L-ornithine (green map, contoured at 4.8σ) bound in the active site of LmARG. Atoms are color-coded as follows: yellow (protein) or grey (inhibitor) for C, blue for N, red for O, pink spheres for Mn2+ ions, and red spheres for solvent. Metal coordination and hydrogen bond interactions are represented by blue and green dashed lines, respectively.
LmARG-L-Ornithine Complex
The structure of LmARG complexed with the amino acid product of catalysis, L-ornithine, is essentially identical to that of unliganded LmARG (r.m.s. deviation = 0.52 A for 310 Cα atoms). The binding orientation, conformation, and intermolecular interactions of L-ornithine are similar to those observed in the human arginase I-L-ornithine complex (PDB entry 3GMZ) [58]. However, it is curious that while the Mn2+A-bound water molecule remains in place as observed in the unliganded enzyme, accepting a hydrogen bond from the side chain of L-ornithine and donating a hydrogen bond to the side chain of E288, no electron density is observed for the metal-bridging hydroxide ion. The metal-bridging hydroxide ion was, however, observed in the structure of the human arginase I-L-ornithine complex [58]. It is possible that the structure of the LmARG-L-ornithine complex represents a step in catalysis that follows dissociation of product urea but precedes the binding of another water molecule to the binuclear manganese cluster that would complete the catalytic cycle. A simulated annealing omit map of the LmARG-L-ornithine complex is shown in Figure 8b.
Effects of Arginase Inhibitors on Promastigote Proliferation
The consequences of NOHA, nor-NOHA, ABH, or BEC treatment for L. mexicana promastigote proliferation have been reported previously [34]. The EC50 values measured after five days of incubation were 6 mM and 3 mM for NOHA and nor-NOHA, respectively, while ABH and BEC exhibited no toxicity [34]. These observations were surprising since a genetic deletion of arginase is lethal in Leishmania promastigotes [24–26]. Because L. mexicana parasites contain large amounts of L-ornithine (~130 nmol/107 parasites) compared to levels of L-arginine and putrescine (~50 nmol arginine /107 parasites and ~2.5 nmol putrescine/107 parasites, respectively [34]), it is conceivable that inhibition of arginase will have no effect on parasite proliferation until stored pools of L-ornithine have been consumed. To test this hypothesis, we incubated promastigotes for up to four weeks in the presence of NOHA and nor-NOHA (details in the Experimental Procedures section). Although the EC50 values dropped slightly, from 3 or 6 mM to ca. 1 mM (data not shown), the inhibitors still exhibited poor efficacy against promastigote proliferation, suggesting that L-ornithine pools are not responsible for the drug resistance of the parasites. It should be noted that the modest growth inhibition by NOHA and nor-NOHA could be circumvented completely by the addition of 200 μM putrescine (data not shown), demonstrating that the compounds indeed target arginase in intact parasites, albeit only at high concentrations.
Another possible cause for the poor efficacy of the potent arginase inhibitors may be that the compounds do not reach the glycosome, a nutritional microbody unique to Leishmania and related trypanosome species [59, 60] that sequesters arginase [24–26]. To test this conjecture, we evaluated the effects of NOHA and nor-NOHA on genetically modified parasites that expressed arginase in either the glycosome or the cytosol. Previously characterized L. mexicana Δarg parasites, which had been complemented with episomal constructs containing either the wild-type arginase sequence or a Δskl mutant [24], were utilized for these studies. Deletion of the N-terminal SKL signaling peptide causes the arginase to mislocalize to the cytosol, although the enzyme is fully functional in promastigotes [24]. No significant differences were observed in the proliferation of L. mexicana Δarg[pGFP-argΔskl] and L. mexicana Δarg[pGFP-ARG] promastigotes when inhibition with NOHA and nor-NOHA was compared (data not shown). Thus, the most reasonable explanation for the lack of significant efficacy of the arginase inhibitors is poor uptake by the parasites. Our observations support previous claims that the reduction of infectivity of L. major by nor-NOHA in mice is due solely to inhibition of host arginase [30] and raise the possibility that combined inhibition of host and parasite arginase may be even more potent.
Importance of Subcellular Localization of Arginase for In Vivo Infectivity
Previous studies demonstrated that the glycosomal localization of the L. mexicana arginase had no effect on promastigote viability [24]. A study in L. amazonensis likewise found that a mislocalized arginase allowed normal proliferation in promastigotes; however, the infectivity in mice was reduced to levels even lower than those observed with L. amazonensis Δarg parasites [25]. Because the mislocalized arginase was expressed at very low levels [25], it is difficult to assess if the lack of function can be attributed to low expression or mislocalization. In L. mexicana, arginases expressed in the glycosome or cytosol in a L. mexicana Δarg background (L. mexicana Δarg[pGFP-ARG] and Δarg[pGFP-argΔskl]) are both expressed at robust levels, although somewhat lower than in wild-type parasites (Figure 9). Previous infectivity studies in mice had shown that L. mexicana Δarg showed reduced infectivity compared to wild-type parasites and L. mexicana Δarg and Δarg[pGFP-ARG] parasites [53]. Here, we show that L. mexicana Δarg[pGFP-argΔskl] parasites yield smaller lesions than Δarg[pGFP-ARG] parasites, which were similar in size to the lesions of Δarg parasites (Figure 10). Furthermore, 3 of 5 mice infected with wild-type, and 1 of 5 mice infected with Δarg[pGFP-ARG] parasites, developed ulcerated lesions during the period of infection. Because the mislocalized protein is expressed at robust levels, at least in the promastigote stage (Figure 9), the data intimate that the glycosomal milieu in which LmARG resides is essential for enzyme function in the amastigote stage of the parasite. This is particularly surprising because the cellular localization does not affect the polyamine function of arginase in Leishmania promastigotes [24–26]. Our data do not formally discern whether the smaller lesions are due to impaired amastagote proliferation or due to a dimished ability of the intracellular parasite to recruit a cellular infiltrate leading to lesions. Furthermore, we cannot discern whether the impact of mislocalization on enzyme activity is due to impaired polyamine metabolism or some other downstream defect affecting survival or function of the intracellular amastigote. Further studies will be necessary to investigate the molecular cause of this conundrum and how the host environment may influence parasite arginase function.
Figure 9.

Expression levels of arginase in transgenic parasites. Western blot analysis of lysates from wild-type, Δarg, Δarg [pGFP-ARG], and Δarg [pGFP-argΔskl] parasites grown in the presence of 200 μM polyamines (+ polyamines) or absence of polyamines (−polyamines). The blot was probed with an anti-arginase antibody and with an anti-tubulin antibody as a loading control. Because the GFP-ARG is a fusion protein, the molecular weight is larger than that of wild-type LmARG.
Figure 10.
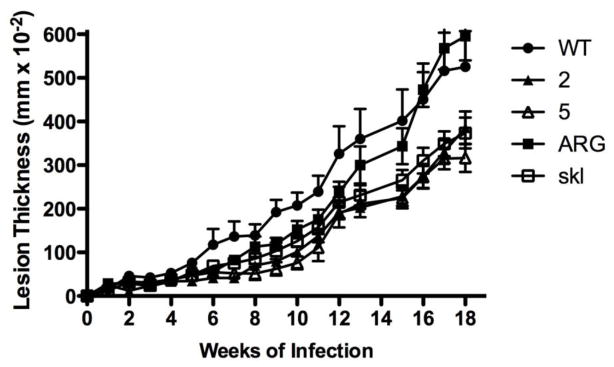
Proper LmARG localization is essential for robust infection in mice. BALB/c mice were infected with L. mexicana wild-type (black circles), two independent Δarg strains (white and black triangles labeled “2” and “5” for clone numbers), Δarg[pGFP-ARG] complemented with wild-type ARG (labeled ARG, black squares), Δarg[pGFP-argΔskl] complemented with a mutant ARG gene (labeled “skl”, white squares). Lesion thickness, measured as the size of the infected footpad minus the size of the uninfected footpad, was measured weekly for 18 weeks. Data show the mean ± standard deviation of lesion thickness in groups of 5 mice inoculated with each parasite strain. These data are representative of two separate experiments. Results for the wild-type, Δarg clone 5, and Δarg[pGFP-ARG] strains have been reported previously [53] and are reproduced with permission (Gaur, U., Roberts, S. C., Dalvi, R. P., Corraliza, I., Ullman, B., Wilson, M.E. (2007) An effect of parasite-encoded arginase on the outcome of murine cutaneous leishmaniasis. J. Immunol. 197, 8446–8453. Copyright © [2007] The American Association of Immunologists, Inc.).
Inhibitor Design Strategy for LmARG
Arginase is a critical enzyme for polyamine biosynthesis and the survival of Leishmania species, as specifically demonstrated in studies of arginase knockout mutants in L. mexicana [24], L. amazonensis [25], and L. major [26, 27]. These studies validate arginase as a potential drug target for the treatment of leishmaniasis, and the search for arginase inhibitors targeting intact Leishmania parasites is an urgent area of investigation. However, it is surprising that even the relatively potent arginase inhibitor nor- NOHA, with Ki ≈ 50 μM [34], exhibits an EC50 value of only 3 mM when incubated with L. mexicana promastigotes [34]; moreover, the EC50 value is only nominally improved to ~1 mM after long-term incubation with nor-NOHA, as described in the preceding section. Accordingly, the relative insensitivity of L. mexicana promastigotes to nor-NOHA cannot be explained by residual L-ornithine pools that might sustain polyamine biosynthesis and parasite survival in short-term studies. Therefore, we conclude that the lack of significant efficacy of the arginase inhibitors nor-NOHA and NOHA is due to poor uptake by the parasites.
With its pKa of 8.1, the hydroxyguanidinium moiety of nor-NOHA or NOHA is predominantly protonated at physiological pH, as shown in Table 1. It is conceivable that uptake of inhibitors bearing a net positive charge is hindered. However, while the inhibitors ABH and BEC are neutral (Table 1), parasites are resistant to these inhibitors as well [34]. The positively charged α-amino group and a negatively charged α-carboxylate group of these zwitterions may similarly hinder diffusion across parasitic membranes. It is possible that the α-amino and αcarboxylate groups of ABH or BEC could be protected by chemical derivatization in the form of a neutral, non-zwitterionic prodrug that, upon incorporation into the glycosome, could be hydrolyzed to yield the free amino acid inhibitor. Based on the crystal structures of LmARG-inhibitor complexes (Figures 7 and 8), the α-amino and α-carboxylate groups of the inhibitor ultimately must be fully deprotected to engage in key hydrogen bond interactions that firmly anchor the inhibitor in the enzyme active site.
Importantly, the crystal structures of LmARG and its inhibitor complexes reveal a new region of the active site that could be targeted by α,α-disubstituted amino acid inhibitors in which the Cα-H group of ABH or nor-NOHA can be replaced with a longer side chain capable of targeting interactions in an anionic cleft specific to LmARG (Figure 11). Crystal structures of human arginase I complexed with α,α-disubstituted amino acid inhibitors such as DFMO, or ABH derivatized with methyl or difluoromethyl substituents, reveal that the additional αsubstituent binds in a corresponding cleft that is not so anionic, the “T136 region” [58]. Structural comparisons with the corresponding inhibitor complexes of P. falciparum arginase show that the shape and electrostatic surface potential of this region differs between the human enzyme and the parasitic enzyme [58]. This region is additionally different between LmARG and human arginase I (Figure 11). Therefore, we suggest that α,α-disubstituted amino acid inhibitors based on the ABH or nor-NOHA scaffolding could be designed with specificity for leishmanial arginase by targeting interactions in the anionic cleft highlighted in Figure 11b with the additional α-substituent. Derivatization with positively-charged groups might facilitate new enzyme-inhibitor salt link interactions in the anionic cleft; the introduction of halogen atoms might allow for other polar interactions with fluorine, chlorine, or bromine capable of enhancing affinity [61–63]. Such substitutents could also provide a route toward modulating the chemical properties of the inhibitor without compromising inhibitor binding interactions in the arginase active site (e.g., side chain metal coordination interactions and hydrogen bond interactions of αamino and α-carboxylate groups as shown in Figures 7 and 8), so as to enhance inhibitor uptake properties by the parasite.
Figure 11.
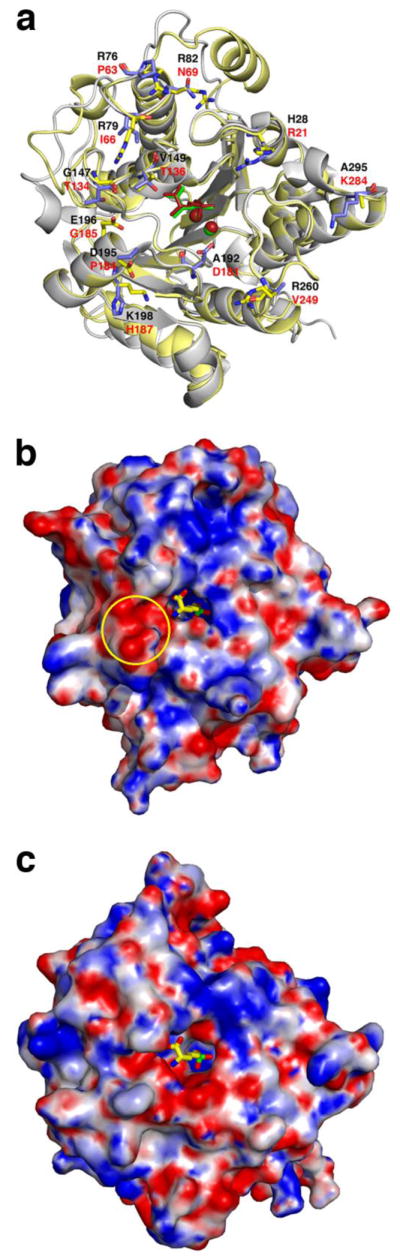
(a) Superposition of the LmARG-ABH complex and the human arginase I-ABH complex (PDB entry 2AEB) [36] highlighting non-identical residues on the protein surface surrounding the active site cleft. Atoms are color-coded as follows: LmARG-ABH complex, yellow for protein and green for ABH and Mn2+ ions; human arginase I-ABH complex, blue-grey for protein and dark red for ABH and Mn2+ ions; residue labels are black for LmARG and red for human arginase I. (b) Electrostatic surface potential of the LmARG-ABH complex calculated at pH 8.5 (the orientation of the protein structure is the same as that in plate (a)). ABH was not included in the calculation but it is superimposed as a stick figure in its original position with atoms color-coded as follows: yellow for carbon, blue for nitrogen, red for oxygen, and green for boron. The electrostatic surface potential ranges from −15kT (red) to +15kT (blue). The anionic cleft in which the second substituent of an α,α-disubstituted amino acid inhibitor based on ABH or nor-NOHA is indicated by the yellow circle. (c) Electrostatic surface potential of the human arginase I-ABH complex calculated at pH 8.5 and color-coded as in (b). ABH was not included in the calculation but it is superimposed as a stick figure in its original position with atoms color-coded as in (b). While the cleft corresponding to that in (b) is generally conserved, it is not as significantly anionic as that in (b).
In conclusion, the crystal structure of LmARG provides important guidance regarding its biological function in polyamine biosynthesis and the design of new inhibitors. Additionally, the crystal structures of LmARG-inhibitor complexes provide a starting point for the design of α,α-disubstituted amino acid inhibitors that may exhibit enhanced biological activity. Future studies in this regard will be reported in due course.
Highlights.
Crystal structures of LmARG and its complexes with inhibitors have been determined
The inhibitor nor-NOHA attenuates parasite proliferation, albeit weakly
LmARG structures will enable the design of selective inhibitors with improved parasite uptake properties
Acknowledgments
We thank the National Synchrotron Light Source at Brookhaven National Laboratory (beamline X29) for access to X-ray crystallographic data collection facilities.
Footnotes
This work was supported by US National Institutes of Health Grants GM49758 to D.W.C., AI41622 to B.U., and AI045540 to M.E.W., Grant 1I01BX001983 from the US Department of Veterans’ Affairs (M.E.W.), and a Medical Research Foundation New Investigator Grant to S.C.R.
Abbreviations: ABH, 2(S)-amino-6-boronohexanoic acid; BEC, S-(2-boronoethyl)-L-cysteine; BME, β-mercaptoethanol; DFMO, D,L-α-difluoromethylornithine; GFP, green fluorescent protein; HEPES, N-(2-hydroxyethyl)piperazine-N′-(2-ethansulfonic acid); HEPPS, N-(2-hydroxyethyl)piperazine-N′-(3-propanesulfonic acid); IPTG, isopropyl β-D-thiogalactopyranoside; LmARG, Leishmania mexicana arginase; MES, 2-(N-morpholino)ethanesulfonic acid; MPD, (+/−)-2-methyl-2,4-pentanediol; NOHA, Nω-hydroxy-L-arginine; nor-NOHA, nor-Nω-hydroxy-L-arginine; PDB, Protein Data Bank; r.m.s., root-mean-square.
Accession Codes
The atomic coordinates and structure factors of LmARG, the LmARG-ABH complex, the LmARG-nor-NOHA complex, the LmARG-BEC complex, and the LmARG-L-ornithine complex have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 4ITY, 4IU0, 4IU1, 4IU4, and 4IU5, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Desjeux P. Leishmaniasis: public health aspects and control. Clin Dermatol. 1996;14:417–423. doi: 10.1016/0738-081x(96)00057-0. [DOI] [PubMed] [Google Scholar]
- 2.Desjeux P. Global control and Leishmania HIV co-infection. Clin Dermatol. 1999;17:317–325. doi: 10.1016/s0738-081x(99)00050-4. [DOI] [PubMed] [Google Scholar]
- 3.Wanasen N, Soong L. L-Arginine metabolism and its impact on host immunity against Leishmania infection. Immunol Res. 2008;41:15–25. doi: 10.1007/s12026-007-8012-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desjeux P. The increase in risk factors for leishmaniasis worldwide. Trans R Soc Tropical Med Hygiene. 2001;95:239–243. doi: 10.1016/s0035-9203(01)90223-8. [DOI] [PubMed] [Google Scholar]
- 5.James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 11. Elsevier/Saunders; Philadelphia, PA: 2011. p. 968. [Google Scholar]
- 6.Antinori S, Gianelli E, Calattini S, Longhi E, Gramiccia M, Corbellino M. Cutaneous leishmaniasis: an increasing threat for travelers. Clin Microbiol Infect. 2005;11:343–346. doi: 10.1111/j.1469-0691.2004.01046.x. [DOI] [PubMed] [Google Scholar]
- 7.Wright NA, Davis LE, Aftergut KS, Parrish CA, Cockerell CJ. Cutaneous leishmaniasis in Texas: a northern spread of endemic areas. J Am Acad Dermatol. 2008;58:650–652. doi: 10.1016/j.jaad.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 8.González C, Wang O, Strutz SE, González-Salazar C, Sánchez-Cordero V, Sarkar S. Climate change and risk of leishmaniasis in North America: predictions from ecological niche models of vector and reservoir species. PLoS Negl Trop Dis. 2010;4:e585. doi: 10.1371/journal.pntd.0000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heby O, Roberts SC, Ullman B. Polyamine biosynthetic enzymes as drug targets in parasitic protozoa. Biochem Soc Trans. 2003;31:415–419. doi: 10.1042/bst0310415. [DOI] [PubMed] [Google Scholar]
- 10.Heby O, Persson L, Rentala M. Targeting the polyamine biosynthetic enzymes: a promising approach to therapy of African sleeping sickness, Chagas’ disease, and leishmaniasis. Amino Acids. 2007;33:359–366. doi: 10.1007/s00726-007-0537-9. [DOI] [PubMed] [Google Scholar]
- 11.Bacchi CJ, Nathan HC, Hutner SH, McCann PP, Sjoerdsma A. Polyamine metabolism: a potential therapeutic target in trypanosomes. Science. 1980;210:332–334. doi: 10.1126/science.6775372. [DOI] [PubMed] [Google Scholar]
- 12.Grishin NV, Osterman AL, Brooks HB, Phillips MA, Goldsmith EJ. X-ray structure of ornithine decarboxylase from Trypanosoma brucei: the native structure and the structure in complex with α-difluoromethylornithine. Biochemistry. 1999;38:15174–15184. doi: 10.1021/bi9915115. [DOI] [PubMed] [Google Scholar]
- 13.Kaur K, Emmett K, McCann PP, Sjoerdsma A, Ullman B. Effects of DL-α-difluoromethylornithine on Leishmania donovani promastigotes. J Protozool. 1986;33:518–521. doi: 10.1111/j.1550-7408.1986.tb05654.x. [DOI] [PubMed] [Google Scholar]
- 14.Reguera RM, Fouce RB, Cubría JC, Bujidos ML, Ordóñez D. Fluorinated analogues of L-ornithine are powerful inhibitors of ornithine decarboxylase and cell growth of Leishmania infantum promastigotes. Life Sci. 1995;56:223–230. doi: 10.1016/0024-3205(94)00916-3. [DOI] [PubMed] [Google Scholar]
- 15.Gradoni L, Iorio MA, Gramiccia M, Orsini S. In vivo effect of eflornithine (DFMO) and some related compounds on Leishmania infantum. Farmaco. 1989;44:1157–1166. [PubMed] [Google Scholar]
- 16.Mukhopadhyay R, Madhubala R. Effect of a bis(benzyl)polyamine analogue, and DL-α-difluoromethylornithine on parasite suppression and cellular polyamine levels in golden hamster during Leishmania donovani infection. Pharmacol Res. 1993;28:359–365. doi: 10.1006/phrs.1993.1138. [DOI] [PubMed] [Google Scholar]
- 17.Olenyik T, Gilroy C, Ullman B. Oral putrescine restores virulence of ornithine decarboxylase-deficient Leishmania donovani in mice. Mol Biochem Parasitol. 2011;176:109–111. doi: 10.1016/j.molbiopara.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boitz JM, Yates PA, Kline C, Gaur U, Wilson ME, Ullman B, Roberts SC. Leishmania donovani ornithine decarboxylase is indispensable for parasite survival in the mammalian host. Infect Immun. 2009;77:756–763. doi: 10.1128/IAI.01236-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilroy C, Olenyik T, Roberts SC, Ullman B. Spermidine synthase is required for virulence of Leishmania donovani. Infect Immun. 2011;79:109–111. doi: 10.1128/IAI.00073-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ash DE, Cox JD, Christianson DW. Arginase: a binuclear manganese metalloenzyme. Manganese and Its Role in Biological Processes. 1999;37:407–428. [PubMed] [Google Scholar]; Sigel A, Sigel H, editors. Metal Ions in Biological Systems. M. Dekker; New York: [Google Scholar]
- 21.Christianson DW. Arginase: structure, mechanism, and physiological role in male and female sexual arousal. Acc Chem Res. 2005;38:191–201. doi: 10.1021/ar040183k. [DOI] [PubMed] [Google Scholar]
- 22.Vincendeau P, Gobert AP, Daulouède S, Moynet D, Mossalayi MD. Arginases in parasitic diseases. Trends Parasitol. 2003;19:9–12. doi: 10.1016/s1471-4922(02)00010-7. [DOI] [PubMed] [Google Scholar]
- 23.Das P, Lahiri A, Chakravortty D. Modulation of the arginase pathway in the context of microbial pathogenesis: a metabolic enzyme moonlighting as an immune modulator. PLoS Pathogens. 2010;6:e10000899. doi: 10.1371/journal.ppat.1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts SC, Tancer MJ, Polinsky MR, Gibson KM, Heby O, Ullman B. Arginase plays a pivotal role in polyamine precursor metabolism in Leishmania. J Biol Chem. 2004;279:23668–23678. doi: 10.1074/jbc.M402042200. [DOI] [PubMed] [Google Scholar]
- 25.da Silva MF, Zampieri RA, Muxel SM, Beverley SM, Floeter-Winter LM. Leishmania amazonensis arginase compartmentalization in the glycosome is important for parasite infectivity. PLoS One. 2012;7:e34022. doi: 10.1371/journal.pone.0034022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reguera RM, Balaña-Fouce R, Showalter M, Hickerson S, Beverley SM. Leishmania major lacking arginase (ARG) are auxotrophic for polyamines but retain infectivity to susceptible BALB/c mice. Mol Biochem Parasitol. 2009;165:48–56. doi: 10.1016/j.molbiopara.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muleme HM, Reguera RM, Berard A, Azinwi R, Jia P, Okwor IB, Beverley S, Uzonna JE. Infection with arginase-deficient Leishmania major reveals a parasite number-dependent and cytokine-independent regulation of host cellular arginase activity and disease pathogenesis. J Immunol. 2009;183:8068–8076. doi: 10.4049/jimmunol.0803979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Custot J, Moali C, Brollo M, Boucher JL, Delaforge M, Mansuy D, Tenu JP, Zimmermann JL. The new α-amino acid Nω-hydroxy-nor-L-arginine: a high-affinity inhibitor of arginase well adapted to bind to its manganese cluster. J Am Chem Soc. 1997;119:4086–4087. [Google Scholar]
- 29.Iniesta V, Gómez-Nieto LC, Corraliza I. The inhibition of arginase by Nω-hydroxy-L-arginine controls the growth of Leishmania inside macrophages. J Exp Med. 2001;193:777–783. doi: 10.1084/jem.193.6.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kropf P, Fuentes JM, Fähnrich E, Arpa L, Herath S, Weber V, Soler G, Celada A, Modolell M, Müller I. Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo. FASEB J. 2005;19:1000–1002. doi: 10.1096/fj.04-3416fje. [DOI] [PubMed] [Google Scholar]
- 31.Iniesta V, Carcelén J, Molano I, Peixoto PM, Redondo E, Parra P, Mangas M, Monroy I, Campo ML, Nieto CG, Corraliza I. Arginase I induction during Leishmania major infections mediates the development of disease. Infect Immun. 2005;73:6085–6090. doi: 10.1128/IAI.73.9.6085-6090.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, Gardner MJ, Gingle A, Grant G, Harb OS, Heiges M, Hertz-Fowler C, Houston R, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Logan FJ, Miller JA, Mitra S, Myler PJ, Nayak V, Pennington C, Phan I, Pinney DF, Ramasamy G, Rogers MB, Roos DS, Ross C, Sivam D, Smith DF, Srinivasamoorthy G, Stoeckert CJ, Jr, Subramanian S, Thibodeau R, Tivey A, Treatman C, Velarde G, Wang H. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010;38:D457–D462. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.da Silva ER, da Silva MFL, Fischer H, Mortara RA, Mayer MG, Framesqui K, Silber AM, Floeter-Winter LM. Biochemical and biophysical properties of a highly active recombinant arginase from Leishmania (Leishmania) amazonensis and subcellular location of native enzyme. Mol Biochem Parasitol. 2008;159:104–111. doi: 10.1016/j.molbiopara.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 34.Riley E, Roberts SC, Ullman B. Inhibition profile of Leishmania mexicana arginase reveals differences with human arginase I. Int J Parasitol. 2011;41:545–552. doi: 10.1016/j.ijpara.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baggio R, Elbaum D, Kanyo ZF, Carroll PJ, Cavalli RC, Ash DE, Christianson DW. Inhibition of Mn2+2-arginase by borate leads to the design of a transition state analogue inhibitor, 2(S)-amino-6-boronohexanoic acid. J Am Chem Soc. 1997;119:8107–8108. [Google Scholar]
- 36.Di Costanzo L, Sabio G, Mora A, Rodriquez PC, Ochoa AC, Centeno F, Christianson DW. Crystal structure of human arginase I at 1.29-Å resolution and exploration of inhibition in the immune response. Proc Natl Acad Sci U S A. 2005;102:13058–13063. doi: 10.1073/pnas.0504027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim NN, Cox JD, Baggio RF, Emig FA, Mistry SK, Harper SL, Speicher DW, Morris SM, Ash DE, Traish A, Christianson DW. Probing erectile function: S-(2-Boronoethyl)-L-cysteine binds to arginase as a transition state analogue and enhances smooth muscle relaxation in human penile corpus cavernosum. Biochemistry. 2001;40:2678–2688. doi: 10.1021/bi002317h. [DOI] [PubMed] [Google Scholar]
- 38.Di Costanzo L, Ilies M, Thorn KJ, Christianson DW. Inhibition of human arginase I by substrate and product analogues. Arch Biochem Biophys. 2010;496:101–108. doi: 10.1016/j.abb.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 41.Yeates TO. Detecting and overcoming crystal twinning. Methods Enzymol. 1997;276:344–358. [PubMed] [Google Scholar]
- 42.Brünger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 43.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 44.Collaborative Computational Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.D’Antonio EL, Christianson DW. Crystal structures of complexes with cobalt-reconstituted human arginase I. Biochemistry. 2011;50:8018–8027. doi: 10.1021/bi201101t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 47.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 48.Kleywegt GJ, Zou JY, Kjeldgaard M, Jones TA. Around O. In: Rossmann MG, Arnold E, editors. International Tables of Crystallography. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2001. pp. 353–356.pp. 366–367. [Google Scholar]
- 49.DeLano WL. MacPyMOL: A PyMOL-based molecular graphics application for MacOS X. Palo Alto, California: DeLano Scientific LLC; 2007. [Google Scholar]
- 50.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dolinsky TJ, Nielson JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olsson MH, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 53.Gaur U, Roberts SC, Dalvi RP, Corraliza I, Ullman B, Wilson ME. An effect of parasite-encoded arginase on the outcome of murine cutaneous leishmaniasis. J Immunol. 2007;179:8446–8453. doi: 10.4049/jimmunol.179.12.8446. [DOI] [PubMed] [Google Scholar]
- 54.Di Costanzo L, Pique ME, Christianson DW. Crystal structure of human arginase I complexed with thiosemicarbazide reveals an unusual thiocarbonyl μ-sulfide ligand in the binuclear manganese cluster. J Am Chem Soc. 2007;129:6388–6389. doi: 10.1021/ja071567j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 56.Cox JD, Kim NN, Traish AM, Christianson DW. Arginase-boronic acid complex highlights a physiological role in erectile function. Nat Struct Biol. 1999;6:1043–1047. doi: 10.1038/14929. [DOI] [PubMed] [Google Scholar]
- 57.Dowling DP, Ilies M, Olszewski KL, Portugal S, Mota MM, Llinás M, Christianson DW. Crystal structure of arginase from Plasmodium falciparum and implications for L-arginine depletion in malarial infection. Biochemistry. 2010;49:5600–5608. doi: 10.1021/bi100390z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ilies M, Di Costanzo L, Dowling DP, Thorn KJ, Christianson DW. Binding of α,α-disubstituted amino acids to arginase suggests new avenues for inhibitor design. J Med Chem. 2011;54:5432–5443. doi: 10.1021/jm200443b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Opperdoes FR. Compartmentation of carbohydrate metabolism in trypanosomes. Annu Rev Microbiol. 1987;41:127–151. doi: 10.1146/annurev.mi.41.100187.001015. [DOI] [PubMed] [Google Scholar]
- 60.Michels PA, Bringaud F, Herman M, Hannaert V. Metabolic functions of glycosomes in trypanosomatids. Biochim Biophys Acta. 2006;1763:1463–1477. doi: 10.1016/j.bbamcr.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 61.Kim CY, Chandra PP, Jain A, Christianson DW. Fluoroaromatic-fluoroaromatic interactions between inhibitors bound in the crystal lattice of human carbonic anhydrase II. J Am Chem Soc. 2001;123:9620–9627. doi: 10.1021/ja011034p. [DOI] [PubMed] [Google Scholar]
- 62.Auffinger P, Hays FA, Westhof E, Ho PS. Halogen bonds in biological molecules. Proc Natl Acad Sci USA. 2004;101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem. 2013;56:1363–1388. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- 64.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins: Struct, Funct, Genet. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 65.Heinig M, Frishman D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004;32:W500–W502. doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gouet P, Courcelle E, Stuart DI, Métoz F. ESPript: analysis of multiple sequence alignments in postscript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]