

Abstract
The enzyme phenylalanine hydroxylase catalyzes the hydroxylation of excess phenylalanine in the liver to tyrosine. The enzyme is regulated allosterically by phenylalanine and by phosphorylation of Ser16. Hydrogen/deuterium exchange monitored by mass spectrometry has been used to gain insight into any structural change upon phosphorylation. Peptides in both the catalytic and regulatory domains show increased deuterium incorporation into the phosphorylated protein. Deuterium is incorporated into fewer peptides than when the enzyme is activated by phenylalanine, and the incorporation is slower. This establishes that the conformational change upon phosphorylation of phenylalanine hydroxylase is different from and less extensive than that upon phenylalanine activation.
Keywords: phenylalanine hydroxylase, phosphorylation, regulation, hydrogen-deuterium exchange, allostery, structure
INTRODUCTION
The physiological role of the enzyme phenylalanine hydroxylase (PheH) in the liver is to convert excess phenylalanine in the diet to tyrosine, using tetrahydrobiopterin as the source of the electrons for this hydroxylation reaction [1, 2]. To maintain a proper balance between catabolism and the biosynthetic requirements for phenylalanine, the enzyme is heavily regulated [3]. The best-characterized regulatory mechanism is allosteric, in that the activity of the enzyme exhibits positive cooperativity versus phenylalanine [4]. Activation by phenylalanine involves the discrete regulatory domain of the enzyme [5], which contains a separate phenylalanine binding site [6, 7]. While there are no structures of either the intact protein or the isolated regulatory domain with phenylalanine bound, the structure of the enzyme in the absence of ligands shows that the N-terminus of the regulatory domain reaches across the active site in the catalytic domain [8]. This structure is the basis for the present understanding of the mechanism of phenylalanine activation, in that binding of phenylalanine to the regulatory site is proposed to result in a large conformational change that opens up the active site [9, 10]. The proposed conformational change upon phenylalanine binding is supported by a number of indirect experimental approaches [11–15]. More recently, analysis of the effect phenylalanine on incorporation of deuterium from solvent into PheH has provided direct evidence that phenylalanine binding disrupts interactions between the regulatory and catalytic domains, consistent with a reorientation of the regulatory domain [16].
PheH is also regulated by phosphorylation of Ser16 in the regulatory domain. The kinase responsible is generally considered to be cAMP-dependent protein kinase A (PKA), since this kinase will phosphorylate Ser16 in vitro and treatment of liver cells with activators of PKA such as glucagon results in increased phosphorylation of PheH [17, 18]. The increase in the activity of PheH upon phosphorylation is much less than that due to phenylalanine activation [19, 20], and the phosphorylated enzyme can be activated further by phenylalanine [21, 22]. Rather than directly activating the enzyme, phosphorylation has been proposed to allow activation of PheH at lower concentrations of phenylalanine [19]. The effect of phosphorylation on the structure of PheH is not known. Structures of both the unphosphorylated and the phosphorylated rat enzyme have been determined [8]. However, in both structures the N-terminal 18 residues are not visible, and there are no detectable differences in the structures of the two forms of the enzyme. The lack of a difference has been attributed to either the structural change being coupled to phenylalanine binding, so that no change occurs in the absence of phenylalanine [8], or the effect of phosphorylation being localized to amino acid residues near Ser16 [23]. To gain more direct insight into the effect of Ser16 phosphorylation on the structure of PheH, we have now analyzed the effects of phosphorylation on the kinetics of incorporation of deuterium from solvent into the protein by mass spectrometry.
MATERIALS AND METHODS
Materials
ATP and phosphatase inhibitor cocktail 1 were purchased from Sigma-Aldrich Chemical Co. (Milwaukee, WI). Phenyl-Sepharose™ CL-4B was purchased from Amersham Biosciences (Sweden). Porcine stomach pepsin A was from Worthington Biochemical Co. (Lakewood, NJ). Deuterium oxide (D2O, 99% D) was from Cambridge Isotope Laboratories (Andover, MA). Pepstatin A and leupeptin were from Peptides Institute, Inc (Osaka, Japan). [γ-32P]-ATP (10 µCi/µl) was from Perkin Elmer Life and Analytical Science (Boston).
Protein Purification and Preparation
Wild-type rat PheH was expressed in E. coli and purified as previously described [24, 25]. The apo-enzyme was reconstituted with ferrous iron under argon immediately before use [16]. The catalytic subunit of cAMP-dependent protein kinase (PKA) was purified from bovine heart as described by Flockhart [26].
For preparation of phosphorylated PheH, ~12 µM PheH was incubated with 0.3 mM ATP, 10 mM MgCl2, 0.8 µM PKA, and 50 mM Hepes, pH 7.0, at 22 °C for 1 h in a total volume of 65 ml. After the first 15 min, additional PKA and ATP were added for final concentrations of 1 µM PKA and 0.4 mM ATP. The phosphorylated PheH was isolated by FPLC using a HiPrep 16/10 Q XL column (GE Healthcare Life Science) in 50 mM Hepes, 5% glycerol, pH 7.0, with a gradient of 0–0.3 M KCl. The pooled column fractions were dialyzed against 15% glycerol, 50 µM EDTA, 60 µl phosphatase inhibitor cocktail 1, 1 µM pepstatin A, 1 µM leupeptin, 50 mM Hepes, pH 7.0, at 4 °C, concentrated to about 200 µM using an Amicon Ultracel centrifugal filter, and stored at −80 °C. To determine if phosphorylation was complete, a small phosphorylation reaction was performed with the phosphorylated enzyme using [γ-32P]-ATP. No incorporation of radioactivity was observed, establishing that the stoichiometry of phosphorylation was >0.95/subunit. The site of phosphorylation was confirmed by mass spectrometric analysis of peptides at the Institutional Mass Spectrometry Laboratory at the University of Texas Health Science Center.
H/D Exchange
The protocol for the H/D exchange reactions was as described previously [16]. Enzyme (15 µl) was added under argon to 0.3 ml 50 mM Hepes, pD 7.0, in D2O at 25 °C. Samples (20 µl) were removed through a septum with an air-tight syringe at different time points and quenched with 20 µl ice-cold 100 mM sodium citrate, pH 2.4. After mixing, the sample was immediately frozen in liquid nitrogen and stored at −80 °C. For analysis, each freshly thawed sample was incubated with an equal mass of pepsin for 5 min on ice before being injected onto a Vydac C18 HPLC column (2.1 mm × 150 mm) connected to a Thermo Finnigan LCQ DECA XP ion-trap mass spectrometer. All the peptides analyzed in this study eluted after 6 to 12 min and with m/z values of 400 to 2000. Singly, doubly and triply charged peptides were analyzed. Tandem mass spectrometry under the same conditions but with H2O instead of D2O was used for peptide identification. The TurboSEQUEST software from Thermo Finnigan, version 3.1, was used to process the data. The mass spectrometry experiments were all conducted in the Protein Chemistry Laboratory at Texas A&M University. The extent of back-exchange was determined from lyophilized peptides that had been allowed to equilibrate in D2O as previously described [16].
Data Processing
The data were exported using Thermo Finnigan Xcalibur software into HX-Express, a Microsoft Excel-based software for generating a deuterium incorporation curve [27]. The kinetics of exchange were fit to equation 1 or equation 2 using KaleidaGraph (Synergy), where N is the total fraction of exchange over the course of the reaction and A and B are the fractions of amide hydrogens exchanging with rate constants k1 and k2, respectively.
| (1) |
| (2) |
RESULTS
Peptide identification and sequence coverage
To prepare phosphorylated PheH, the enzyme was incubated with cAMP-dependent protein kinase before conducting the H/D exchange, the identities of peptides generated by pepsin digestion of phosphorylated PheH were determined by tandem mass spectrometry. A total of 23 peptides of phosphorylated PheH could routinely be identified covering 69% of the protein sequence (Figure 1). This is slightly less than was seen with the unphosphorylated enzyme, for which a total of 30 peptides could be routinely identified, covering 80% of the protein sequence.
Figure 1.

Sequence coverage of PheH. The peptic peptides in phosphorylated PheH analyzed in this study are indicated by solid arrows and those in the unphosphorylated enzyme by dashed arrows. Ser16 is indicated by an asterisk.
H/D exchange of phosphorylated PheH
Phosphorylated PheH was reconstituted with ferrous iron immediately before carrying out the exchange reaction to ensure that the active site iron was in the active form. In addition, the exchange reaction was carried out under argon to prevent oxidation of the iron. To initiate the exchange reaction, a concentrated sample of phosphorylated PheH was diluted 20-fold into buffer in D2O at pD 7 and 25 °C. Aliquots were taken out over the course of 4 h and mixed with an equal volume of ice-cold buffer at pH 2.4 to quench the exchange. To determine the extent of deuterium incorporation, samples were mixed with pepsin for 5 min before being injected onto the HPLC. After a brief reverse-phase column, the peptides were injected directly into the mass spectrometer. To correct for any loss of deuterium during the digestion and chromatography, a sample of enzyme was digested with pepsin, lyophilized, and dissolved in D2O to obtain the fully exchanged peptides. The amount of deuterium remaining in these peptides after the chromatography, typically 60–70%, was then used to calculate the extent of back-exchange.
The kinetics of deuterium incorporation from solvent into individual peptides could be fit with either equation 1 or 2. The exchange of most of the peptides could be described by equation 1, which describes an exchange with two kinetic components, a very rapid one that was complete before the first time point, 30 s, and a second slower phase. Equation 2 describes a reaction with three components, a very rapid phase and two slower phases. Figure 2A shows the extent of exchange of individual peptides after 8 min, at which time the exchange of most peptides has reached an endpoint. For simplicity, the peptides are divided into three classes: those showing less than 33% exchange, those showing 33–67% exchange, and those showing >67% exchange. Figure 2B shows similar results for the unphosphorylated enzyme. The phosphorylated enzyme exhibits greater exchange in several peptides; these are all on the surface of the protein rather than in the more stable core. These results suggest that phosphorylation results in increased flexibility throughout the protein.
Figure 2.

Comparison of the deuterium incorporation into phosphorylated (A) and unphosphorylated (B) PheH after 8 minutes in D2O: green, less than 33% incorporation; yellow, 33-66% incorporation; red, greater than 66% incorporation. The active site iron is indicated by an orange sphere. The regulatory domain is on the right and the catalytic domain on the left. The figure was drawn with the pdb file 2PHM.
Binding of phenylalanine to PheH results in significant increases in the incorporation of deuterium from solvent into 8 peptides [16]. Five of the eight peptides were also seen in the peptic digests of the phosphorylated enzyme, whereas peptides 220–227, 241–248 and 370–376 were not detected. Figure 3 compares the effects of phosphorylation and phenylalanine binding on the deuterium incorporation kinetics of these five peptides. Two peptides, 249–259 and 306–317, display exchange kinetics similar to those in unphosphorylated PheH in the absence of phenylalanine. In contrast, peptides 39–59, 82–91 and 164–174 show patterns that differ from both the resting enzyme and the enzyme with phenylalanine bound. For peptides 82–91 and 164–174, the extent of exchange at the end of 4 hours was the same for phosphorylated PheH and phenylalanine-bound PheH, but deuterium incorporation into the phosphorylated enzyme was slower. For peptide 39–59, the extent of incorporation into the phosphorylated enzyme was less than into the phenylalanine-bound enzyme.
Figure 3.
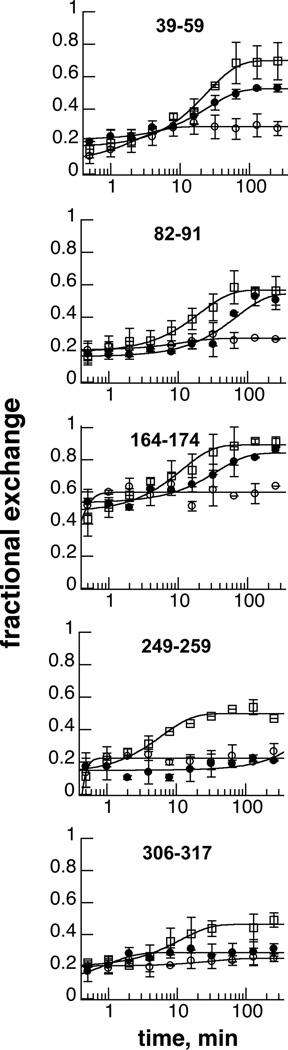
Effect of phosphorylation and phenylalanine binding on kinetics of incorporation of deuterium into selected PheH peptides; open circles, unphosphorylated enzyme; filled circles, phosphorylated enzyme; squares, unphosphorylated enzyme in the presence of 5 mM phenylalanine. The data are averages of duplicate analyses.
DISCUSSION
PheH was identified as a substrate for PKA several decades ago [18], and the phosphorylation site was identified soon after [28]. The physiological relevance of phosphorylation to the regulation of the activity of PheH in the liver has been established in studies of cells and whole animals [19, 29, 30]. Phosphorylation of the enzyme results in an increase in activity under certain conditions, although not as much as occurs upon activation by phenylalanine [19, 20]. To explain these results, Shiman et al. [19] proposed that phosphorylation of PheH does not activate the enzyme directly but instead allows it to be activated by lower concentrations of phenylalanine. This model was based on data for the rat enzyme, but later experiments with human PheH support a similar model [20].
The details of the structural changes that accompany activation of PheH by either phenylalanine or phosphorylation remain unclear. There is no high-resolution structure available of PheH with just phenylalanine bound, and structures with both an amino acid and tetrahydrobiopterin bound lack the regulatory domain [31]. Lower resolution methods have established that there is a significant conformational change when the enzyme is activated by phenylalanine [11, 14, 16, 32–34]. These are consistent with the proposal, based on the structure of the combined regulatory and catalytic domains of rat PheH, that this conformational change involves a movement of residues in the N-terminal regulatory domain away from the active site [8, 9]. The lack of any differences seen in the crystal structures between the phosphorylated and unphosphorylated enzyme raised the possibility that any conformational change upon phosphorylation is localized to the vicinity of Ser16, since the structures lack the first 18 residues [8].
The results of the deuterium exchange analyses described here suggest that phosphorylation of Ser16 of PheH affects more of the structure than the immediate vicinity of the phosphorylation site. Comparison of the extent of deuterium exchange into the phosphorylated and unphosphorylated protein (Figure 2) is consistent with phosphorylation increasing the overall dynamics of the protein. The more detailed comparison of the kinetics of deuterium incorporation into peptides affected by phenylalanine binding (Figure 3) establishes that the effect of phosphorylation on the structure is different from the effect of phenylalanine. Deuterium incorporation from solvent into the amide bonds of folded proteins typically exhibits EX2 kinetics, for which the rate constant(s) for exchange reflect an equilibrium constant between exchange-competent and non-exchanging local conformations [35]. Consequently, a change in the kinetics of exchange without a change in the extent of exchange suggests that a similar conformational change is occurring, but the exchange-competent form makes up a smaller subpopulation of the protein structural landscape. Such a pattern of exchange kinetics, as is seen for peptides 82–91 and 164–174, would be consistent with a model in which phosphorylation results in a structure that is more readily activated by phenylalanine but is not itself activated. The two peptides that do not show altered exchange kinetics upon phosphorylation, 249–259 and 306–317, lie at the interface between the catalytic and regulatory domains. The lack of change in their exchange establishes that a large reorientation of the two domains, as appears to occur upon phenylalanine binding[10, 16], does not occur when PheH is phosphorylated.
The present results suggest that phosphorylation of PheH does indeed affect much less of the structure than does binding of phenylalanine to the allosteric site. Even for the regions of the enzyme that are affected by phosphorylation, the extent of any conformational change is significantly less than occurs upon phenylalanine binding. Such changes would not necessarily be found in a crystal structure, especially one determined from crystals frozen at 100 K. They could still be sufficient to shift the equilibrium between the activated and inhibited conformations of the protein, thereby allowing activation at lower concentrations of phenylalanine as previously proposed [19].
Highlights.
Phenylalanine hydroxylase is regulated by binding of phenylalanine and by phosphorylation of Ser16.
The effect of phosphorylation on the incorporation of deuterium from solvent was determined.
The effect of phosphorylation was d ifferent from the effect of phenylalanine binding.
ACKNOWLEDGMENTS
This research was supported in part by NIH grant R01 GM098140. We thank Dr. Lawrence Dangott of the Protein Chemistry Lab at Texas A&M University for assistance with the mass spectroscopy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Shiman R, Blakley RL, Benkovic SJ, editors. Folates and Pterins. Vol. 2. New York: John Wiley & Sons; 1985. pp. 179–249. [Google Scholar]
- 2.Fitzpatrick PF. Ann. Rev. Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 3.Fitzpatrick PF. Arch. Biochem. Biophys. 2012;519:194–201. doi: 10.1016/j.abb.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shiman R, Gray DW. J. Biol. Chem. 1980;255:4793–4800. [PubMed] [Google Scholar]
- 5.Daubner SC, Hillas PJ, Fitzpatrick PF. Arch. Biochem. Biophys. 1997;348:295–302. doi: 10.1006/abbi.1997.0435. [DOI] [PubMed] [Google Scholar]
- 6.Shiman R. J. Biol. Chem. 1980;255:10029–10032. [PubMed] [Google Scholar]
- 7.Li J, Ilangovan U, Daubner SC, Hinck AP, Fitzpatrick PF. Arch. Biochem. Biophys. 2011;505:250–255. doi: 10.1016/j.abb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobe B, Jennings IG, House CM, Michell BJ, Goodwill KE, Santarsiero BD, Stevens RC, Cotton RGH, Kemp BE. Nat. Struct. Biol. 1999;6:442–448. doi: 10.1038/8247. [DOI] [PubMed] [Google Scholar]
- 9.Jennings IG, Teh T, Kobe B. FEBS Lett. 2001;488:196–200. doi: 10.1016/s0014-5793(00)02426-1. [DOI] [PubMed] [Google Scholar]
- 10.Jaffe EK, Stith L, Lawrence SH, Andrake M, Dunbrack RL., Jr Arch. Biochem. Biophys. 2013;530:73–82. doi: 10.1016/j.abb.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiman R, Gray DW, Pater A. J. Biol. Chem. 1979;254:11300–11306. [PubMed] [Google Scholar]
- 12.Koizumi S, Tanaka F, Kaneda N, Kano K, Nagatsu T. Biochemistry. 1988;27:640–646. doi: 10.1021/bi00402a022. [DOI] [PubMed] [Google Scholar]
- 13.Abita J-P, Parniak M, Kaufman S. J. Biol. Chem. 1984;259:14560–14566. [PubMed] [Google Scholar]
- 14.Phillips RS, Parniak MA, Kaufman S. Biochemistry. 1984;23:3836–3842. doi: 10.1021/bi00312a007. [DOI] [PubMed] [Google Scholar]
- 15.Knappskog PM, Haavik J. Biochemistry. 1995;34:11790–11799. doi: 10.1021/bi00037a017. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Dangott LJ, Fitzpatrick PF. Biochemistry. 2010;49:3327–3335. doi: 10.1021/bi1001294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donlon J, Kaufman S. J. Biol. Chem. 1978;253:6657–6659. [PubMed] [Google Scholar]
- 18.Abita JP, Milstien S, Chang N, Kaufman S. J. Biol. Chem. 1976;251:5310–5314. [PubMed] [Google Scholar]
- 19.Shiman R, Mortimore GE, Schworer CM, Gray DW. J. Biol. Chem. 1982;257:11213–11216. [PubMed] [Google Scholar]
- 20.Doskeland AP, Martinez A, Knappskog PM, Flatmark T. Biochem. J. 1996;313:409–414. doi: 10.1042/bj3130409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kowlessur D, Yang X-J, Kaufman S. Proc. Natl. Acad. Sci. USA. 1995;92:4743–4747. doi: 10.1073/pnas.92.11.4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miranda FF, Teigen K, Thorolfsson M, Svebak RM, Knappskog PM, Flatmark T, Martinez A. J. Biol. Chem. 2002;277:40937–40943. doi: 10.1074/jbc.M112197200. [DOI] [PubMed] [Google Scholar]
- 23.Miranda FF, Thórólfsson M, Teigen K, Sanchez-Ruiz JM, Martínez A. Protein Sci. 2004;13:1219–1226. doi: 10.1110/ps.03595904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daubner SC, Hillas PJ, Fitzpatrick PF. Biochemistry. 1997;36:11574–11582. doi: 10.1021/bi9711137. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Fitzpatrick PF. Arch. Biochem. Biophys. 2008;475:164–168. doi: 10.1016/j.abb.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flockhart DA, Corbin JD. In: Brain Receptor Methodologies, Part A. Maranos PJ, Campbell IC, Cohen RM, editors. New York: Academic Press; 1984. pp. 209–215. [Google Scholar]
- 27.Weis DD, Engen JR, Kass IJ. J. Am. Soc. Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 28.Wretborn M, Humble E, Ragnarsson U, Engstrom L. Biochem. Biophys. Res. Commun. 1980;93:403–408. doi: 10.1016/0006-291x(80)91091-8. [DOI] [PubMed] [Google Scholar]
- 29.Fisher MJ, Pogson CI. Biochem. J. 1984;219:79–85. doi: 10.1042/bj2190079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisher MJ, Santana MA, Pogson CI. Biochem. J. 1984;219:87–90. doi: 10.1042/bj2190087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersen OA, Stokka AJ, Flatmark T, Hough E. J. Mol. Biol. 2003;333:747–757. doi: 10.1016/j.jmb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Phillips RS, Iwaki M, Kaufman S. Biochem. Biophys. Res. Commun. 1983;110:919–925. doi: 10.1016/0006-291x(83)91050-1. [DOI] [PubMed] [Google Scholar]
- 33.Davis MD, Parniak MA, Kaufman S, Kempner E. Arch. Biochem. Biophys. 1996;325:235–241. doi: 10.1006/abbi.1996.0029. [DOI] [PubMed] [Google Scholar]
- 34.Stokka AJ, Flatmark T. Biochem. J. 2003;369:509–518. doi: 10.1042/BJ20021009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Busenlehner LS, Armstrong RN. Arch. Biochem. Biophys. 2005;433:34–46. doi: 10.1016/j.abb.2004.09.002. [DOI] [PubMed] [Google Scholar]