

Abstract
Gemcitabine is a deoxycytidine analog that is widely used in the chemotherapy of many solid tumors. However, acquired tumor cell resistance often limits its use. Previously, we discovered that 4-(N)-stearoyl gemcitabine solid lipid nanoparticles (4-(N)-GemC18-SLNs) can overcome multiple acquired gemcitabine resistance mechanisms, including RRM1 overexpression. The present study was designed to elucidate the mechanisms underlying the 4-(N)-GemC18-SLNs' ability to overcome gemcitabine resistance. The 4-(N)-GemC18 in the 4-(N)-GemC18-SLNs entered tumor cells due to clathrin-mediated endocytosis of the 4-(N)-GemC18-SLNs into the lysosomes of the cells, whereas the 4-(N)-GemC18 alone in solution entered cells by diffusion. We substantiated that it is the way the 4-(N)-GemC18-SLNs deliver the 4-(N)-GemC18 into tumor cells that allows the gemcitabine hydrolyzed from the 4-(N)-GemC18 to be more efficiently converted into its active metabolite, gemcitabine triphosphate (dFdCTP), and thus more potent against gemcitabine-resistant tumor cells than 4-(N)-GemC18 or gemcitabine alone. Moreover, we also showed that the RRM1-overexpressing tumor cells were also cross-resistant to cytarabine, another nucleoside analog commonly used in cancer therapy, and 4-(N)-stearoyl cytarabine carried by solid lipid nanoparticles can also overcome the resistance. Therefore, formulating the long-chain fatty acid amide derivatives of nucleoside analogs into solid lipid nanoparticles may represent a platform technology to increase the antitumor activity of the nucleoside analogs and to overcome tumor cell resistance to them.
Keywords: ribonucleotide reductase, endocytosis, lysosome, hydrolysis, cell uptake, cytarabine, western blotting
1. INTRODUCTION
Gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) is a deoxycytidine analog with antitumor activity against a wide variety of solid tumors. Unfortunately, tumor cells often develop resistance to gemcitabine, which limits its clinical efficacy [1]. The mechanism of gemcitabine resistance is closely related to its mechanism of action and intracellular metabolism [2]. Gemcitabine is transported into cells by nucleoside transporters such as the human equilibrative nucleoside transporter 1 (hENT1) [3]. Upon entering cells, gemcitabine is phosphorylated by deoxycytidine kinase (dCK) to gemcitabine monophosphate (dFdCMP) and subsequently by nucleotide kinase to its active metabolites, gemcitabine diphosphate (dFdCDP) and gemcitabine triphosphate (dFdCTP) [4]. The dFdCTP is incorporated into DNA to inhibit DNA synthesis [5], and dFdCDP inhibits ribonucleotide reductase (RR), an enzyme that is required for the synthesis of deoxynucleotides (dNTPs) [6]. Because dFdCTP competes with deoxycytidine triphosphate (dCTP) for incorporation into DNA, a decrease in cellular concentration of dNTPs (i.e., dNTP pool) further increases the incorporation of dFdCTP into DNA [4, 5]. Therefore, tumor cells that have decreased expression of nucleoside transporters and/or dCK or increased expression of the large and/or small subunits of the RR (RRM1 and/or RRM2) are resistant to gemcitabine [7, 8].
Recently, there is increasing evidence that certain gemcitabine nanoparticles can overcome tumor cells resistance to gemcitabine [9–14]. Previously we discovered that the 4-(N)-stearoyl gemcitabine solid lipid nanoparticles (4-(N)-GemC18-SLNs) developed in our laboratory are more cytotoxic than gemcitabine in gemcitabine resistant tumor cells that are deficient in hENT1 or dCK [11]. More importantly, the 4-(N)-GemC18-SLNs can overcome gemcitabine resistance caused by the overexpression of RRM1, both in vitro and in vivo [11]. This finding is clinically relevant considering that data from numerous clinical studies have documented the inverse correlation between RRM1 expression in tumor cells and their sensitivity to gemcitabine. Clinical studies in non-small cell lung and pancreatic cancer patients who received gemcitabine-based therapy revealed that patients with low levels of RRM1 expression showed a better response and a longer survival than those with high RRM1 levels [15–18]. Moreover, the treatment benefit from gemcitabine in pancreatic cancer patients with disease recurrence was observed only in patients with low RRM1 [19]. A strong expression of RRM1 was also detected in biliary tract cancer patients and intrahepatic cholangiocarcinoma patients who were resistant to gemcitabine [20, 21]. Finally, recent data in patients with advanced nasopharyngeal carcinoma also revealed that the progression-free survival of patients with RRM1-positive expression is shorter than patients with RRM1-negative expression [22].
In the present study, we identified the mechanisms by which our 4-(N)-GemC18-SLNs overcome gemcitabine resistance caused by RRM1 overexpression.
2. MATERIALS AND METHODS
2.1. Cell line
Mouse lung cancer cell line (TC-1, ATCC # CRL-2785) were cultured in complete RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml of penicillin and 100 μg/ml of streptomycin, all from Invitrogen. The previously established TC-1-GR cells were cultured in similar RPMI 1640 medium further supplemented with 1 μM of gemcitabine HCl [11].
2.2. Syntheses of 4-(N)-stearoyl gemcitabine (4-(N)-GemC18), 3′-(O)-stearoyl gemcitabine (3′-(O)-GemC18), 4-(N)-octyl gemcitabine (4-(N)-GemC8), and 4-(N)-stearoyl cytarabine (4-(N)-Ara-C-C18) and the incorporation of them into nanoparticles
The 4-(N)-GemC18 was synthesized as previously described by us [23]; 3′-(O)-GemC18, 4-(N)-GemC8, and 4-(N)-Ara-C-C18 were synthesized according to a literature protocol with slight modifications [24, 25] (see Supplement for detailed methods and Fig. S1 for structures). The compounds were incorporated into solid lipid nanoparticles (SLNs) as previously described to prepare 4-(N)-GemC18-SLNs, 3′-(O)-GemC18-SLNs, 4-(N)-GemC8-SLNs, and 4-(N)-Ara-C-C18-SLNs, respectively [23]. The 4-(N)-GemC18-incorporated poly (lactic-co-glycolic) acid nanoparticles (4-(N)-GemC18-PLGA-NPs) were prepared using a solvent displacement method as previously described [26]. The concentration of 4-(N)-GemC18 in the nanoparticles was determined using a HPLC method as previously described [27]. Table S1 includes diameters, zeta potentials, and polydispersity indices of the nanoparticles.
2.3. Hydrolysis of gemcitabine from 4-(N)-GemC18-SLNs and 3′-(O)-GemC18-SLNs in cell culture medium
The 4-(N)-GemC18-SLNs or 3′-(O)-GemC18-SLNs (10 μM of GemC18) were incubated in 1 ml of RPMI 1640 medium supplemented with 10% FBS at 37°C, 5% CO2. At predetermined time points, medium was collected and analyzed for gemcitabine concentration using an HPLC method as previously described [28].
2.4. Western blot analysis
Immunoblotting for RRM1 protein was performed as previously described [11]. β-Actin (mouse monoclonal antibody) was used as a control.
2.5. Quantification of intracellular dNTPs, NTPs, and dFdCTP
Intracellular dNTP, NTP, and dFdCTP were extracted and analyzed (using HPLC) as previously described with slight modifications [29–31] (see Supplement methods). An Agilent 1260 Infinity Quaternary Liquid Chromatographic System equipped with an Aglient ZORBAX Eclipse Plus C18 column (3.5 μm, 4.6 mm × 150 mm) was used for the analysis for all compounds. For dNTPs and NTPs analyses, the mobile phase consisted of two solutions: 10 mM KH2PO4/10 mM tetrabutylammonium chloride (TBACl) (pH 7.0) with 0.25% methanol (A) and 50 mM KH2PO4/5.6 mM TBACl (pH 7.0) : methanol (70:30, v/v) (B). Run was started at 60% A followed by a linear gradient to 40% A over 30 min and held at 40% A for 40 min. The flow rate was 1.0 ml/min. The injection volume was 40 μl. The detection wavelength was 254 nm. Figs. S2B–C show the HPLC histograms of dNTPs and NTPs in TC-1 and TC-1-GR cells, respectively. For dFdCTP analysis, two solutions were used: 10 mM KH2PO4/10 mM TBACl (pH 7.0) with 0.25% methanol (A) and 250 mM KH2PO4/10 mM TBACl (pH 7.0) : methanol (85:15, v/v) (B). They were mixed at 50:50 (v/v). The flow rate was 1.2 ml/min. The injection volume was 20 μl. The detection wavelength was 271 nm.
2.6. In vitro cytotoxicity assay
TC-1 and TC-1-GR cells were seeded into 96-well plates (3 × 103 cells/well). After overnight incubation at 37°C, 5% CO2, cells were treated with various concentrations of gemcitabine HCl, cytarabine (Ara-C), gemcitabine derivatives in nanoparticles, or the Ara-C derivative in nanoparticles for 48 h. Cell viability was determined using an MTT assay [11].
2.7. Inhibition of RRM1 expression by siRNA silencing
TC-1-GR cells were transfected with RRM1 siRNA or control siRNA complexed with Lipofectamine™ RNAiMAX (Invitrogen) following the manufacturer's instruction [11]. The siRNA-transfected cells were re-seeded (3 × 104 cells/well) into 96-well plates 48 h after transfection and incubated overnight at 37°C, 5% CO2. Cells were then treated with Ara-C for 48 additional hours, and the cytotoxicity was evaluated using an MTT assay.
2.8. In vitro cellular uptake assay
Cellular uptake was performed as previously described [27]. To inhibit endocytosis, cell uptake was carried out as described above but at 4°C [23]. To inhibit specific endocytosis mechanisms, cells were pretreated with chlorpromazine (5 μg/ml), filipin (2.5 μg/ml), wortmannin (3 μg/ml), or cytochalasin B (20 ng/ml) in RPMI 1640 medium for 30 min at 37°C before performing the uptake study. Chlorpromazine, filipin, wortmannin, and cytochalasin B are inhibitors of clathrin-mediated endocytosis, caveolae-mediated endocytosis, macropinocytosis, and phagocytosis, respectively [32–34]. The concentrations of the inhibitors were the highest concentrations that did not affect the viability of TC-1-GR cells in 2.5 h (Fig. S3).
2.9. Fluorescence microscopy
TC-1-GR (1.5 × 105 cells/well) were seeded in a 35 mm poly-D-lysine-coated glass-bottom dish (Mattek Corporation, Ashland, MA) and incubated overnight at 37°C, 5% CO2. Cells were incubated with 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)-fluorescein-labeled SLNs (100 μg/ml of DOPE-fluorescein) for 2 h [23]. The nanoparticle-containing medium was then replaced with fresh medium and incubated for 0, 2, or 6 additional hours. Intracellular localization of fluorescein-labeled SLNs was monitored as previously described [27].
2.10. Quantitation of GemC18 in lysosomes
The lysosomal fraction was prepared using a cell fractionation method described previously with slight modifications [35, 36] (see Supplement for more details). The activity of cathepsin B in the fraction was confirmed to be significantly higher than in the cytoplasmic fraction. The concentration of GemC18 in the fraction was determined using HPLC [27]. An Agilent 1260 Infinity Quaternary Liquid Chromatographic System with an Aglient ZORBAX Eclipse Plus C18 column (5 μm, 4.6 mm × 150 mm) was used for HPLC analysis. The mobile phase was methanol. The flow rate was 1 ml/min, and the detection wavelength was 248 nm.
2.11. Determination of the intracellular stability of 4-(N)-GemC18
Intracellular degradation of the 4-(N)-GemC18 was determined as previously described [27, 31]. To inhibit lysosomal acidification, cells were pre-incubated with 10 mM NH4Cl for 10 min before 6 additional hours of incubation with 4-(N)-GemC18 in solution or in 4-(N)-GemC18-SLNs, still in the presence of NH4Cl (10 mM). To evaluate the intracellular degradation of the 4-(N)-GemC18, the 4-(N)-GemC18-containing medium was replaced with fresh medium containing either 0 mM or 10 mM of NH4Cl. After 16 additional hours of incubation, the level of 4-(N)-GemC18 that remained in the cells was determined using HPLC [27].
2.12. In vitro release of gemcitabine derivatives from nanoparticles
The release of 4-(N)-GemC8 or 4-(N)-GemC18 from nanoparticles was determined as previously described [23]. The concentrations of 4-(N)-GemC18 and 4-(N)-GemC8 in the release medium were determined using HPLC [27].
2.13. In vivo tumor growth inhibition assay
All animal procedures were performed following National Institutes of Health guidelines for humane treatment of animals. Animal protocol was approved by the Institutional Animal care and Use Committee at the University of Texas at Austin. Female Nu/Nu mice (18–20 g) were from Charles River Laboratories (Wilmington, MA). TC-1 or TC-1-GR tumors were established in the right flank of mice by subcutaneous (s.c.) injection of 5 × 105 cells. When tumor diameters reached 3–4 mm, mice were randomized and injected via the tail vein with 4-(N)-GemC18-SLNs, 3′-(O)-GemC18-SLNs, or sterile mannitol (5% w/v) as a vehicle control on days 6 and 13 (after tumor implantation). The dose of 4-(N)-GemC18 or 3′-(O)-GemC18 was 1 mg per mouse per injection. Tumor size was measured using a caliper in two perpendicular diameters every other day, and tumor volume was calculated based on the following equation: tumor volume (mm3) = 1/2 [length × (width)2].
2.14. Determination of the concentration of 4-(N)-GemC18 or 3′-(O)-GemC18 in tumor tissues and plasma samples
When TC-1-GR tumors in Nu/Nu mice grew to ~1 cm3, mice were injected via the tail vein with 4-(N)-GemC18-SLNs or 3′-(O)-GemC18-SLNs. The dose of 4-(N)-GemC18 or 3′-(O)-GemC18 was 1 mg per mouse. Mice were euthanized 6 h later. Blood was collected into heparinized tubes to isolate plasma. Tumor tissues were harvested, weighed, and stored at −80°C until further analysis. The concentrations of 4-(N)-GemC18 and 3′-(O)-GemC18 in tumor tissues and in plasma were determined after extraction [27].
2.15. Statistical analysis
Statistical analyses were carried out using a one-way ANOVA followed by a Bonferroni/Dunn post-hoc comparison test. Difference is considered significant if P ≤ 0.05.
3. RESULTS
3.1. The 4-(N)-GemC18-SLNs down-regulate RRM1 expression and increase the concentration of dFdCTP in the RRM1-overexpressing TC-1-GR cells
We previously discovered that our 4-(N)-GemC18-SLNs overcome gemcitabine resistance caused by RRM1 overexpression [11]. As shown in Fig. 1A, 4-(N)-GemC18 alone or physically mixed with blank SLNs were not as cytotoxic as 4-(N)-GemC18-SLNs against TC-1-GR cells, indicating that the incorporation of the 4-(N)-GemC18 into SLNs is critical for it to overcome the resistance. Given that one of mechanisms underlying the antitumor activity of gemcitabine is its ability to inhibit RR and deplete cellular dNTP pools [37, 38], we evaluated the effect of the 4-(N)-GemC18-SLNs on RRM1 expression in TC-1-GR cells. Treatment of TC-1-GR cells with 4-(N)-GemC18-SLNs significantly decreased the level of RRM1 protein (relative to treatment with PBS) (Fig. 1B), indicating that the 4-(N)-GemC18-SLNs' ability to down-regulate RRM1 overexpression is related to their ability to overcome gemcitabine resistance caused by RRM1 overexpression. The 4-(N)-GemC18-SLNs' ability to down-regulate RRM1 expression is not likely related to the C18 (stearic acid) itself because neither C18 alone nor C18 incorporated into SLNs (i.e., C18-NPs) significantly affected RRM1 expression in TC-1-GR cells (Fig. S4).
Fig. 1. 4-(N)-GemC18-SLNs down-regulate RRM1 expression and increase dFdCTP level in the RRM1-overexpressing TC-1-GR cells.

(A) The IC50 values of gemcitabine HCl, 4-(N)-GemC18 in DMSO, 4-(N)-GemC18 mixed with blank SLNs, and the 4-(N)-GemC18-SLNs in TC-1 and TC-1-GR cells (n ≥ 3). aP < 0.05. (B) Representative western blot of RRM1 protein levels in TC-1-GR cells after treatment with 4 μM of gemcitabine HCl or the molar equivalent concentration of 4-(N)-GemC18 in DMSO (GemC18), or 4-(N)-GemC18-SLNs for 48 h (n ≥ 3). (C) The intrinsic levels of dCTP and dATP in TC-1 and TC-1-GR cells (n = 3). bP < 0.001, cP < 0.01, TC-1 vs. TC-1-GR. (D) dCTP level in TC-1-GR cells 24 h after treatment with 10 μM of gemcitabine HCl, 4-(N)-GemC18 alone, or 4-(N)-GemC18-SLNs (n = 3). dP < 0.05 vs. PBS. (E) dFdCTP levels in TC-1 and TC-1-GR cells after 24 h of incubation with gemcitabine HCl (10 μM) (n = 3). eP < 0.01. (F) dFdCTP level in TC-1-GR cells after 24 h of incubation with 10 μM of gemcitabine HCl, 4-(N)-GemC18 alone, or 4-(N)-GemC18-SLNs (n = 3). gP < 0.01; fP < 0.001 vs. gemcitabine.
Overexpression of RRM1 can lead to the expansion of intracellular dNTP pools [39, 40]. We therefore compared the intrinsic dNTP levels in TC-1 and TC-1-GR cells. As shown in Fig. 1C, the intracellular concentrations of dCTP and deoxyadenosine triphosphate (dATP) in TC-1-GR cells were significantly higher than in TC-1 cells. Treatment of TC-1-GR cells with the 4-(N)-GemC18-SLNs significantly decreased the intracellular dCTP level (Fig. 1D), without affecting dATP level (data not shown). We then determined the intracellular levels of dFdCTP in TC-1 and TC-1-GR cells after they were treated with gemcitabine HCl (10 μM) for 24 h. The intracellular dFdCTP level in TC-1-GR cells was only 18% of that in TC-1 cells (Fig. 1E), which explains why the TC-1-GR cells are resistant to gemcitabine. Importantly, treatment of TC-1-GR cells with our 4-(N)-GemC18-SLNs significantly increased the intracellular level of dFdCTP, as compared to treatment with gemcitabine HCl or 4-(N)-GemC18 alone (Fig. 1F), which explains why our 4-(N)-GemC18-SLNs were more cytotoxic to the RRM1-overexpressing TC-1-GR cells than gemcitabine or 4-(N)-GemC18 alone (Fig.1A).
3.2. The way the 4-(N)-GemC18 enters cells determines its activity against TC-1-GR cells
The different activities of 4-(N)-GemC18 in solution and in 4-(N)-GemC18-SLNs prompted us to test whether the way the 4-(N)-GemC18 enters cells is important for it to overcome gemcitabine resistance caused by RRM1 overexpression. As shown in Fig. 2A, when TC-1-GR cells were incubated at 4°C, the uptake of 4-(N)-GemC18 in solution was significantly decreased, compared to when incubated at 37°C. In contrast, the uptake of 4-(N)-GemC18 in the 4-(N)-GemC18-SLNs by TC-1-GR cells was almost completely inhibited at 4°C (Fig. 2A), suggesting that 4-(N)-GemC18-SLNs entered TC-1-GR cells by endocytosis, whereas 4-(N)-GemC18 in solution entered cells by passive diffusion. To further confirm this, we determined the uptake of 4-(N)-GemC18 in solution or in 4-(N)-GemC18-SLNs by TC-1-GR cells in the presence of specific endocytosis inhibitors. As shown in Fig. 2B, the uptake of the 4-(N)-GemC18-SLNs was significantly inhibited by chlorpromazine, but not by filipin, wortmannin, or cytochalasin B, indicating that the 4-(N)-GemC18-SLNs entered cells primarily by clathrin-mediated endocytosis. As expected, the uptake of 4-(N)-GemC18 in solution was not affected by any of the endocytosis inhibitors (Fig. 2B). Therefore, the fact that the 4-(N)-GemC18-SLNs deliver the 4-(N)-GemC18 into TC-1-GR cells by endocytosis is likely related to their ability to overcome gemcitabine resistance caused by RRM1 overexpression.
Fig. 2. In vitro uptake of 4-(N)-GemC18 in solution or in 4-(N)-GemC18-SLNs by TC-1-GR cells, and the effect of the neutralization of lysosomes with NH4Cl on the intracellular degradation/hydrolysis of 4-(N)-GemC18 and the cytotoxicity of 4-(N)-GemC18 in solution or in 4-(N)-GemC18-SLNs in TC-1-GR cells.

(A) The level of 4-(N)-GemC18 in TC-1-GR cells after the cells were incubated with 40 μM of 4-(N)-GemC18 in solution or 4-(N)-GemC18-SLNs at either 4°C or 37°C for up to 6 h (n = 3). (B) The level of 4-(N)-GemC18 in TC-1-GR cells after the cells were incubated with 40 μM of 4-(N)-GemC18 in solution or 4-(N)-GemC18-SLNs for 2 h in the presence of various endocytosis inhibitors. Data shown are normalized to the levels of 4-(N)-GemC18 in TC-1-GR cells after 2 h of incubation in the absence of any inhibitors. aP < 0.01, 4-(N)-GemC18 vs. 4-(N)-GemC18-SLNs when chlorpromazine was used. (C) Co-localization of fluorescein-labeled SLNs (green) and lysosomes (red) at 0, 2 and 6 h after the removal of the nanoparticle-containing medium. Cell nuclei are in blue. (D) The level of 4-(N)-GemC18 detected in the lysosomal fraction of TC-1-GR cells after treatment with 40 μM of 4-(N)-GemC18 in solution or 4-(N)-GemC18-SLNs for 3 h (n = 3). Values reported are the % of 4-(N)-GemC18 detected in the lysosomal fraction divided by the total 4-(N)-GemC18 internalized by the cells within 3 h. bP < 0.05. (E) The percentage of 4-(N)-GemC18 remaining in TC-1-GR cells after it was delivered into the cells as 4-(N)-GemC18 alone or as 4-(N)-GemC18-SLNs and then incubated in the presence or absence of NH4Cl for 16 h. cP < 0.001, 0 mM vs. 10 mM NH4Cl for 4-(N)-GemC18-SLNs. (F) The IC50 values of 4-(N)-GemC18 in solution and 4-(N)-GemC18-SLNs in TC-1-GR cells after 24 h of treatment in the presence or absence of 10 mM NH4Cl (n = 5). dP < 0.01, 0 mM vs. 10 mM NH4Cl for 4-(N)-GemC18-SLNs.
After endocytosis, 4-(N)-GemC18-SLNs are expected to enter the endo-lysosomes. We investigated the intracellular location of 4-(N)-GemC18-SLNs using fluorescein-labeled nanoparticles. After 2 h of incubation, an almost complete overlap between the nanoparticles and the LysoTracker Red, a lysosomal marker, was observed (Fig. 2C). The fluorescein-labeled-SLNs remained in the lysosomes 2 h after internalization. However, the fluorescence intensity of nanoparticles was significantly decreased 6 h later, suggesting that the 4-(N)-GemC18-SLNs that were taken up by endocytosis were targeted to the lysosomes for degradation/destruction. To further confirm that the 4-(N)-GemC18-SLNs are targeted to lysosomes after endocytosis, we determined the concentration of 4-(N)-GemC18 in lysosomes after TC-1-GR cells were incubated with 4-(N)-GemC18 in solution or with 4-(N)-GemC18-SLNs for 3 h. Data in Fig. 2D showed that a significantly higher percentage of the 4-(N)-GemC18 delivered into cells by the 4-(N)-GemC18-SLNs was recovered in the lysosomes, compared to when the 4-(N)-GemC18 entered cells by passive diffusion.
We then investigated whether the alkalization of the lysosomal pH with NH4Cl alters the uptake and intracellular degradation of 4-(N)-GemC18. As shown in Fig. 2E, alkalization of lysosomal pH did not affect the uptake and intracellular degradation of 4-(N)-GemC18 when it was taken up as 4-(N)-GemC18 alone in solution. However, when cells were incubated with the 4-(N)-GemC18-SLNs, alkalization of lysosomal pH significantly inhibited the intracellular degradation of 4-(N)-GemC18 (Fig. 2E), strongly indicating that the acidic lysosomal environment is important for the hydrolysis of 4-(N)-GemC18 when the 4-(N)-GemC18 was internalized by the endocytosis of the 4-(N)-GemC18-SLNs.
To further confirm that the acidic condition in the lysosomes and the hydrolysis of 4-(N)-GemC18 in lysosomes are important for the 4-(N)-GemC18-SLNs to inhibit TC-1-GR cell growth, we determined the cytotoxicity of the 4-(N)-GemC18-SLNs and 4-(N)-GemC18 in solution when lysosomal pH was alkalized. As shown in Fig. 2F, alkalization of lysosomes significantly decreased the cytotoxicity of the 4-(N)-GemC18-SLNs, but not the 4-(N)-GemC18 in solution, in TC-1-GR cells. Taken together, data in Fig. 2 show that the delivery of the 4-(N)-GemC18 into the endolysosomes of the tumor cells by the endocytosis of the 4-(N)-GemC18-SLNs and the subsequent degradation/hydrolysis of the 4-(N)-GemC18 to release the gemcitabine in the lysosomes are critical for the 4-(N)-GemC18-SLNs to overcome gemcitabine resistance caused by RRM1 overexpression.
3.3. The stearic acid amidification of gemcitabine is critical for the 4-(N)-GemC18-SLNs to overcome gemcitabine resistance caused by RRM1 overexpression
To investigate whether the amide bond between gemcitabine and the stearoyl group is critical for the 4-(N)-GemC18-SLNs to overcome gemcitabine resistance caused by RRM1 overexpression, we synthesized another stearoyl gemcitabine derivative, 3′-(O)-GemC18, by conjugating the stearoyl group with gemcitabine on the 3′-OH position of the gemcitabine to form an ester (Fig. S4). The ester bond on the 3′-(O)-GemC18 is expected to be more susceptible to hydrolysis at physiological pH, compared to the amide bond on the 4-(N)-GemC18 [41]. The 3′-(O)-GemC18 was then incorporated into SLNs [23]. The 3′-(O)-GemC18-SLNs and the 4-(N)-GemC18-SLNs were similar in size and zeta potential (Supplement Table S1). We first compared the cytotoxic activities of the 3′-(O)-GemC18-SLNs and 4-(N)-GemC18-SLNs in TC-1 and TC-1-GR cells. In TC-1 cells, gemcitabine HCl displayed the strongest cytotoxicity, followed by 3′-O-GemC18-SLNs, and then by 4-(N)-GemC18-SLNs (Fig. 3A). In contrast, in TC-1-GR cells, 4-(N)-GemC18-SLNs were the most cytotoxic, followed by 3′-(O)-GemC18-SLNs, and then by gemcitabine HCl (Fig. 3B).
Fig. 3. In vitro and in vivo antitumor activity of 4-(N)-GemC18-SLNs and 3′-(O)-GemC18-SLNs against TC-1 or TC-1-GR tumors.

(A) The viability of TC-1 cells after incubation with various concentrations of gemcitabine HCl, 3′-(O)-GemC18-SLNs, or 4-(N)-GemC18-SLNs for 48 h (n = 4). aP < 0.001, 3′-(O)-GemC18-SLNs or 4-(N)-GemC18-SLNs vs. gemcitabine HCl; bP < 0.05, cP < 0.001, 4-(N)-GemC18-SLNs vs. 3′-(O)-GemC18-SLNs. (B) Viability of TC-1-GR cells after incubation with various concentrations of gemcitabine HCl, 3′-(O)-GemC18-SLNs, or 4-(N)-GemC18-SLNs for 48 h (n = 4). dP < 0.001, 4-(N)-GemC18-SLNs vs. gemcitabine HCl; eP < 0.05, 4-(N)-GemC18-SLNs vs. 3′-(O)-GemC18-SLNs. (C) The hydrolysis of gemcitabine from 3′-(O)-GemC18-SLNs or 4-(N)-GemC18-SLNs when incubated in RPMI 1640 medium at 37°C (n = 4). fP < 0.001, 3′-(O)-GemC18-SLNs vs. 4-(N)-GemC18-SLNs. (D) The levels of 3′-(O)-GemC18 or 4-(N)-GemC18 detected in TC-1-GR cells after incubation with 40 μM of 3′-(O)-GemC18-SLNs or 4-(N)-GemC18-SLNs at 37°C for up to 6 h (n = 3). (E) In vivo antitumor activity of 3′-(O)-GemC18-SLNs and 4-(N)-GemC18-SLNs against TC-1 tumors in Nu/Nu mice (n = 4–5). gP < 0.05 hP < 0.01 iP < 0.001 vs. mannitol control. (F) In vivo antitumor activity of 3′-(O)-GemC18-SLNs and 4-(N)-GemC18-SLNs against TC-1-GR tumors in Nu/Nu mice (n = 4–5). jP < 0.05, kP < 0.01, 4-(N)-GemC18-SLNs vs. mannitol; lP < 0.05, 4-(N)-GemC18-SLNs vs. 3′-(O)-GemC18-SLNs. (G) Levels of 3′-(O)-GemC18 or 4-(N)-GemC18 in TC-1-GR tumor tissues 6 h after i.v. injection of either 3′-(O)-GemC18-SLNs or 4-(N)-GemC18-SLNs (n = 3–4). mP < 0.01. (H) Plasma concentrations of 3′-(O)-GemC18 or 4-(N)-GemC18 in TC-1-GR tumor-bearing mice 6 h after i.v. injection of either 3′-(O)-GemC18-SLNs or 4-(N)-GemC18-SLNs (n = 3–4). nP < 0.05. (I) Representative Western blot of RRM1 protein in TC-1-GR tumor tissues 19 days after tumor cell injection. Mice were i.v. injected with 4-(N)-GemC18-SLNs or 3′-(O)-GemC18-SLNs 6 and 13 days after tumor cell injection. The dose of the GemC18 was 1 mg per injection per mouse (n = 3).
To explain why the 3′-(O)-GemC18-SLNs were more cytotoxicity in TC-1 cells, but the 4-(N)-GemC18-SLNs were more cytotoxic in the gemcitabine-resistant TC-1-GR cells, we determined the rates of the hydrolysis of gemcitabine from 3′-(O)-GemC18-SLNs and 4-(N)-GemC18-SLNs by measuring the concentration of gemcitabine in the medium when the nanoparticles were incubated in complete RPMI 1640 medium supplemented with 10% FBS at 37°C (without cells). The rate at which gemcitabine was hydrolyzed from the 3′-(O)-GemC18-SLNs was significantly higher than from the 4-(N)-GemC18-SLNs (Fig. 3C), clearly demonstrating that the ester bond between the gemcitabine and the stearic acid in 3′-(O)-GemC18-SLNs is significantly more sensitive to hydrolysis than the amide bond in the 4-(N)-GemC18-SLNs. The results from the uptake of 3′-(O)-GemC18 and 4-(N)-GemC18 by TC-1-GR cells also demonstrated that at the early time points (i.e., in the first hour), the level of 3′-(O)-GemC18 detected in TC-1-GR cells incubated with the 3′-(O)-GemC18-SLNs was similar to that in cells incubated with the 4-(N)-GemC18-SLNs (Fig. 3D). Thereafter, the level of 3′-(O)-GemC18 detected in cells incubated with the 3′-(O)-GemC18-SLNs remained unchanged, whereas the level of 4-(N)-GemC18 detected in TC-1-GR cells incubated with the 4-(N)-GemC18-SLNs continued to increase, likely because the 3′-(O)-GemC18 in 3′-(O)-GemC18-SLNs was more sensitive to hydrolysis than the 4-(N)-GemC18 in 4-(N)-GemC18-SLNs in the cell culture medium (and after the nanoparticles were endocytosed into the TC-1-GR cells).
We then compared the antitumor activity of the 3′-(O)-GemC18-SLNs and 4-(N)-GemC18-SLNs in mice with pre-established TC-1 or TC-1-GR tumors. As shown in Fig. 3E, TC-1 tumors grew aggressively in mice; both 3′-(O)-GemC18-SLNs and 4-(N)-GemC18-SLNs significantly delayed the tumor growth, although the 4-(N)-GemC18-SLNs were ultimately more effective than 3′-(O)-GemC18-SLNs (Fig. 3E). Conversely, for TC-1-GR tumors, only the 4-(N)-GemC18-SLNs significantly inhibited the tumor growth (Fig. 3F). Six hours after the nanoparticles were i.v. injected into TC-1-GR tumor-bearing mice, the concentration of the 4-(N)-GemC18 accumulated in tumor tissues was significantly higher than the concentration of the 3′-(O)-GemC18 (Fig. 3G). In addition, the concentration of 4-(N)-GemC18 that remained in mouse plasma 6 h after injection was about 6-fold higher than 3′-(O)-GemC18 (Fig. 3H). More importantly, Western blot analysis also revealed that the RRM1 protein level in TC-1-GR tumors in mice treated with the 4-(N)-GemC18-SLNs was significantly lower than in tumor tissues in control mice, whereas the RRM1 level in TC-1-GR tumors in mice treated with the 3′-(O)-GemC18-SLNs was not significantly different from that in tumors in control mice (Fig. 3I). The high concentration of 4-(N)-GemC18 in TC-1-GR tumors and the down-regulation of RRM1 expression in TC-1-GR tumors after treatment with 4-(N)-GemC18-SLNs may explain why only the 4-(N)-GemC18-SLNs significantly inhibited TC-1-GR tumor growth in mice (Fig. 3F).
3.4. The 4-(N)-GemC18 incorporated in polymeric PLGA nanoparticles and 4-(N)-GemC8 incorporated in SLNs do not overcome gemcitabine resistance caused by RRM1 overexpression
To test whether the 4-(N)-GemC18 in nanoparticles other than the SLNs can also overcome gemcitabine resistance caused by RRM1 overexpression, we prepared 4-(N)-GemC18 in polymeric PLGA nanoparticles. The mean particle size of the 4-(N)-GemC18-PLGA-NPs was 212 ± 20 nm with a zeta potential of −42.4 ± 1.2 mV. In addition, we also synthesized a shorter chain fatty acid amide derivative of gemcitabine, 4-(N)-GemC8, and incorporated it into the SLNs to prepare 4-(N)-GemC8-SLNs. The mean particle size of the 4-(N)-GemC8-SLNs was 168 ± 19 with a zeta potential of −45.3 ± 0.5 mV. The cytotoxicities of the 4-(N)-GemC18-PLGA-NPs and 4-(N)-GemC8-SLNs were determined in TC-1-GR cells. Neither the 4-(N)-GemC18-PLGA-NPs, nor the 4-(N)-GemC8-SLNs, were as effective as 4-(N)-GemC18-SLNs in inhibiting TC-1-GR tumor cell growth (Figs. 4A–B). In fact, the 4-(N)-GemC18-PLGA-NPs were even less cytotoxic than gemcitabine alone against the TC-1-GR cells (Fig. 4B). Data in Fig. 4C showed the release of the 4-(N)-GemC18 from the 4-(N)-GemC18-PLGA-NPs and the release of 4-(N)-GemC8 from the 4-(N)-GemC8-SLNs were significantly faster than the release of GemC18 from the 4-(N)-GemC18-SLNs. We suspected that the faster release of the 4-(N)-GemC18 from the 4-(N)-GemC18-PLGA-NPs and the 4-(N)-GemC8 from the 4-(N)-GemC8-SLNs may explain, at least partially, why those nanoparticles were not as cytotoxic as the 4-(N)-GemC18-SLNs against the gemcitabine-resistant TC-1-GR tumor cells.
Fig. 4. The in vitro cytotoxicity of 4-(N)-GemC8-SLNs and 4-(N)-GemC18-PLGA-NPs in TC-1-GR cells.
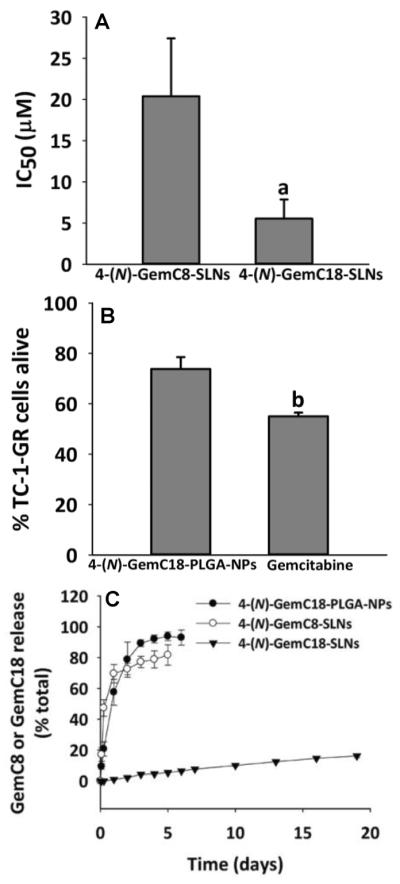
(A) The IC50 values of the 4-(N)-GemC8-SLNs and 4-(N)-GemC18-SLNs in TC-1-GR cells (n = 4). aP < 0.01. (B) The cytotoxicity of the 4-(N)-GemC18-PLGA-NPs in TC-1-GR cells after 48 h of incubation (n = 5). bP < 0.001. (C) The release profiles of GemC18 or GemC8 from 4-(N)-GemC18-PLGA-NPs, 4-(N)-GemC18-SLNs, or 4-(N)-GemC8-SLNs (n = 2–3).
3.5. The RRM1-overexpressing TC-1-GR tumor cells are also resistant to cytarabine, and 4-(N)-stearoyl Ara-C in solid lipid nanoparticles also overcome the resistance
To further confirm that conjugation of a fatty acid group at the 4-amino group of gemcitabine is critical for the antitumor activity of 4-(N)-GemC18-SLNs against the RRM1-overexpressing TC-1-GR tumor cells, we investigated the cytotoxicity of Ara-C in TC-1-GR cells. Ara-C is structurally and functionally related to gemcitabine [4]. It was previously shown that gemcitabine-resistant cell lines display cross-resistance to Ara-C [42, 43]. As shown in Fig. 5A, TC-1-GR cells displayed significant resistance to Ara-C, and silencing the RRM1 expression using RRM1-specific siRNA restored the susceptibility of TC-1-GR cells to Ara-C (Fig. 5B), indicating that the RRM1 overexpression was related to the cross-resistance of TC-1-GR cells to Ara-C. We then test whether the similar 4-(N)-stearoyl gemcitabine in SLNs strategy can be used to overcome the resistance of the TC-1-GR cells to Ara-C. The 4-(N)-stearoyl Ara-C was synthesized and incorporated into SLNs in a manner similar to the preparation of the 4-(N)-GemC18-SLNs. The mean size of the 4-(N)-Ara-C-C18-SLNs was 186 ± 13 nm with a zeta potential of −33.4 ± 0.9 mV. The IC50 value of the 4-(N)-Ara-C-C18-SLNs was about twice that of Ara-C in the TC-1 cells (Fig. 5C). However, in the TC-1-GR cells, the IC50 of the 4-(N)-Ara-C-C18-SLNs was only about 20% of that of the Ara-C (Fig. 5C).
Fig. 5. The TC-1-GR tumor cells that overexpress RRM1 are also resistant to Ara-C, and 4-(N)-Ara-CC18-SLNs overcome the resistance.
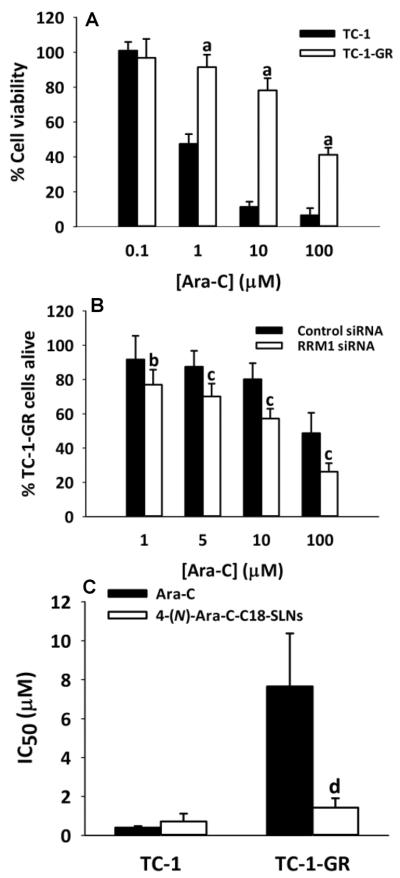
(A) The cytotoxicity of Ara-C in TC-1 and TC-1-GR cells in culture (n = 8). aP < 0.001. (B) The cytotoxicity of Ara-C in TC-1-GR cells that were transfected with RRM1-specific siRNA or control siRNA (n = 7–10). bP < 0.05, cP < 0.001. (C) The IC50 values of Ara-C and 4-(N)-Ara-C-C18-SLNs in TC-1-GR cells (n = 4). dP < 0.01. In A–C, cells were incubated with Ara-C or 4-(N)-Ara-C-C18-SLNs for 48 h.
4. DISCUSSION
Data from numerous clinical studies showed that RRM1 expression in tumor cells is inversely correlated to the sensitivity of the tumor cells to gemcitabine therapy [17, 18, 20, 22]. Previously, we discovered that our TC-1-GR cells, derived from the TC-1 cells, are resistant to gemcitabine because they overexpress RRM1 [11]. In the present study, we showed that the dCTP and dATP levels in the TC-1-GR cells were significantly higher than in TC-1 cells (Fig. 1C), demonstrating that the overexpression of RRM1 increases cellular dNTPs, which is in agreement with previous reports [39, 40]. After incubation with gemcitabine HCl, the level of dFdCTP in the TC-1-GR cells was significantly lower than in TC-1 cells (Fig. 1E), which explains why the TC-1-GR cells are ~4000-fold less sensitive to gemcitabine than TC-1 cells [11]. Our 4-(N)-GemC18-SLNs significantly decreased the level of RRM1 and increased the level of dFdCTP in TC-1-GR cells (Fig. 1), which explains why they can overcome gemcitabine resistance caused by RRM1 overexpression. Proteasome-mediated protein degradation is an important mechanism involved in regulating RRM1 protein level [44]. Although we did not measure the effect of 4-(N)-GemC18-SLNs on the intracellular dFdCDP level, it is likely that the 4-(N)-GemC18-SLNs have supplied an adequate amount of dFdCDP to bind to the excess amount of RRM1 in TC-1-GR cells and triggered the degradation of RRM1 protein.
Our data in Fig. 2 indicated that how the 4-(N)-GemC18 enters the TC-1-GR cells is critical in determining whether it can overcome gemcitabine resistance. Gemcitabine is hydrophilic; it is transported into cells by nucleoside transporters such as hENT1 [3]. The 4-(N)-GemC18 is highly lipophilic, and likely enters cells by passive diffusion. In fact, it was previously reported that another similar lipophilized gemcitabine, 4-(N)-trisnorsqualenoyl gemcitabine, also enters cells by passive diffusion [35]. In contrast, due to their particulate nature, our 4-(N)-GemC18-SLNs enter cells by clathrin-mediated endocytosis (Fig. 2B). Fluorescence confocal microscopic results revealed that the 4-(N)-GemC18-SLNs internalized by endocytosis were targeted to the lysosomal compartment, and the nanoparticles disappeared from the lysosomes within 6 h (Fig. 2C). By alkalizing lysosomes with NH4Cl, we confirmed that the acidic condition in lysosomes is critical for the intracellular degradation/hydrolysis of the 4-(N)-GemC18 and for the 4-(N)-GemC18-SLNs' cytotoxicity against the gemcitabine-resistant TC-1-GR cells (Figs. 2E–F). In contrast, alkalization of lysosomal pH with NH4Cl did not affect the intracellular degradation and cytotoxicity of the 4-(N)-GemC18 in solution. Therefore, it appears that the 4-(N)-GemC18-SLNs' ability to deliver 4-(N)-GemC18 into lysosomes and the acidic condition in the lysosomes are critical for the 4-(N)-GemC18 to overcome gemcitabine resistance.
Lysosomes contain enzymes (e.g., cathepsin B and D) capable of catalyzing the hydrolysis of amides [45]. Previously, it was shown that cathepsin B and D facilitate the hydrolysis of 4-(N)-GemC18 [13]. Moreover, it was recently reported that human equilibrative nucleoside transporter 3 (hENT3) is located on lysosome membrane [46], likely responsible for exporting nucleosides recycled from DNA and RNA degraded in lysosomes from the lumen of the lysosomes to the cytoplasm, in where the recycled nucleosides are utilized for the “salvage” synthesis of nucleotides [47]. It is likely that because our 4-(N)-GemC18-SLNs deliver the 4-(N)-GemC18 into the lysosomes of the tumor cells, and the lysosomes are rich in enzymes responsible for the metabolisms of lipids and nucleic acids, 4-(N)-GemC18 is hydrolyzed by enzymes in the lysosomes to release gemcitabine, which is then exported out of the lysosomes to the cytoplasm by nucleoside transporters, such as hENT3. The gemcitabine in the cytoplasm is then efficiently converted into its active metabolite, dFdCTP, to inhibit DNA synthesis. It happens that our 4-(N)-GemC18-SLNs `channeled' the 4-(N)-GemC18 into a `natural' pathway that has evolved for cells to efficiently recycle nucleic acids from within the cells or internalized from outside of the cells (e.g., DNA and RNA in bacteria or viruses taken up by macrophages). It is likely that, due to potential compartmentation of enzymes responsible for “salvage” nucleotide synthesis, gemcitabine that is exported out of lysosomes by nucleoside transporters may have been directly delivered to enzymes such as dCK and more efficiently phosphorylated into dFdCTP, while minimizing its deamination by cytoplasmic deoxycytidine deaminase (Fig. 6).
Fig. 6. A cartoon depicting how the uptake of gemcitabine, 4-(N)-GemC18 in solution, or 4-(N)-GemC18-SLNs influence their intracellular metabolism and activity against tumor cells.
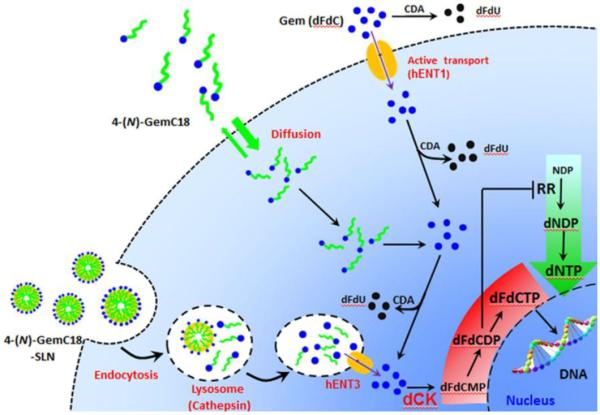
CDA, (deoxy)cytidine deaminase; dCK, deoxycytidine kinase; dFdC, gemcitabine; dFdCMP, gemcitabine monophosphate; dFdCDP, gemcitabine diphosphate; dFdCTP, gemcitabine trisphosphate; dNDP, deoxyribonucleoside diphosphate; dNTP, deoxyribonucleotide triphosphate; hENT, human equilibrative nucleoside transporter; NDP, ribonucleoside diphosphates; RR, ribonucleotide reductase
In contrast, the intracellular fate and metabolism of 4-(N)-GemC18 when it enters cells by passive diffusion are expected to be different. Data from a previous study showed that the lipophilic gemcitabine derivative mainly accumulates in the perinuclear endoplasmic reticulum (ER) after diffusing into cells [35]. The passive diffusion of 4-(N)-GemC18 into cells was faster, and even more extensive, than the endocytosis of 4-(N)-GemC18-SLNs (Fig. 2A). The 4-(N)-GemC18 that diffused into cells and accumulated in the intracellular membrane may be hydrolyzed by enzymes such as fatty acid amide hydrolases (FAAH), which facilitate the hydrolysis of fatty acid amide and are mainly localized in the ER [48], to release the gemcitabine into the cytoplasm. Unlike the gemcitabine that are exported from the lumen of lysosomes to the cytoplasm by nucleoside transporters, gemcitabine that is hydrolyzed from the 4-(N)-GemC18 outside of the lysosomes may not be as efficiently `channeled' to enzymes such as dCK for phosphorylation, while at the same time is subjected to extensive deamination (Fig. 6). This may explain why the same 4-(N)-GemC18 is more cytotoxic against the RRM1-overexpressing TC-1-GR cells when delivered into cells by the endocytosis of the 4-(N)-GemC18-SLNs than when passively diffused into TC-1-GR cells. In fact, this same mechanism may also explain why our 4-(N)-GemC18-SLNs are significantly more cytotoxic than 4-(N)-GemC18 in solution to tumor cells that have other mechanisms of gemcitabine resistance, such as hENT1 or dCK deficiency [28].
Indeed we have more evidence to support the above hypothesis. For example, the 3′-(O)-GemC18 ester has the same molecular weight as the 4-(N)-GemC18 amide, but was unable to overcome gemcitabine resistance caused by RRM1 overexpression, even when carried by the same SLNs (Fig. 3). The ester bond on the 3′-(O)-GemC18 is more sensitive to hydrolysis than the amide bond on the 4-(N)-GemC18 (Fig 3C), which may explain why the uptake of the 3′-(O)-GemC18 in 3′-(O)-GemC18-SLNs by TC-1-GR cells stopped increasing, and even slightly decreased, after 1 h of incubation, whereas the uptake of the 4-(N)-GemC18 in 4-(N)-GemC18-SLNs by TC-1-GR cells continued to gradually increase (Fig 3D). A significant percentage of the 3′-(O)-GemC18 in the 3′-(O)-GemC18-SLNs may have been hydrolyzed in the cell culture medium even before entering cells. Similarly, the higher sensitivity of the 3′-(O)-GemC18 to hydrolysis may also explain why the 3′-(O)-GemC18-SLNs, unlike the 4-(N)-GemC18-SLNs, were unable to inhibit the growth of the gemcitabine-resistant TC-1-GR cells in mice (Fig. 3F).
We have also discovered that the same 4-(N)-GemC18 in polymeric PLGA nanoparticles, 4-(N)-GemC18-PLGA-NPs, and the shorter-chain fatty acid amide gemcitabine derivative 4-(N)-GemC8 in the same solid lipid nanoparticles, 4-(N)-GemC8-SLNs, were not as cytotoxic as the 4-(N)-GemC18-SLNs to TC-1-GR cells (Figs. 4A–B). We speculate that the faster release of the 4-(N)-GemC18 from the 4-(N)-GemC18-PLGA-NPs and the 4-(N)-GemC8 from the 4-(N)-GemC8-SLNs explains, at least partially, why they are not as cytotoxic as the 4-(N)-GemC18-SLNs against the TC-1-GR cells. The fast release of the gemcitabine derivatives from their corresponding nanoparticles may have prevented them from entering cells by the endocytosis of the nanoparticles that carried them. Of course, factors other than the release rate may have also contributed to their weak cytotoxicity against the gemcitabine resistant TC-1-GR cells. Data from a previous study showed that long-chain fatty acid gemcitabine derivatives such as 4-(N)-GemC18 and 4-(N)-GemC12 are more stable than short-chain fatty acid gemcitabine derivatives such as 4-(N)-GemC5 and 4-(N)-GemC7 [25]. Moreover, the acidic condition within the PLGA nanoparticles may have also made the 4-(N)-GemC18 less stable than in 4-(N)-GemC18-SLNs [49]. As to why the release of 4-(N)-GemC18 from the 4-(N)-GemC18-SLNs was significantly slower than the release of 4-(N)-GemC8 from the 4-(N)-GemC8-SLNs, and the 4-(N)-GemC18 from the 4-(N)-GemC18-PLGA-NPs, we speculate that it is because the diffusion of the 4-(N)-GemC8 out of the 4-(N)-GemC8-SLNs, and the 4-(N)-GemC18 out of the 4-(N)-GemC18-PLGA-NPs, was faster than the diffusion of the 4-(N)-GemC18 from the 4-(N)-GemC18-SLNs, considering that 4-(N)-GemC8 is less lipophilic than 4-(N)-GemC18, and also the affinity of the highly lipophilic 4-(N)-GemC18 to the PLGA polymer in the 4-(N)-GemC18-PLGA-NPs is likely significantly less than to the lecithin and glycerol monostearate in the 4-(N)-GemC18-SLNs.
We have noticed that in the gemcitabine-sensitive TC-1 cells in culture, the 4-(N)-GemC18-SLNs are significantly less cytotoxic than free gemcitabine (Fig. 1A), which agrees with our previous data [23, 28]. We speculate that the slow hydrolysis of the gemcitabine from the 4-(N)-GemC18-SLNs is related to the relatively lower cytotoxicity of the 4-(N)-GemC18-SLNs in the gemcitabine-sensitive TC-1 cells. We have previously shown that when TC-1 tumor cells were incubated with the 4-(N)-GemC18-SLNs for 48 h, the percent of the dead TC-1 cells reached a level similar to that when the TC-1 cells were incubated with the gemcitabine HCl for 24 h, indicating that it simply takes a longer time for the 4-(N)-GemC18-SLNs to kill tumor cells [23]. IN addition, we also noticed that the 4-(N)-GemC18, alone or physically mixed with blank SLNs, is also more cytotoxic than free gemcitabine to TC-1-GR cells in culture, although it is significantly less cytotoxic than the 4-(N)-GemC18-SLNs (Fig. 1A). This is likely related to the slow hydrolysis of the gemcitabine from the 4-(N)-GemC18 as well, which helps prevent the deamination of gemcitabine. Of course, the fact that 4-(N)-GemC18 in solution diffuses into cells, and its unique distribution intracellularly due to its lipophilicity, may be also related to its higher cytotoxicity to TC-1-GR cells (relative to free gemcitabine). Therefore, it is clear that the 4-(N)-GemC18-SLNs' ability to overcome gemcitabine resistance caused by RRM1 overexpression is not simply due to the 4-(N)-GemC18 per se, but the 4-(N)-GemC18 is still critical. It is worth emphasizing that the data shown in Fig. 1A were from cell culture. In animal studies, our 4-(N)-GemC18-SLNs were significantly more effective than free gemcitabine in controlling the growth of both TC-1 and TC-1-GR cells [11, 23], and the 4-(N)-GemC18 in solution was not significantly more effective than free gemcitabine [23, 27].
Finally, we found that the similar strategy of incorporating a long-chain fatty acid amide derivative of a nucleoside analog into our SLNs is applicable in overcoming tumor cell resistance to cytarabine (Ara-C) as well. Our gemcitabine-resistant TC-1-GR cells displayed cross-resistance to Ara-C (Fig. 5A), and RRM1 overexpression was related, at least partially, to the cross-resistance of the TC-1-GR cells towards Ara-C (Fig. 5B). Since the metabolism and mechanism of action of Ara-C are similar to that of gemcitabine, an increase in total dNTP level in tumor cells due to the overexpression of RRM1 may have decreased the ratio of Ara-C triphosphate (Ara-CTP) to dNTPs, and thus the chance for the Ara-CTP to effectively compete with dCTP to inhibit DNA repair or synthesis [50]. The fact that the 4-(N)-Ara-C-C18-SLNs were able to overcome the cross-resistance of TC-1-GR cells to Ara-C highlights the potential of using our strategy of incorporating a long-chain fatty acid amide derivative of a nucleoside analog into SLNs as a platform technology to overcome tumor cell resistance to the nucleoside analog. It is however noted that unlike gemcitabine, Ara-C does not regulate RR [6]. Therefore, mechanisms other than down-regulating the overexpression of RRM1 in TC-1-GR cells were likely responsible for the greater cytotoxicity of 4-(N)-Ara-C-C18-SLNs to the TC-1-GR cells than Ara-C. We speculate that it is because the 4-(N)-Ara-C-C18-SLNs delivered 4-(N)-Ara-C-C18 into the lysosomes of the TC-1-GR cells by endocytosis. In addition to Ara-C, RRM1 overexpression in tumor cells significantly affects the cytotoxicity of other nucleoside analogs such as 5-fluorouracil [39]. Incorporating long chain lipophilic amide prodrugs of those cytotoxic agents into our SLNs may also overcome tumor cell resistance to them as well.
5. CONCLUSION
For a nucleoside analog such as gemcitabine to effectively kill resistant tumor cells, it is not enough for them to just enter the tumor cells. How they enters cells is as important, if not more important. Our 4-(N)-GemC18-SLNs overcome gemcitabine resistance, including resistance caused by RRM1 overexpression (more efficiently relative to 4-(N)-GemC18 in solution), because they deliver the 4-(N)-GemC18 into the lysosomes of tumor cells by endocytosis, allowing the gemcitabine hydrolyzed from the 4-(N)-GemC18 in the lysosomes to be more efficiently converted into the active metabolite, dFdCTP, to inhibit DNA synthesis. Our strategy of incorporating the long-chain fatty acid amide derivative of a nucleoside analog into our solid lipid nanoparticles may represent a platform technology to increase the antitumor activity of the nucleoside analog and to overcome tumor cell resistance.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Prabhakar Risbood at the National Cancer Institute for kindly providing the gemcitabine triphosphate sample and Dr. Yue Li at the Microscopy Core Facility at the Dell Pediatric Research Institute at The University of Texas at Austin for assistance in acquiring the confocal microscopic images. This work was supported in part by a National Institutes of Health grant (CA135274) and a fund from the University of Texas at Austin, College of Pharmacy (to Z Cui).
Abbreviations
- 4-(N)-GemC18
4-(N)-stearoyl gemcitabine
- 4-(N)-GemC18-SLNs
4-(N)-stearoyl gemcitabine solid lipid nanoparticles
- 3'-(O)-GemC18
3'-(O)-stearoyl gemcitabine
- 3'-(O)-GemC18-SLNs
3'-(O)-stearoyl gemcitabine solid lipid nanoparticles
- 4-(N)-GemC8
4-(N)-octyl gemcitabine
- 4-(N)-GemC8-SLNs
4-(N)-octyl gemcitabine solid lipid nanoparticles
- Ara-C
cytarabine
- 4-(N)-Ara-C-C18
4-(N)-stearoyl cytarabine
- 4-(N)-Ara-C-C18-SLNs
4-(N)-stearoyl cytarabine solid lipid nanoparticles
- CDA
(deoxy)cytidine deaminase
- dATP
deoxyadenosine triphosphate
- dCK
deoxycytidine kinase
- dFdC
gemcitabine
- dCTP
deoxycytidine triphosphate
- dFdCMP
gemcitabine monophosphate
- dFdCDP
gemcitabine diphosphate
- dFdCTP
gemcitabine triphosphate
- dNTPs
deoxynucleotide triphosphates
- DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- FAAH
fatty acid amide hydrolases
- hENT1
human equilibrative nucleoside transporter 1
- NTPs
ribonucleoside triphosphates
- PLGA-NPs
poly (lactic-co-glycolic) acid nanoparticles
- RRM1
ribonucleotide reductase M1
- SLNs
solid lipid nanoparticles
- TBACl
tetrabutylammonium chloride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Long J, Zhang Y, Yu X, Yang J, LeBrun DG, Chen C, Yao Q, Li M. Overcoming drug resistance in pancreatic cancer. Expert Opin Ther Targets. 2011;15:817–828. doi: 10.1517/14728222.2011.566216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nakano Y, Tanno S, Koizumi K, Nishikawa T, Nakamura K, Minoguchi M, Izawa T, Mizukami Y, Okumura T, Kohgo Y. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer. 2007;96:457–463. doi: 10.1038/sj.bjc.6603559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mackey JR, Yao SY, Smith KM, Karpinski E, Baldwin SA, Cass CE, Young JD. Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J Natl Cancer Inst. 1999;91:1876–1881. doi: 10.1093/jnci/91.21.1876. [DOI] [PubMed] [Google Scholar]
- [4].Heinemann V, Hertel LW, Grindey GB, Plunkett W. Comparison of the cellular pharmacokinetics and toxicity of 2',2'-difluorodeoxycytidine and 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1988;48:4024–4031. [PubMed] [Google Scholar]
- [5].Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W. Action of 2',2'-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51:6110–6117. [PubMed] [Google Scholar]
- [6].Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2',2'-difluorodeoxycytidine. Mol Pharmacol. 1990;38:567–572. [PubMed] [Google Scholar]
- [7].Ferrandina G, Mey V, Nannizzi S, Ricciardi S, Petrillo M, Ferlini C, Danesi R, Scambia G, Del Tacca M. Expression of nucleoside transporters, deoxycitidine kinase, ribonucleotide reductase regulatory subunits, and gemcitabine catabolic enzymes in primary ovarian cancer. Cancer Chemother Pharmacol. 2010;65:679–686. doi: 10.1007/s00280-009-1073-y. [DOI] [PubMed] [Google Scholar]
- [8].Gong W, Zhang X, Wu J, Chen L, Li L, Sun J, Lv Y, Wei X, Du Y, Jin H, Dong J. RRM1 expression and clinical outcome of gemcitabine-containing chemotherapy for advanced non-small-cell lung cancer: A meta-analysis. Lung Cancer. 2012;75:374–80. doi: 10.1016/j.lungcan.2011.08.003. [DOI] [PubMed] [Google Scholar]
- [9].Palakurthi S, Yellepeddi VK, Vangara KK. Recent trends in cancer drug resistance reversal strategies using nanoparticles. Expert Opin Drug Deliv. 2012;9:287–301. doi: 10.1517/17425247.2012.665365. [DOI] [PubMed] [Google Scholar]
- [10].Dong X, Mumper RJ. Nanomedicinal strategies to treat multidrug-resistant tumors: current progress. Nanomedicine (Lond) 2010;5:597–615. doi: 10.2217/nnm.10.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chung WG, Sandoval MA, Sloat BR, Lansakara PD, Cui Z. Stearoyl gemcitabine nanoparticles overcome resistance related to the over-expression of ribonucleotide reductase subunit M1. J Control Release. 2012;157:132–140. doi: 10.1016/j.jconrel.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Couvreur P, Stella B, Reddy LH, Hillaireau H, Dubernet C, Desmaele D, Lepetre-Mouelhi S, Rocco F, Dereuddre-Bosquet N, Clayette P, Rosilio V, Marsaud V, Renoir JM, Cattel L. Squalenoyl nanomedicines as potential therapeutics. Nano Lett. 2006;6:2544–2548. doi: 10.1021/nl061942q. [DOI] [PubMed] [Google Scholar]
- [13].Reddy LH, Dubernet C, Mouelhi SL, Marque PE, Desmaele D, Couvreur P. A new nanomedicine of gemcitabine displays enhanced anticancer activity in sensitive and resistant leukemia types. J Control Release. 2007;124:20–27. doi: 10.1016/j.jconrel.2007.08.018. [DOI] [PubMed] [Google Scholar]
- [14].Rejiba S, Reddy LH, Bigand C, Parmentier C, Couvreur P, Hajri A. Squalenoyl gemcitabine nanomedicine overcomes the low efficacy of gemcitabine therapy in pancreatic cancer. Nanomedicine. 2011;7:841–849. doi: 10.1016/j.nano.2011.02.012. [DOI] [PubMed] [Google Scholar]
- [15].Rosell R, Danenberg KD, Alberola V, Bepler G, Sanchez JJ, Camps C, Provencio M, Isla D, Taron M, Diz P, Artal A. Ribonucleotide reductase messenger RNA expression and survival in gemcitabine/cisplatin-treated advanced non-small cell lung cancer patients. Clin Cancer Res. 2004;10:1318–1325. doi: 10.1158/1078-0432.ccr-03-0156. [DOI] [PubMed] [Google Scholar]
- [16].Zhang GB, Chen J, Wang LR, Li J, Li MW, Xu N, Wang Y, Shentu JZ. RRM1 and ERCC1 expression in peripheral blood versus tumor tissue in gemcitabine/carboplatin-treated advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2012;69:1277–1287. doi: 10.1007/s00280-012-1834-x. [DOI] [PubMed] [Google Scholar]
- [17].Nakahira S, Nakamori S, Tsujie M, Takahashi Y, Okami J, Yoshioka S, Yamasaki M, Marubashi S, Takemasa I, Miyamoto A, Takeda Y, Nagano H, Dono K, Umeshita K, Sakon M, Monden M. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int J Cancer. 2007;120:1355–1363. doi: 10.1002/ijc.22390. [DOI] [PubMed] [Google Scholar]
- [18].Xie H, Jiang W, Jiang J, Wang Y, Kim R, Liu X. Predictive and prognostic roles of ribonucleotide reductase M1 in resectable pancreatic adenocarcinoma. Cancer. 2013;119:173–81. doi: 10.1002/cncr.27715. [DOI] [PubMed] [Google Scholar]
- [19].Akita H, Zheng Z, Takeda Y, Kim C, Kittaka N, Kobayashi S, Marubashi S, Takemasa I, Nagano H, Dono K, Nakamori S, Monden M, Mori M, Doki Y, Bepler G. Significance of RRM1 and ERCC1 expression in resectable pancreatic adenocarcinoma. Oncogene. 2009;28:2903–2909. doi: 10.1038/onc.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ohtaka K, Kohya N, Sato K, Kitajima Y, Ide T, Mitsuno M, Miyazaki K. Ribonucleotide reductase subunit M1 is a possible chemoresistance marker to gemcitabine in biliary tract carcinoma. Oncol Rep. 2008;20:279–286. [PubMed] [Google Scholar]
- [21].Wakai T, Shirai Y, Sakata J, Takamura M, Matsuda Y, Korita PV, Muneoka K, Sasaki M, Ajioka Y, Hatakeyama K. Ribonucleotide reductase M1 expression in intrahepatic cholangiocarcinoma. Hepatogastroenterology. 2011;58:1659–1663. doi: 10.5754/hge11175. [DOI] [PubMed] [Google Scholar]
- [22].Zhao LP, Xue C, Zhang JW, Hu ZH, Zhao YY, Zhang J, Huang Y, Zhao HY, Zhang L. Expression of RRM1 and its correlation with sensitivity to gemcitabine-based chemotherapy in advanced nasopharyngeal carcinoma. Chin J Cancer. 2012;31:476–83. doi: 10.5732/cjc.012.10029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sloat BR, Sandoval MA, Li D, Chung WG, Lansakara PD, Proteau PJ, Kiguchi K, DiGiovanni J, Cui Z. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int J Pharm. 2011;409:278–288. doi: 10.1016/j.ijpharm.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Guo Zw Z, Gallo JM. Selective Protection of 2',2'-Difluorodeoxycytidine (Gemcitabine) J Org Chem. 1999;64:8319–8322. doi: 10.1021/jo9911140. [DOI] [PubMed] [Google Scholar]
- [25].Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release. 2004;100:331–346. doi: 10.1016/j.jconrel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- [26].Mondal N, Halder KK, Kamila MM, Debnath MC, Pal TK, Ghosal SK, Sarkar BR, Ganguly S. Preparation, characterization, and biodistribution of letrozole loaded PLGA nanoparticles in Ehrlich Ascites tumor bearing mice. Int J Pharm. 2010;397:194–200. doi: 10.1016/j.ijpharm.2010.06.049. [DOI] [PubMed] [Google Scholar]
- [27].Zhu S, Lansakara PD, Li X, Cui Z. Lysosomal delivery of a lipophilic gemcitabine prodrug using novel acid-sensitive micelles improved its antitumor activity. Bioconjug Chem. 2012;23:966–980. doi: 10.1021/bc2005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lansakara PD, Rodriguez BL, Cui Z. Synthesis and in vitro evaluation of novel lipophilic monophosphorylated gemcitabine derivatives and their nanoparticles. Int J Pharm. 2012;429:123–134. doi: 10.1016/j.ijpharm.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Decosterd LA, Cottin E, Chen X, Lejeune F, Mirimanoff RO, Biollaz J, Coucke PA. Simultaneous determination of deoxyribonucleoside in the presence of ribonucleoside triphosphates in human carcinoma cells by high-performance liquid chromatography. Anal Biochem. 1999;270:59–68. doi: 10.1006/abio.1999.4066. [DOI] [PubMed] [Google Scholar]
- [30].Losa R, Sierra MI, Gion MO, Esteban E, Buesa JM. Simultaneous determination of gemcitabine di- and triphosphate in human blood mononuclear and cancer cells by RP-HPLC and UV detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;840:44–49. doi: 10.1016/j.jchromb.2006.04.036. [DOI] [PubMed] [Google Scholar]
- [31].Zhu S, Wonganan P, Lansakara PD, O'Mary HL, Li Y, Cui Z. The effect of the acid-sensitivity of 4-(N)-stearoyl gemcitabine-loaded micelles on drug resistance caused by RRM1 overexpression. Biomaterials. 2013;34:2327–2339. doi: 10.1016/j.biomaterials.2012.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rejman J, Bragonzi A, Conese M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol Ther. 2005;12:468–474. doi: 10.1016/j.ymthe.2005.03.038. [DOI] [PubMed] [Google Scholar]
- [33].Greulich C, Diendorf J, Simon T, Eggeler G, Epple M, Koller M. Uptake and intracellular distribution of silver nanoparticles in human mesenchymal stem cells. Acta Biomater. 2011;7:347–354. doi: 10.1016/j.actbio.2010.08.003. [DOI] [PubMed] [Google Scholar]
- [34].Peng B, Koga K, Cardenas I, Aldo P, Mor G. Phagocytosis of apoptotic trophoblast cells by human endometrial endothelial cells induces proinflammatory cytokine production. Am J Reprod Immunol. 2010;64:12–19. doi: 10.1111/j.1600-0897.2010.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bildstein L, Dubernet C, Marsaud V, Chacun H, Nicolas V, Gueutin C, Sarasin A, Benech H, Lepetre-Mouelhi S, Desmaele D, Couvreur P. Transmembrane diffusion of gemcitabine by a nanoparticulate squalenoyl prodrug: an original drug delivery pathway. J Control Release. 2010;147:163–170. doi: 10.1016/j.jconrel.2010.07.120. [DOI] [PubMed] [Google Scholar]
- [36].Garcia-Garcia E, Andrieux K, Gil S, Kim HR, Le Doan T, Desmaele D, d'Angelo J, Taran F, Georgin D, Couvreur P. A methodology to study intracellular distribution of nanoparticles in brain endothelial cells. Int J Pharm. 2005;298:310–314. doi: 10.1016/j.ijpharm.2005.03.030. [DOI] [PubMed] [Google Scholar]
- [37].Baker CH, Banzon J, Bollinger JM, Stubbe J, Samano V, Robins MJ, Lippert B, Jarvi E, Resvick R. 2'-Deoxy-2'-methylenecytidine and 2'-deoxy-2',2'-difluorocytidine 5'-diphosphates: potent mechanism-based inhibitors of ribonucleotide reductase. J Med Chem. 1991;34:1879–1884. doi: 10.1021/jm00110a019. [DOI] [PubMed] [Google Scholar]
- [38].Heinemann V, Schulz L, Issels RD, Plunkett W. Gemcitabine: a modulator of intracellular nucleotide and deoxynucleotide metabolism. Semin Oncol. 1995;22:11–18. [PubMed] [Google Scholar]
- [39].Zhou BS, Hsu NY, Pan BC, Doroshow JH, Yen Y. Overexpression of ribonucleotide reductase in transfected human KB cells increases their resistance to hydroxyurea: M2 but not M1 is sufficient to increase resistance to hydroxyurea in transfected cells. Cancer Res. 1995;55:1328–1333. [PubMed] [Google Scholar]
- [40].Jordheim LP, Guittet O, Lepoivre M, Galmarini CM, Dumontet C. Increased expression of the large subunit of ribonucleotide reductase is involved in resistance to gemcitabine in human mammary adenocarcinoma cells. Mol Cancer Ther. 2005;4:1268–1276. doi: 10.1158/1535-7163.MCT-05-0121. [DOI] [PubMed] [Google Scholar]
- [41].D'Souza AJ, Topp EM. Release from polymeric prodrugs: linkages and their degradation. J Pharm Sci. 2004;93:1962–1979. doi: 10.1002/jps.20096. [DOI] [PubMed] [Google Scholar]
- [42].Bergman AM, Pinedo HM, Ruiz van Haperen VW, Veerman G, Kuiper CM, Peters GJ. Cross-resistance of the gemcitabine resistant human ovarian cancer cell line AG6000 to standard and investigational drugs. Adv Exp Med Biol. 1998;431:587–590. doi: 10.1007/978-1-4615-5381-6_113. [DOI] [PubMed] [Google Scholar]
- [43].Davidson JD, Ma L, Flagella M, Geeganage S, Gelbert LM, Slapak CA. An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res. 2004;64:3761–3766. doi: 10.1158/0008-5472.CAN-03-3363. [DOI] [PubMed] [Google Scholar]
- [44].Mitsuno M, Kitajima Y, Ohtaka K, Kai K, Hashiguchi K, Nakamura J, Hiraki M, Noshiro H, Miyazaki K. Tranilast strongly sensitizes pancreatic cancer cells to gemcitabine via decreasing protein expression of ribonucleotide reductase 1. Int J Oncol. 2010;36:341–349. [PubMed] [Google Scholar]
- [45].Thomssen C, Schmitt M, Goretzki L, Oppelt P, Pache L, Dettmar P, Janicke F, Graeff H. Prognostic value of the cysteine proteases cathepsins B and cathepsin L in human breast cancer. Clin Cancer Res. 1995;1:741–746. [PubMed] [Google Scholar]
- [46].Baldwin SA, Yao SY, Hyde RJ, Ng AM, Foppolo S, Barnes K, Ritzel MW, Cass CE, Young JD. Functional characterization of novel human and mouse equilibrative nucleoside transporters (hENT3 and mENT3) located in intracellular membranes. J Biol Chem. 2005;280:15880–15887. doi: 10.1074/jbc.M414337200. [DOI] [PubMed] [Google Scholar]
- [47].Hsu CL, Lin W, Seshasayee D, Chen YH, Ding X, Lin Z, Suto E, Huang Z, Lee WP, Park H, Xu M, Sun M, Rangell L, Lutman JL, Ulufatu S, Stefanich E, Chalouni C, Sagolla M, Diehl L, Fielder P, Dean B, Balazs M, Martin F. Equilibrative nucleoside transporter 3 deficiency perturbs lysosome function and macrophage homeostasis. Science. 2012;335:89–92. doi: 10.1126/science.1213682. [DOI] [PubMed] [Google Scholar]
- [48].Arreaza G, Deutsch DG. Deletion of a proline-rich region and a transmembrane domain in fatty acid amide hydrolase. FEBS Lett. 1999;454:57–60. doi: 10.1016/s0014-5793(99)00774-7. [DOI] [PubMed] [Google Scholar]
- [49].Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric systems for controlled drug release. Chem Rev. 1999;99:3181–3198. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- [50].Cooper T, Ayres M, Nowak B, Gandhi V. Biochemical modulation of cytarabine triphosphate by clofarabine. Cancer Chemother Pharmacol. 2005;55:361–368. doi: 10.1007/s00280-004-0906-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.