

Abstract
17β-estradiol (E2), as the main circulating estrogen hormone, plays critical roles in the physiology and pathophysiology of various tissues. The E2 information is primarily conveyed by the transcription factors, estrogen receptors (ERs) α and β. ERs share similar structural and functional features. Experimental studies indicate that upon binding to E2, ERs directly or indirectly intERαct with DNA and regulate gene expressions with ERα being more potent transregulator than ERβ. However, studies also showed that ERβ induces alterations in phenotypic features of cancer cell lines independent of E2. These observations suggested that the manner in which the unliganded ERβ induces phenotypic alterations in cancer cell models differs from that of ERα. Studies demonstrated that while requiring E2 for function at low levels of synthesis, the unliganded ERα at augmented concentrations modulates gene expressions and cellular growth. We, therefore, anticipated that heightened levels of ERβ synthesis could similarly circumvent the dependency on E2 leading to gene transcriptions and cellular proliferation. To test this prediction, we used adenovirus-infected cancer cell lines in which ERs were shown to induce genomic and cellular responses. We found that while ERβ at low levels of synthesis was dependent upon E2 for function, the receptor at high levels regulated gene expression and cellular proliferation independent of E2. We then addressed whether ERs at comparable levels that require E2 for function differentially alter gene expressions and cellular responses. We found that ERs mediate the effects of E2 on gene expression, cellular proliferation, apoptosis, and motility with an overlapping pattern. However, ERα was more potent regulator than ERβ in inducing cellular responses. Our results suggest that differences in potencies to regulate the expression of genes are a critical feature of the ER subtypes in mediating E2 signaling in cancer cell lines.
Keywords: estrogen, estrogen receptor, gene expression, proliferation, motility, apoptosis
Introduction
17β-estradiol (E2) is the main circulating estrogen hormone and plays critical roles in the regulation of many tissue functions including breast tissue [1, 2]. E2 also contributes to the initiation and development of target tissue malignancies [1, 2]. The E2 information is primarily conveyed by estrogen receptors (ERs) α and β [1, 2]. Two distinct genes encode ERs, which are expressed in the same tissue as well as different tissues with varying levels [1, 2]. However, ERs share structural characteristics that are responsible for similar functional features [1, 2]. The binding of E2 induces conformational alterations in ERs that convert the functionally inactive receptors to an active state. IntERαctions of E2–ERs with specific DNA sequences, estrogen responsive elements (EREs) [1, 3], or intERαctions with transcription factors bound to their cognate regulatory elements on DNA [4, 5] constitute nuclear estrogen signaling pathways. E2–ERs bound directly or indirectly to DNA then recruit an ensemble of multi-subunit complexes responsible for the alteration of local chromatin structure and the intERαction with the basal transcription machinery [1, 3]. The combinatorial effects of these complexes initiate the transcription of responsive genes that results in the regulation of cellular proliferation, apoptosis, and motility [6–10]. Although ERβ, like ERα, is largely dependent on E2 for transcription, studies also showed that the unliganded ERβ in contrast to ERα induces profound alterations in phenotypic characteristics of various cancer cell models independent of tissue of origin [11–18]. These observations suggested that the unliganded ERβ utilizes a different mechanism to alter cellular growth [8, 11, 12, 17, 19].
In contrast, we recently reported that both ER subtypes require E2 to induce genomic and cellular responses [20, 21]. Underlying reasons for the effects of unliganded ERβ on cellular responses are unclear. Insightful studies showed that augmented levels of unliganded ERα through mechanisms distinct from those initiated by the ligand binding regulate the expression of responsive genes and expend the repertoire of target genes that result in alterations in cellular growth [22, 23]. These observations raise the possibility that ERβ at high levels of synthesis could circumvent the dependency on E2 to modulate gene expression and cellular proliferation. To examine this issue, we used recombinant adenovirus-infected MDAMB-231 and U-2OS cells that derived from a breast adenocarcinoma and an osteosarcoma, respectively. In these cells exogenously introduced ERs were shown to modulate genomic and cellular responses [8, 9, 11, 15, 24–32]. We found that at high levels of synthesis, ERβ, as ERα, regulated the transcription of some of the responsive genes independently of E2 that resulted in the alteration of cellular growth. We then addressed whether at comparable levels of synthesis that are dependent upon E2 for functions ER subtypes differ in inducing genomic and cellular responses. We observed that ERs in the presence of physiological concentration of E2 induce gene expression and phenotypic alterations with an overlapping pattern. However, ERα was a more potent transregulator than ERβ in mediating the effects of E2 on cellular responses. We, therefore, suggest that differences in potencies to induce cellular responses are the defining feature of the ER subtypes.
Results
Effects of increasing levels of ERs on gene expressions and cellular growth ERα and ERβ are largely dependent upon E2 to modulate transcriptional responses in heterologous expression systems in which E2–ERα alters responsive gene expression more potently than E2–ERβ. However, the unliganded ERβ in contrast to ERα has been also shown to alter phenotypic features of cell models, including MDA-MB-231 and U-2OS cells [11–14, 16–18]. Although underling reasons unclear, these results suggested that ERβ utilizes a mode of action that differs from that of ERα to induce phenotypic changes [8, 11, 12, 17, 19]. In contrast, we recently reported that ERβ, like ERα, requires E2 to induce genomic and cellular responses [20, 21]. Studies also showed that augmented levels of the unliganded ERα have the capacity to mediate responsive gene expressions and expend the target gene repertoire that alter the growth of model cells through mechanisms independent of ligand binding [22, 23]. We, therefore, wanted to initially ask whether ERβ at high levels of synthesis could circumvent the dependency on E2 to regulate the expression of genes and consequently cellular growth. To address this issue, we assessed the effects of increasing concentrations of ERs on the growth of MDA-MB-231 cells. We used recombinant adenovirus infection as an efficient transgene delivery system [20, 21, 29, 33]. Infection with varying concentrations of recombinant adenoviruses also provides a means to readily adjust the level of protein synthesis in cells. We produced viruses bearing no cDNA (Ad5) or cDNA for human ERα or ERβ. The ER cDNAs contain sequences that encode a Flag epitope at the aminoterminus, which does not affect functional features of ERs [33] and allows the biochemical characterization of both ER subtypes. We infected ER-negative MDA-MB-231 cells at various multiplicity of infections (MOIs) of recombinant adenoviruses in the absence or presence of a physiological level of E2 (10–9 M) and examined the effects of receptor subtypes on cellular growth (Fig. 1). The level of ER synthesis, assessed by western blot (WB) using an antibody specific to the Flag epitope (Fig. 1a), was correlated with the repression of cell growth (Fig. 1b). At lower levels of synthesis, the effects of ERs were dependent on E2 to reduce cellular proliferation. However, ERs at high levels suppressed the growth in the absence of E2, which was further repressed when E2 was present. Thus, augmented levels of ERβ, as shown for ERα [22, 23], alter cellular growth independent of E2.
Figure 1.

Fig. 1 Effects of increasing levels of ER subtypes on gene expression and cellular growth in the absence or presence of E2. a and b MDA-MB-231 cells were infected with various MOIs of recombinant adenovirus expressing none (Ad5) or a cDNA for human ERα or ERβ. The total adenovirus concentration was equalized with the inclusion of an appropriate MOI of the parent Ad5 as indicated. a Cell extracts (10 μg) at 48 h post-infection were subjected to 8–16% SDS-PAGE for WB using a horseradish peroxidase-conjugated monoclonal Flag antibody specific to the Flag epitope present at the amino-terminus of each ER subtype. Molecular mass in KDa is indicated. b MDA-MB-231 cells infected with recombinant adenoviruses at indicated MOIs without (−) or with 10−9 M E2 were subjected to cell counting at day 6. Changes in cell numbers are given as percent change compared to cells infected with the parent Ad5 (Ad5), which was set to 100. Superscript (a) denotes responses that are significantly different from those observed with cells infected with Ad5 in the absence of E2. Superscript (b) indicates responses that are significantly different from responses to ERs in the absence of E2 at the corresponding concentration. c MDA-MB-231 cells were infected with recombinant adenoviruses in the absence or presence of 10−9 M E2 for 48 h. The total amount of recombinant adenovirus expressing ERα cDNA (ERα) at low (100) or high (250) MOI was brought to 600 or 1500 by the addition of 500 or 1250 MOI of Ad5, respectively. Cells were then subjected to totalRNAextraction for qPCR. Results for the expression of the CTSD, CDKN1A, HBEGF, RARA, TGFB2, and MYC genes are the mean ± SEM of three independent determinations in duplicate. Responses are given as fold change compared to responses observed with cells infected with the parent Ad5 (600 MOI) in the absence of E2, which were set to one. Superscript (a) indicates responses that are significantly different from those observed with cells infected with Ad5 in the absence of E2. Superscript (b) denotes responses that are significantly different from responses to ERs in the absence of E2 at the corresponding concentration.
E2 treatment of stably transfected or adenovirus-infected MDA-MB-231 cells synthesizing ERs regulates gene expressions [8, 9, 20, 21]. We then examined whether the repression of cellular growth induced by the unliganded ERs at high concentrations results from the abilities of ERs to modulate the transcription of estrogen responsive genes independent of E2. We previously found, using global gene expression profiling and qPCR approaches, that ERs in the absence or presence of E2 do not affect the expression of GAPDH (Glyceraldehyde-3-phosphate dehydrogenase), PGR (Progesterone receptor), CCND1 (Cyclin D1), or MYC (c-myc) gene in MDA-MB-231 cells [20, 21]. However, E2–ERs regulate the transcription the CTSD (Cathepsin D), CDKN1A (Cyclin-dependent kinase inhibitor 1; p21, WAF), HBEGF (Heparin-binding EGF-like growth factor), and RARA (Retinoic acid receptor α) genes [20, 21]. On the other hand, the TGFB2 (Transforming growth factor, β2) gene expression is responsive to only ERα and when E2 is present [20]. To examine the effects of low levels of ERs on endogenous gene expression, we used Ad5–ERα at 100 and Ad5–ERβ at 600 MOIs. Whereas, 250 MOI of Ad5–ERα and 1500 MOI of Ad5–ERβ were used for high levels of receptor synthesis that affected cellular growth independent of E2 (Fig. 1b). The parent recombinant adenovirus (Ad5) was used at 600 or 1500 MOI. In infections, the total Ad5–ERα concentration was equalized to 600 or 1500 MOI with the inclusion of an appropriate amount of the parent Ad5. We found that at low levels of receptor synthesis, the transcriptional regulation of the CTSD, CDKN1A, HBEGF, and RARA genes by both ER subtypes was dependent upon the presence of E2 (Fig. 1c), whereas only ERα in the presence of E2 modulated the expression of TGFB2. ERs at low levels of synthesis were without an effect on the expression of the MYC gene whether or not cells were treated with E2 (Fig. 1c), as the receptors had no effect on the expression of GAPDH, PGR, or CCND1 gene (data not shown). On the other hand, the unliganded ERs at augmented levels effectively modulated the expression of CTSD, CDKN1A, and HBEGF genes, which were further modified with the presence of E2. At the high level of synthesis ERα altered the expression of the TGFB2 gene in the absence or presence of E2. Interestingly, at the high level of synthesis, ERβ gained the ability to regulate the transcription of TGFB2 whether or not cells were treated with E2. We also found that at high levels of synthesis, both ERs in response to E2 altered the expression of the MYC gene (Fig. 1c), without affecting the GAPDH, PGR, or CCND1 gene transcription (data not shown). Thus, unliganded ERs when present at high concentrations can regulate the expression of estrogen responsive genes and also have the potential to broaden their repertoire of gene targets.
Genomic and cellular responses at comparable levels of synthesis of ER subtypes
We then wanted to assess whether at a similar level of receptor synthesis that is dependent on E2 for function ERs would differentially affect genomic and cellular responses as an indication of a subtype-specific mode of action. To examine this issue, we used Ad5 at 600 MOI, Ad5–ERα at 100 MOI, which was brought to 600 MOI by supplementing with 500 MOI of Ad5, and Ad5–ERβ at 600 MOI in subsequent experiments to comparatively assess the role of E2 on ER subtype-mediated responses of MDA-MB-231 cells. We analyzed functional protein synthesis in a timedependent manner with WB and electrophoretic mobility shift assay (EMSA) (Fig. 2a, b). Although the synthesis of ERβ is somewhat delayed compared to that observed with ERα at early time points examined, both ERs reached to comparable levels at 48 h post-infection (Fig. 2a). This was reflected in EMSA, which showed that ERα and ERβ also interacted in vitro similarly with a 32P-end labeled DNA fragment bearing the consensus ERE (Fig. 2b). Moreover, ERs were localized in the nuclei of infected cells such that at 48 h post-infection nearly all cells showed nuclear staining for the receptors (Fig. 2c). Furthermore, 3H-E2 was similarly retained in cells infected with Ad5–ERα and Ad5–ERβ but not the parent Ad5 (Fig. 2d). The cellular retention of 3H-E2 is ER-specific, because the incubation of cells with ER antagonist ICI effectively reduced the retention of 3H-E2 in cells synthesizing an ER subtype. Thus, MDA-MB-231 cells infected with these selected concentrations of recombinant adenoviruses synthesize comparable levels of functional ERs.
Figure 2.

Fig. 2 Synthesis of functional ERs. For Western blot (WB) (a) and Electrophoretic Mobility Shift Assay (EMSA) (b), MDA-MB-231 cells were infected with the parent recombinant adenovirus (Ad5) at MOI 600, a recombinant adenovirus bearing ERβ cDNA at 600 MOI or ERα cDNA at 100 MOI together with 500 MOI of Ad5 to equalize the total adenovirus concentration. a Infected cells were collected at indicated times, and cell extracts (10 μg) were subjected to WB. Molecular mass in KDa is indicated. b For EMSA, 10 μg extracts of infected cells for the indicated times were incubated with radiolabeled DNA fragment containing the consensus ERE sequence for 1 h in the absence or presence of a flag antibody. Reactions were subjected to 5% non-denaturing PAGE. The gel was dried and exposed to PhosphorImager. Free depicts unbound ERE. ER–ERE indicates the radiolabeled ERE-bound-ERs in the absence or presence of the antibody. c For immunocytochemistry, infected cells were probed with a fluorescein-conjugated Flag antibody (FITC) at 48 h postinfection. DAPI was used to stain nuclei. d For in situ 3H-E2 Binding Assay, cells were infected with adenoviruses in the absence E2 for 48 h. Cells were then incubated in fresh medium containing 10−7 M of 3H-E2 for 1 h. Cells were collected, and the radioactivity retained in cells was quantified by a scintillation counter. The specific retention of 3H-E2 in cells by ERs in cells was assessed by the incubation of cells with 10−6 M ICI. The graph represents the mean ± SEM of three independent experiments performed in duplicate. e MDA-MB-231 cells were infected with recombinant adenoviruses in the absence of E2 for 48 h. Cells were then incubated in fresh medium with (E2) or without (−) 10−9 M E2 for 24h. Total RNA was subjected to qPCR. Results for the expression of the CCNA1 (Cyclin A1), TGFA (Transforming growth factor α), CRISPLD2 (Cysteine-rich secretory protein LCCL domain-containing 2), C3 (Complement component 3), TFF1 (Trefoil Factor 1), CTGF (Connective tissue growth factor), PLAT (tissue-type plasminogen activator), IL1B (Interleukin 1b), DKK1 (Dickkopf-1), and HAS2 (Hyaluronan synthase-2) genes are the mean ± SEM of three independent determinations in duplicate. Responses are given as fold change compared to response observed with cells infected with the parent Ad5 (600 MOI) in the absence of E2, which was set to one. Superscript (a) denotes responses that are significantly different from responses observed with cells infected with Ad5 in the absence of E2. Superscript (b) indicates responses that are significantly different from those observed with cells synthesizing ERs in the absence of E2. Superscript (c) indicates responses that are significantly different from responses to ERβ + E2
To examine the effects of ER subtypes on the transcription of estrogen responsive genes, we infected MDAMB-231 cells with recombinant adenoviruses in the absence of E2 for 48 h to allow the synthesis of ERs to reach to similar levels (Fig. 2a). We then treated cells with 10−9 M E2 for 24 h. Total RNA was subjected to quantitative PCR (qPCR). Results revealed that in addition to genes described in Fig. 1c, ERs in response to E2 induced the expression of the CCNA1 (Cyclin A1), TGFA (Transforming growth factor α), CRISPLD2 (Cysteine-rich secretory protein LCCL domain-containing 2), C3 (Complement component 3), and TFF1 (Trefoil Factor 1) genes with ERα being more potent transregulator than ERβ (Fig. 2e). While both receptors in the presence of E2 repressed the expression of the DKK1 (Dickkopf-1) and HAS2 (Hyaluronan synthase-2) genes to a similar extent, the expression of the CTGF (Connective tissue growth factor), PLAT (tissue-type plasminogen activator), or IL1B (Interleukin 1β) gene was more pronounced with E2–ERα than E2–ERβ. Thus, while ERs at comparable levels that are dependent upon the presence of E2 for function regulate gene expression similarly, ERα is a more potent regulator than ERβ.
Effects of E2–ERs on the proliferation and the cell cycle distribution of infected MDA-MB-231 cells
To address how these E2–ER-mediated genomic responses are correlated with alterations in phenotypic features of infected cells, we initially examined the effects of E2 on cellular proliferation mediated by ERs. MDA-MB-231 cells were infected with recombinant adenoviruses in the absence or presence of 10−9 M E2. Cellular proliferation as a function of time was assessed by cell counting (Fig. 3a) and MTT assay (Fig. 3b). The E2 treatment had no effect on the growth of MDA-MB-231 cells infected with the parent Ad5. On the other hand, both ERα and ERβ in response to E2 repressed the cell growth in a time-dependent manner. As observed with gene expression (Fig. 2), E2–ERα was more effective than E2–ERβ in attenuating cellular growth.
Figure 3.

Fig. 3 ERs in the presence of E2 alter the proliferation of MDA-MB-231 cells. Cells were infected with the parent recombinant adenovirus (Ad5) at MOI 600, a recombinant adenovirus bearing ERβ cDNA at 600 MOI or ERα cDNA at 100 MOI together with 500 MOI Ad5 to equalize the total adenovirus concentration in the absence or presence of 10−9 M E2 for various durations of time. At termination, cells were counted using a hemacytometer (a) or subjected to the MTT assay (b). The graphs represent the mean of three independent experiments performed in duplicate. Superscript (a) indicates significant difference from the parent Ad5 in the absence of E2 at the corresponding time point. Superscript (b) indicates responses that are significantly different from ERs in the absence of E2 at the corresponding time point. Superscript (c) denotes responses that are significantly different from those observed with ERβ + E2 at the corresponding time point. c and d Infected MDA-MB-231 cells were subjected to fluorescenceactivated cell sorting (FACS) as a function of time. c Shown are histograms at 48 h post-infection, with the mean ± SEM that indicates percentage of cells in G1, S, and G2 phases from three independent determinations. d Graph depicts the percentage of cells in G1 phase at various time points. Superscript (a) indicates responses that are significantly different from those observed with cells infected with Ad5 in the absence of E2 at the corresponding time point. Superscript (b) denotes responses that are significantly different from responses to ERs in the absence of E2 at the corresponding time point. Superscript (c) indicates responses that are significantly different from those observed with ERβ + E2
The impairment of cell growth by E2–ERα and E2–ERβ was expected to reflect alterations in cell cycle progression and/or apoptosis. To examine this issue, MDA-MB-231 cells were infected with adenoviruses and treated with or without 10−9 M E2 for various durations of time (Fig. 3c, d). Cells were then subjected to fluorescence-activated cell sorting (FACS) for the analysis of cell cycle progression. Results are summarized as cell numbers distributed in G1 phase as a function of time (Fig. 3d) for which representative histograms depicting the percentage of cells accumulated in G1, S, and G2 phases at 48 h post-infection are also shown (Fig. 3c). We found that both ERs only in response to 10−9 M E2 increased the number of cells accumulated in G1 phase. This was inversely correlated with a decrease in cell numbers in S and G2 phases (data not shown). As observed with cellular growth, E2–ERα was more potent than E2–ERβ in altering progression through cell cycle phases. responses, we used a TUNEL assay, which is designed to detect the fragmented genomic DNA as late stages of apoptosis [34]. MDA-MB-231 cells infected with Ad5 or a recombinant adenovirus expressing an ER subtype cDNA were maintained in the absence or presence of 10−9 M E2 for various lengths of time. At termination, cells were subjected to the TUNEL assay (Fig. 4). DAPI staining is used to define the cell nucleus, while the incorporation of FITC-conjugated nucleotide into DNA indicates the fragmented genome. Shown are images at 96 h post-infection (Fig. 4a) and depicted also as number of cells stained with FITC as a function of time (Fig. 4b). Results demonstrated that ERs only in the presence of E2 induced the fragmentation of genomic DNA. We found that E2–ERα was more effective than E2–ERβ in inducing apoptosis. Thus, these results indicate that the ER-mediated repression of cellular proliferation involves alterations in cell cycle progression and apoptosis, the extent of which are more pronounced with E2–ERα than E2–ERβ.
Figure 4.
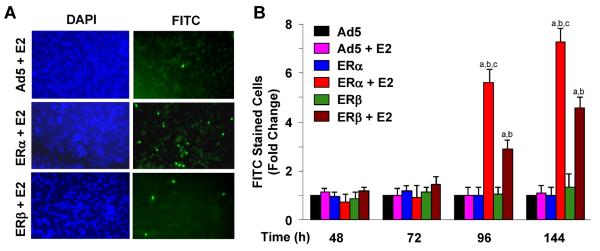
Fig. 4 ERs in the presence of E2 induce apoptosis of MDA-MB-231 cells. Cells were infected with recombinant adenoviruses as a function of time in the absence or presence of 10−9 M E2. Infected cells were subjected to a TUNEL assay. a Representative TUNEL images at 96 h post-infection are shown. FITC indicates apoptotic cells that incorporated the FITC-conjugated dUTP into the fragmented DNA. DAPI was used to stain cell nuclei. b Graph, which is the mean ± SEM of three independent experiments, depicts the number of cells stained with FITC at different time points. Superscript (a) denotes responses that are significantly different from those observed with cells infected with the parent Ad5 in the absence of E2. Superscript (b) indicates responses that are significantly different from responses to ERs in the absence of E2 at the corresponding time point. Superscript (c) denotes responses that are significantly different from responses to ERβ + E2
Effects of E2–ERs on the proliferation and the cell cycle distribution of infected U-2OS cells
The differences in potencies of ER subtypes to alter cellular growth were also evident in U-2OS cells. Infection with increasing concentrations of recombinant adenovirus led to increasing amounts of ER synthesis assessed by WB (Fig. 5a). We found that ERs at low levels of synthesis required E2 to induce a repression in cellular growth (Fig. 5b). On the other hand, unliganded ERs at high levels of synthesis effectively suppressed proliferation, which was further repressed with the presence of 10−9 M E2. To examine the effects of comparable levels of ERs (Fig. 5c) that relied on E2 for function to affect cellular growth, we infected cells with the parent adenovirus (Ad5) and Ad5–ERβ at 60 MOI. We used Ad5–ERα at 40 MOI, which was brought to 60 MOI by supplementing with 20 MOI of Ad5. Our results further revealed that at 48 h postinfection ERα in the presence of E2 was more potent in repressing S and G2 phases with a corresponding increase in G1 phase than the E2–ERβ complex (Fig. 5d, e). This was reflected in cellular growth, which was robustly repressed by E2–ERα than E2–ERβ at day 6 of postinfection (Fig. 5f).
Figure 5.

Fig. 5 ERs repress the growth of U-2OS cells. a Cells were infected with various concentrations of adenoviruses. In all infections, the total recombinant adenovirus concentration was brought to 80 MOI with the inclusion of an appropriate MOI of the parent Ad5 as indicated. Cells extracts (10 μg) were subjected to 8–16% SDS-PAGE for WB using the horsERαdish peroxidase-conjugated monoclonal Flag antibody. Molecular mass in KDa is indicated. b Infected cells were treated with or without 10−9 M E2. At day 6, cells were subjected to cell counting. The graph represents the mean ± SEM of three independent experiments performed in duplicate. Superscript (a) denotes responses that are significantly different from those observed with cells infected with the parent Ad5 in the absence of E2. Superscript (b) indicates responses that are significantly different from responses to ERs in the absence of E2. c U-2OS cells were infected with Ad5 and Ad5–ERβ at 60 MOI, whereas Ad5–ERα was used at 40 MOI, which was supplemented with 20 MOI of Ad5 to equalize the total adenovirus concentration. At 48 h post-infection, cells were subjected to WB. Molecular mass in KDa is indicated. d and e Infected cells in the absence or presence of 10−9 M E2 were also subjected to FACS. d Histograms depict the distribution of cell cycle phases at 48 h post-infection in the presence of E2. e Quantitative analysis of histograms is depicted as graph, in which the mean ± SEM indicates percent cells in G1, S, and G2 phases from three independent determinations performed in duplicate. Superscript (a) denotes responses that are significantly different from those observed with cells infected with the parent Ad5 in the absence of E2. Superscript (b) indicates responses that are significantly different from responses to ER in the absence of E2. Superscript (c) denotes responses that are significantly different from ERβ in the presence of E2. f Infected cells in the absence or presence of 10−9 M E2 as described in legend of c were subjected to cell counting at day 6 postinfection. Changes in cell numbers are given as the percent change compared to cells infected with the parent Ad5, which was set to 100. Shown is the mean ± SEM of three independent experiments performed in duplicate. Superscript (a) indicates responses that are significantly different from those observed with cells infected with Ad5 in the absence of E2. Superscript (b) indicates responses that are significantly different from responses to ER in the absence of E2. Superscript (c) denotes responses that are significantly different from ERβ in the presence of E2
ER-induced changes in cellular motility
We [20, 21] and others [11, 35] showed that ER-negative MDA-MB-231 cells that synthesize ectopically introduced ERs are less motile and invasive than the parent cells. To comparatively assess the effects of E2 on ER-mediated cellular motility, we used wound-healing and invasion assays. For the wound-healing, MDA-MB-231 cells infected with recombinant adenoviruses in the absence or presence of 10−9 M E2 were maintained for 48 h to allow cells to reach confluence. A wound was then created, and the closure of wound was observed 24-h intervals in the absence or presence of 10−9 M E2 (Fig. 6a, b). We found that the unliganded ERs delayed the wound closure compared to Ad5, which was further delayed by the E2 treatment. However, ERα was more effective in delaying the wound closure than ERβ. Since the wound-healing assay encompasses both cell proliferation and migration, we also used an invasion assay to independently verify that E2–ERs affect cell motogenesis. The invasion assay evaluates the capacity of cells to migrate through a reconstituted basement membrane (BM). MDA-MB-231 cells were infected withrecombinant adenoviruses in the absence or presence of 10−9 M E2 for 48 h. Cells were then subjected to the invasion assay for 24 h (Fig. 6c, d). Results revealed that both ERs in the presence of E2 reduced the invasive ability of infected cells. Moreover, E2–ERβ was less effective than E2–ERα in decreasing the invasiveness of the infected cells.
Figure 6.
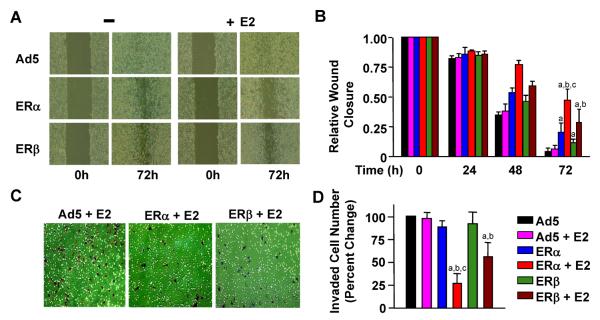
Fig. 6 Effects of ERs on the motility of infected MDA-MB-231 cells. Infected cells were incubated in the absence or presence of 10−9 M E2 for 48 h to allow cells to reach confluence. A wound was then created and images were captured at 0 and 24-h intervals. a Representative images of wound closure in response to Ad5, ERα, or ERβ in the absence (−) or presence of E2 (+E2) at 72 h are shown. b Graph represents the quantitative analysis of images that depict the wound closure at different time points relative to 0 h. Superscript (a) indicates responses that are significantly different from those observed with cells infected with Ad5 in the absence of E2. Superscript (b) denotes responses that are significantly different from responses to ERs in the absence of E2, while superscript (c) indicates responses that are significantly different from responses to ERβ + E2. c MDA-MB-231 cells were infected with recombinant adenoviruses in the absence or presence of 10−9 M E2 for 48 h. Cells were collected and counted. The same number of cells from each experimental group was then seeded on the invasion chamber membrane. After 24 h incubation, cells with invasive capabilities on the bottom of chamber membrane were stained (brown) and imaged. d Cells counted on the bottom of the invasion chamber are given as the percent change in cell numbers relative those infected with the parent Ad5, which is set to 100. Each data point represents the mean ± SEM of three independent experiments performed in duplicate. Superscript (a) indicates responses that are significantly different from those observed with cells infected with Ad5 in the absence of E2. Superscript (b) denotes responses that are significantly different from those observed with cells synthesizing ERs in the absence of E2. Superscript (c) denotes responses that are significantly different from responses to ERβ + E2
Discussion
The regulation of responsive gene expression by E2–ERα is critical for cellular proliferation, motility, and death [6–10,15]. ERβ, like ERα, is largely dependent upon E2 to modulate gene expressions in experimental systems [11,16, 18]. However, the unliganded ERβ was also shown to induce phenotypic alterations in cancer cell models [11–15,17]. These findings suggested that ERβ induces cellular responses in a manner that differs from ERα [8, 11–13, 17,19]. Studies have indicated that elevated levels of the unliganded ERα can regulate gene expression and cellular proliferation [22, 23]. We, therefore, predicted that augmented levels of ERβ synthesis could underlie the ability of the unliganded ERβ to alter gene transcriptions and cellular proliferation. Our results here indicate that high levels of ERβ modulate the expression of genes and cellular proliferation by circumventing the dependency of the receptor on E2.
The underlying mechanism(s) by which unliganded ERs mediate gene expression is unclear. The ligand-binding domains (LBDs) of ERs share a high degree of amino-acid identity that results in an analogous tertiary and quaternary architecture [36, 37]. The LBDs display a fold with 12 a helices, numbered H1–H12, and one b-turn, which are arranged as a three-layered anti-parallel topology. The two parallel outer layers sandwich a central layer. This arrangement of helices forms the ligand-binding cavity. In addition to E2 binding, the LBDs are involved in dimerization and transactivation function of the receptors. In the unliganded state, ERs exhibit an open ligand-binding cavity while the highly mobile H12 extends away from the LBD [38–40]. The binding of E2 to ERs is accompanied by a major reorganization of the tertiary structure of the LBD. The H12 rotates back on the ligand-binding cavity, thereby effectively burying E2 [38, 40]. Studies suggest that this E2-mediated re-positioning of H12 together with conformational changes in H3, 6 and 11 of, at least ERα, is critical for the enhanced affinity of the receptor for coregulatory proteins [41, 42] as well as the stability [43, 44] and in situ interactions of the ERα dimer with DNA [33, 45–47]. Various in vitro and in situ approaches have also showed that unliganded ERα and ERβ interact, albeit inefficiently, with DNA and co-regulatory proteins [33, 41, 45, 46, 48], likely facilitated by the positional variability of the highly mobile H12 resembling to that induced by E2 binding [41]. However, the DNA bound unliganded ERα complex is transcriptionally silent due to the inability of the complex to recruit some components of the basal transcription factors and RNA polymERαse II [49]. It is, therefore, possible that at augmented levels of receptor synthesis, mass action could enhance the efficiency and/or rate of direct or indirect interactions of the unliganded ERβ, as ERα, with DNA and co-regulatory proteins as suggested [22, 23], and also the basal transcription machinery and RNA polymERαse II. This could result in the expression of responsive genes, although at levels lower than those observed with liganded ERs. Moreover, elevated levels of ERs in response to E2 could also expand the range of responsive genes by binding to low affinity DNA binding sites and modulating the transcriptional output, as exemplified here with the MYC gene by both ERs [50]. Regardless of the mechanism(s), our results indicate that the unliganded ERs at high levels of synthesis similarly modulate gene expression and cellular growth. Augmented levels of ERs could pose challenges for the endocrine treatment of breast or bone tissue disorders, as ER-positive breast tumors expressing the high levels of ERα are associated with poor prognosis and decrease responses to endocrine therapies [22, 23, 51, 52]. Alternative, or additional, approaches that target ER synthesis and/or turnover could be novel avenues in combating estrogen target tissue malignancies. ERs at levels that required E2 for function also induced phenotypic alterations with a similar pattern. E2 regulates cell proliferation through key processes that control the entry into and progression through G1 phase [53–55]. Our results demonstrate that ERs in response to a physiological level of E2 decreased cell proliferation by repressing S and G2 phases, which was manifested as increased number of cells accumulated in G1 phase. The decrease in cell proliferation was associated with an increase in programmed cell death as ERs in response to E2-induced DNA fragmentation. Thus, the regulation of cell growth by E2–ERs involves similar mode of actions that encompass both proliferation and apoptosis.
The invasion process plays a crucial role in tumor metastasis. This includes the adhesion of tumor cells to the BM, enzymatic digestion of the BM by proteolytic enzymes followed by migration through the extracellular matrix, and the subsequent growth and proliferation of cells at a new site [54]. Studies indicate that E2 signaling in ER positive cells represses the cellular motility [54, 55]. Extending our previous observations [20, 21], we show here that E2–ERs similarly repress migration and invasiveness of infected cells. Although ERα and ERβ induced similar genomic responses and phenotypic alterations, our results also indicate that the extent of changes is ER subtype-specific in that ERα is a more potent regulator than ERβ. Studies using global gene expression approaches indicated that the transcription of genes mediated by ERα and ERβ shows an overlapping as well as a distinct pattern [7, 8, 10, 16, 20, 21]. A differential regulation of gene expression is certainly one explanation for the differences in the regulatory potential of ERs in inducing cellular alterations. Differences in the magnitude of transcription, as we observed here with a limited number of model genes, could also be critical for the extent to which an ER subtype evokes cellular responses. One conical E2 signaling pathway involves the interaction of ERs with EREs. This pathway is referred to as the ERE-dependent signaling pathway. Studies have established that E2–ERβ is a weaker transregulator than E2–ERα in heterologous reporter systems that emulate the ERE-dependent signaling pathway [41, 56–60]. We previously showed that the expression of the E2 responsive C3, CCNA1, CDKN1A, CTSD, IL1B, RARA, TGFA, and TGFB2 genes requires ER–ERE intERαctions in MDAMB- 231 cells as the ERE binding defective ERs do not affect transcription of these genes [20, 21]. We found here that ERα also regulated the endogenous gene expression through ERE-dependent signaling pathway more potently than ERβ whether or not E2 was present.
What could account for the difference in transcriptional potencies of human ER subtypes? ERs are modular in nature such that isolated structural domains function independently [1, 3]. It is unlikely that the conserved LBDs are responsible for the differences in transcriptional potencies of ER subtypes. This is because the activities of the LBDs of ERs from the ERE-dependent signaling pathway appear to be comparable due to the abilities of the receptors to interact with co-regulatory proteins with similar affinities [56, 57, 59, 60]. In addition to the LBDs, the DNA-binding domains (DBDs) are highly conserved which show a near identical (97%) amino-acid homology that allows ERs to bind to the same spectrum of ERE sequences with similar affinities [41] and utilizing the same nucleotides [41, 61]. It appears that the least conserved (17% amino-acid homology) amino-terminal A/B domains of ERs contribute to the ability of ER subtypes to interact with and subsequently regulate transcription from an ERE in situ [33, 62, 63]. The A/B domain of ERα contains an activation function (AF1) that opERαtes independently as well as in cooperation with the carboxyl-terminus in a cell and promoter context-dependent manner [60, 64–68]. The ability of the A/B domain to recruit [41, 69] and exchange [70] co-regulatory proteins is critical for AF1 but also for the functional integration of both AF1 and AF2 of ERα to mediate transcription at full capacity in response to E2 [60, 65, 71]. In contrast to ERα, the amino-terminus of human ERβ impairs the receptor–ERE interactions [33, 62], lacks an activation function [56, 59, 60, 68], and is incapable of interacting with the carboxyl-terminus [60]. Thus, the distinct amino-terminus appears to define the differences in the magnitude of genomic responses mediated through ERE-dependent E2–ER signaling pathway. Although ERs interact with the same spectrum of EREs utilizing the same nucleotides, ERβ, nevertheless, binds to an ERE with a two-fold lower affinity than ERα [41]. In addition to the functional differences in the amino-termini, differences in affinities to a responsive element could also contribute to the observed ER subtype-selectivity of some of the estrogen responsive genes as exemplified here with the expression of the TGFB2 gene, which we previously showed to be regulated by ERα through an ERE-dependent signaling pathway [21]. We observed here that the transcription of TGFB2 was responsive to ERα at both low and high concentrations, while ERβ mediated the expression only at high levels (Fig. 1c). The distinct amino-termini of ERs appear also to contribute to the differences in the potencies of ER subtypes to regulate gene transcription mediated through an ERE-independent signaling pathway. In addition to ERE-dependent signaling, E2–ER regulates gene expression through functional interactions with transfactors bound to their cognate regulatory elements on DNA, exemplified by activator protein 1 (AP1) and stimulatory protein 1 (Sp1) [4, 5]. The functional interaction involves the stabilization of transcription factor binding to DNA and/or recruitment of co-regulatory proteins to the complex in an ER subtype, promoter-, and cell-context-dependent manner [4, 5]. Studies in cell models derived from breast adenocarcinoma suggest that the magnitude of responses from heterologous reporter systems to ERα is greater than those observed with ERβ [61, 72, 73]. Similarly, the extent of E2 responsive gene expression mediated by Sp1-ER intERαctions appears to be different for ERs [73–75]. Although cis-elements and trans-acting factors involved in E2 responsiveness remain to be elucidated, we previously reported that ERs in response to E2 regulate the expression of the CRISPLD2, CTGF, DKK1, HAS2, HBEGF, and PLAT genes through an ERE-independent signaling pathway [20, 21]. With the exception of the DKK1 and HAS2 gene expressions, which were regulated by ERs to a similar extent, we observed here that ERα is also more potent regulator than ERβ in modulating the transcription of CRISPLD2, CTGF, HBEGF, and PLAT genes. Thus, differences in the potencies of the E2–ER complexes to modulate the gene expression through the ERE-dependent and ERE-independent signaling pathways could underlie differences in the magnitude of cellular responses to an ER subtype.
In summary, our results suggest that ERs induce genomic and phenotypic changes similarly. Despite the functional commonality, ERs display differences in the potencies in conveying E2 signaling in cells such that ERα is a more effective regulator than ERβ.
Materials and methods
Generation of recombinant adenoviruses
The human ERα and ERβ cDNAs encoding 595 and 530 amino-acid long ERα and ERβ, respectively, were described previously [60, 76]. These cDNAs also bear sequences that encode an amino-terminal Flag epitope [33, 61]. Adenoviruses bearing none, the cDNA for Flag-ERα, or Flag-ERβ were produced using the AdEasy-XL Adenoviral System (Stratagene, La Jolla, CA) as described previously [33]. The purified viruses were titered using an Adeno-X Rapid Titer Kit (BD Biosciences, Palo Alto, CA) to determine MOI. Restriction and DNA modifying enzymes were obtained from New England Bio-Labs (Beverly, MA) and Invitrogen Corp., (Carlsbad, CA).
Cell culture
ER-negative MDA-MB-231 cells derived from a breast adenocarcinoma (ATCC, Manassas, VA) were grown in DMEM containing 10% fetal bovine serum (FBS) (Invitrogen Corp). ER-negative U-2OS cells derived from osteosarcoma were purchased from ATCC (Manassas, VA). U-2OS cells were grown in McCoy's 5a medium (Hyclone, Logan, UT) supplemented with 10% FBS. Cell phenotypic assays were carried out in phenol red-free medium containing 10% charcoal dextran-treated FBS (CD-FBS) (Hyclone, Logan, UT) with or without a physiological concentration (10−9 M) of 17β-estradiol (E2). In all the experiments, media were changed at every third day when appropriate. Media were purchased from Invitrogen Corp. 17β-estradiol (E2) was obtained from Sigma-Aldrich(St. Louis, MO), and ICI 182,780 (ICI) was purchased from Tocris Inc. (Ballwin, MO).
Western blot and electrophoretic mobility shift assay
Cells (100,000 cells/well) were plated into 6-well tissue culture plates in phenol red-free media supplemented with 10% CD-FBS for 24 h before infection. Cells were then infected with various MOIs of recombinant adenoviruses for varying lengths of time. Cells were collected and processed for WB analysis and EMSA as detailed previously [20, 21]. For WB, proteins were probed with the horseradish peroxidase-conjugated monoclonal Flag antibody (M2-HRP, Sigma-Aldrich) using the ECL-Plus Western Blotting kit (Amersham-Pharmacia). For EMSA, we used Flag M2 antibody (Sigma-Aldrich) to assess the specificity of receptor–ERE interactions. Images from WB and EMSA were analyzed by a PhosphorImager (Storm 865, GE Life Sciences, Piscataway, NJ) and were quantified with an ImageQuant (GE Life Sciences).
In situ E2 binding assay and immunocytochemistry
The in situ E2 binding assay and immunocytochemistry (ICC) of infected cells were carried out as described previously [33, 60]. In brief, cells infected with recombinant adenoviruses at various durations were incubated with 10−7 M of [2,4,6,7,16,17–3H] 17β-estradiol (118 Ci/mmol, NEN Life Sciences, Boston,MA) in the absence or presence of 10−6 M ICI for 1 h. Cells were then washed extensively with PBS and collected. Radioactivity remaining in cells was measured in a scintillation counter. For immunocytochemistry, infected cells were probed with the Flag M2 antibody (Sigma-Aldrich) and a fluorescein-conjugated secondary antibody (Santa Cruz) for visualization.
Endogenous gene expression
To examine the effects of low and high concentrations of ER subtypes on endogenous gene expression in the absence or presence of 10–9 M E2, cells (100,000 cells/well) were infected with varying MOIs of recombinant adenoviruses for 48 h. At the termination, cells were collected and subjected to total RNA extraction using the RNeasy Mini Kit (Qiagen, Valencia, CA). Total RNA was processed for and subjected to qPCR, using custom TaqMan Low-Density Arrays (Applied Biosystems, Foster City, CA, USA) as we described previously [20, 21]. Relative Quantitation Analysis was performed using the Comparative CT Method [77]. To assess the effects of E2 on endogenous gene expression mediated by a comparable level of ER subtypes that required E2 for function, infected cells were maintained in the absence of E2 for 48 h, at a time which the synthesis of ERs reaches maximal and similar levels (Fig. 1). Cells were then treated without or with 10−9 M E2 for 24 h. Total RNA was subjected to qPCR.
Cell proliferation and cell cycle analysis
For cell proliferation studies, MDA-MB-231 (5,000 cells/well) or U-2OS cells (2,500 cells/well) were plated into 24-well tissue culture plates in phenol red-free medium containing 10% CD-FBS for 24 h. Cells were then infected with recombinant adenoviruses in the absence or presence of 10–9 M E2 for different durations of time. Cells were then subjected to cell counting or MTT assay [20, 21]. For cell cycle analysis, infected cells (100,000 cells/well) were grown in the absence or presence of 10–9 M E2 for different durations. Cells were subjected to cell cycle analysis as described previously [20, 21]. Cell cycle distribution of cells was determined using EPICS Elite (Coulter Corp, Miami, FL).
TUNEL assay
To study the late stages of apoptosis by examining the fragmentation of genomic DNA, we used a Terminal dUTP Nick-End Labeling (TUNEL) assay utilizing the DeadEnd Fluorometric TUNEL System (Promega). Infected cells (25,000 cells/well of a 24-well plate) at different durations were fixed with 4% formaldehyde and subsequently permeabilized with 0.2% Triton X-100 to carry out the TUNEL reaction [20, 21].
Wound-healing and invasion assays
Wound-healing and invasion assays were carried out as detailed previously [20, 21]. In brief, MDA-MB-231 cells (200,000 cells/well) were plated into 12-well tissue culture plates in phenol red-free DMEM containing 10% CD-FBS for 24 h. Cells were then infected with recombinant adenoviruses in the absence or presence of 10−9 M E2 for 48 h to allow cells to reach confluence. A wound was then created using a 1-ml pipette tip. The gap closure was photographed at every 24 h. Generated wound edges show irregular shapes. To compensate for this, we randomly selected five cross edges to obtain a mean gap measurement for wound-healing. For invasion assay, MDA-MB-231 cells (100,000 cells) infected with recombinant adenoviruses in the absence or presence of 10–9 M E2 were grown for 48 h. Cells were then collected and counted. 25,000 cells from eachChambers (BD Biosciences, San Diego, CA), which contained only phenol red-free DMEM with or without 10−9 M E2. The lower section of the chamber contained phenol red-free DMEM supplemented with 10% CD-FBS and 30 μg/ml fibronectin (Sigma-Aldrich) in the absence or presence of E2. After 24h incubation, cells on the bottom of the chamber membrane as the population capable of invasion were stained with the Diff-Quik Stain Set (Dade Behring, Newark, DE). Images were captured by a light microscope. Stained cells were counted from the images.
Statistical analysis
Results were presented as the mean ± SEM of, at least, three biological replicates. Significance was determined using two-way ANOVA with Tukey's HSD post-test. P<0.05 was considered significant.
Acknowledgment
We thank Michelle Zanche and Meghann McBennett of the Functional Genomic Center at the University of Rochester for the incessant guidance, assistance, and execution of qPCR. We are also grateful to Dr. Tim Bushnell and Matthew Cochran of the Cell Separation and Flow Cytometry Facility of the University of Rochester for the supervision and assistance for FACS studies. We thank Mark Gallagher for the critical reading of the manuscript. This study was supported by NIH CA113682 (MM).
Abbreviations
- E2
17β-estradiol
- ERα
Estrogen Receptor α
- ERβ
Estrogen Receptor β
- ERE
Estrogen Response Element
- LBD
Ligand Binding domain
- AF-1
Activation function-1
- AF-2
Activation function-2
- CMV
Cytomegalovirus
- EMSA
electrophoretic mobility shift assay
- 4-OHT
4-hydroxytamoxifen
- PCR
quantitative PCR
- ACTB
b-actin
- C3
Complement component 3
- CTSD
Cathepsin D
- CCNA1
Cyclin A1
- CCND1
Cyclin D1
- CDKN1A
Cyclin-dependent kinase inhibitor 1A, p21, Cip1
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- IL1B
Interleukin 1β
- MMP1
Matrix metallopeptidase 1, Collagenase
- PGR
Progesterone receptor
- RARA
Retinoic acid receptor α
- TFF1
Trefoil factor 1, pS2
- TGFA
Transforming growth factor α
Footnotes
Conflict of interest The authors have nothing to disclose.
References
- 1.Huang J, Li X, Hilf R, Bambara RA, Muyan M. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005;5:379–396. doi: 10.2174/156800805774912944. [DOI] [PubMed] [Google Scholar]
- 2.Deroo BJ, Korach KS. J. Clin. Invest. 2006;116:561–570. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall JM, Couse JF, Korach KS. J. Biol. Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 4.Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. J. Steroid Biochem. Mol. Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- 5.Safe S. Vitam. Horm. 2001;62:231–252. doi: 10.1016/s0083-6729(01)62006-5. [DOI] [PubMed] [Google Scholar]
- 6.Soulez M, Parker MG. J. Mol. Endocrinol. 2001;27:259–274. doi: 10.1677/jme.0.0270259. [DOI] [PubMed] [Google Scholar]
- 7.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 8.Licznar A, Caporali S, Lucas A, Weisz A, Vignon F, Lazennec G. FEBS Lett. 2003;553:445–450. doi: 10.1016/s0014-5793(03)01090-1. [DOI] [PubMed] [Google Scholar]
- 9.Moggs JG, Murphy TC, Lim FL, Moore DJ, Stuckey R, Antrobus K, Kimber I, Orphanides G. J. Mol. Endocrinol. 2005;34:535–551. doi: 10.1677/jme.1.01677. [DOI] [PubMed] [Google Scholar]
- 10.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Endocrinology. 2006;147:4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 11.Lazennec G, Bresson D, Lucas A, Chauveau C, Vignon F. Endocrinology. 2001;142:4120–4130. doi: 10.1210/endo.142.9.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tonetti DA, Rubenstein R, DeLeon M, Zhao H, Pappas SG, Bentrem DJ, Chen B, Constantinou A, Craig Jordan V. J. Steroid Biochem. Mol. Biol. 2003;87:47–55. doi: 10.1016/j.jsbmb.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Cheng J, Lee EJ, Madison LD, Lazennec G. FEBS Lett. 2004;566:169–172. doi: 10.1016/j.febslet.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 14.Hou YF, Yuan ST, Li HC, Wu J, Lu JS, Liu G, Lu LJ, Shen ZZ, Ding J, Shao ZM. Oncogene. 2004;23:5799–5806. doi: 10.1038/sj.onc.1207765. [DOI] [PubMed] [Google Scholar]
- 15.Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Cancer Res. 2004;64:423–428. doi: 10.1158/0008-5472.can-03-2446. [DOI] [PubMed] [Google Scholar]
- 16.Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, Katzenellenbogen BS. Endocrinology. 2004;145:3473–3486. doi: 10.1210/en.2003-1682. [DOI] [PubMed] [Google Scholar]
- 17.Martineti V, Picariello L, Tognarini I, Carbonell Sala S, Gozzini A, Azzari C, Mavilia C, Tanini A, Falchetti A, Fiorelli G, Tonelli F, Brandi ML. Endocr. Relat. Cancer. 2005;12:455–469. doi: 10.1677/erc.1.00861. [DOI] [PubMed] [Google Scholar]
- 18.Monroe DG, Secreto FJ, Subramaniam M, Getz BJ, Khosla S, Spelsberg TC. Mol. Endocrinol. 2005;19:1555–1568. doi: 10.1210/me.2004-0381. [DOI] [PubMed] [Google Scholar]
- 19.Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. Endocr. Relat. Cancer. 2004;11:537–551. doi: 10.1677/erc.1.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Nott SL, Huang Y, Hilf R, Bambara RA, Qiu X, Yakovlev A, Welle S, Muyan M. J. Mol. Endocrinol. 2008;40:211–229. doi: 10.1677/JME-07-0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nott SL, Huang Y, Li X, Fluharty BR, Qiu X, Welshons WV, Yeh S, Muyan M. J. Biol. Chem. 2009;284:15277–15288. doi: 10.1074/jbc.M900365200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fowler AM, Solodin N, Preisler-Mashek MT, Zhang P, Lee AV, Alarid ET. FASEB J. 2004;18:81–93. doi: 10.1096/fj.03-0038com. [DOI] [PubMed] [Google Scholar]
- 23.Fowler AM, Solodin NM, Valley CC, Alarid ET. Mol. Endocrinol. 2006;20:291–301. doi: 10.1210/me.2005-0288. [DOI] [PubMed] [Google Scholar]
- 24.Garcia M, Derocq D, Freiss G, Rochefort H. Proc. Natl. Acad. Sci. U S A. 1992;89:11538–11542. doi: 10.1073/pnas.89.23.11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang SY, Jordan VC. J. Natl. Cancer Inst. 1992;84:580–591. doi: 10.1093/jnci/84.8.580. [DOI] [PubMed] [Google Scholar]
- 26.Zajchowski DA, Sager R, Webster L. Cancer Res. 1993;53:5004–5011. [PubMed] [Google Scholar]
- 27.Wang M, Dotzlaw H, Fuqua SA, Murphy LC. Breast Cancer Res. Treat. 1997;44:145–151. doi: 10.1023/a:1005753117205. [DOI] [PubMed] [Google Scholar]
- 28.Levenson AS, Jordan VC. J. Steroid Biochem. Mol. Biol. 1994;51:229–239. doi: 10.1016/0960-0760(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 29.Lazennec G, Alcorn JL, Katzenellenbogen BS. Mol. Endocrinol. 1999;13:969–980. doi: 10.1210/mend.13.6.0318. [DOI] [PubMed] [Google Scholar]
- 30.Lazennec G, Katzenellenbogen BS. Mol. Cell. Endocrinol. 1999;149:93–105. doi: 10.1016/s0303-7207(98)00254-8. [DOI] [PubMed] [Google Scholar]
- 31.Barron-Gonzalez A, Castro Romero I. Biochem. Cell Biol. 2004;82:335–342. doi: 10.1139/o03-083. [DOI] [PubMed] [Google Scholar]
- 32.Kian Tee M, Rogatsky I, Tzagarakis-Foster C, Cvoro A, An J, Christy RJ, Yamamoto KR, Leitman DC. Mol. Biol. Cell. 2004;15:1262–1272. doi: 10.1091/mbc.E03-06-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang J, Li X, Maguire CA, Hilf R, Bambara RA, Muyan M. Mol. Endocrinol. 2005;19:2696–2712. doi: 10.1210/me.2005-0120. [DOI] [PubMed] [Google Scholar]
- 34.Gorczyca W, Melamed MR, Darzynkiewicz Z. Methods Mol. Biol. 1998;91:217–238. doi: 10.1385/0-89603-354-6:217. [DOI] [PubMed] [Google Scholar]
- 35.Garcia M, Derocq D, Platet N, Bonnet S, Brouillet JP, Touitou I, Rochefort H. J. Steroid Biochem. Mol. Biol. 1997;61:11–17. doi: 10.1016/s0960-0760(96)00255-5. [DOI] [PubMed] [Google Scholar]
- 36.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 37.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, Moras D, Gronemeyer H. Nat. Struct. Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 39.Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Proc. Natl. Acad. Sci. U S A. 1998;95:5998–6003. doi: 10.1073/pnas.95.11.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pike AC. Best Pract. Res. Clin. Endocrinol. Metab. 2006;20:1–14. doi: 10.1016/j.beem.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Yi P, Driscoll MD, Huang J, Bhagat S, Hilf R, Bambara RA, Muyan M. Mol. Endocrinol. 2002;16:674–693. doi: 10.1210/mend.16.4.0810. [DOI] [PubMed] [Google Scholar]
- 42.Tamrazi A, Carlson KE, Rodriguez AL, Katzenellenbogen JA. Mol. Endocrinol. 2005;19:1516–1528. doi: 10.1210/me.2004-0458. [DOI] [PubMed] [Google Scholar]
- 43.Tamrazi A, Carlson KE, Daniels JR, Hurth KM, Katzenellenbogen JA. Mol. Endocrinol. 2002;16:2706–2719. doi: 10.1210/me.2002-0250. [DOI] [PubMed] [Google Scholar]
- 44.Bai Y, Giguere V. Mol. Endocrinol. 2003;17:589–599. doi: 10.1210/me.2002-0351. [DOI] [PubMed] [Google Scholar]
- 45.Chen HW, Lin RJ, Xie W, Wilpitz D, Evans RM. Cell. 1999;98:675–686. doi: 10.1016/s0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- 46.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 47.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 48.Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Mol. Cell. 2003;11:695–707. doi: 10.1016/s1097-2765(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 49.Metivier R, Penot G, Carmouche RP, Hubner MR, Reid G, Denger S, Manu D, Brand H, Kos M, Benes V, Gannon F. EMBO J. 2004;23:3653–3666. doi: 10.1038/sj.emboj.7600377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dubik D, Shiu RP. Oncogene. 1992;7:1587–1594. [PubMed] [Google Scholar]
- 51.Baehner F, Habel L, Quesenberry C, Capra A, Tang G, Paik S, Wolmark N, Watson D, Shak S. Breast Cancer Res. Treat. 2006;100:S21. [Google Scholar]
- 52.Lopez-Tarruella S, Schiff R. Clin. Cancer Res. 2007;13:6921–6925. doi: 10.1158/1078-0432.CCR-07-1399. [DOI] [PubMed] [Google Scholar]
- 53.Prall OW, Rogan EM, Sutherland RL. J. Steroid Biochem. Mol. Biol. 1998;65:169–174. doi: 10.1016/s0960-0760(98)00021-1. [DOI] [PubMed] [Google Scholar]
- 54.Rochefort H, Platet N, Hayashido Y, Derocq D, Lucas A, Cunat S, Garcia M. J. Steroid Biochem. Mol. Biol. 1998;65:163–168. doi: 10.1016/s0960-0760(98)00010-7. [DOI] [PubMed] [Google Scholar]
- 55.Platet N, Cathiard AM, Gleizes M, Garcia M. Crit. Rev. Oncol. Hematol. 2004;51:55–67. doi: 10.1016/j.critrevonc.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 56.Cowley SM, Parker MG. J. Steroid Biochem. Mol. Biol. 1999;69:165–175. doi: 10.1016/s0960-0760(99)00055-2. [DOI] [PubMed] [Google Scholar]
- 57.McInerney EM, Weis KE, Sun J, Mosselman S, Katzenellenbogen BS. Endocrinology. 1998;139:4513–4522. doi: 10.1210/endo.139.11.6298. [DOI] [PubMed] [Google Scholar]
- 58.Hall JM, McDonnell DP. Endocrinology. 1999;140:5566–5578. doi: 10.1210/endo.140.12.7179. [DOI] [PubMed] [Google Scholar]
- 59.Delaunay F, Pettersson K, Tujague M, Gustafsson JA. Mol. Pharmacol. 2000;58:584–590. doi: 10.1124/mol.58.3.584. [DOI] [PubMed] [Google Scholar]
- 60.Yi P, Bhagat S, Hilf R, Bambara RA, Muyan M. Mol. Endocrinol. 2002;16:1810–1827. doi: 10.1210/me.2001-0323. [DOI] [PubMed] [Google Scholar]
- 61.Li X, Huang J, Yi P, Bambara RA, Hilf R, Muyan M. Mol. Cell. Biol. 2004;24:7681–7694. doi: 10.1128/MCB.24.17.7681-7694.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang J, Li X, Qiao T, Bambara RA, Hilf R, Muyan M. Nucl Recept Signal. 2006;4:e015. doi: 10.1621/nrs.04015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li X, Huang J, Fluharty BR, Huang Y, Nott SL, Muyan M. J. Steroid Biochem. Mol. Biol. 2008;109:266–272. doi: 10.1016/j.jsbmb.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beekman JM, Allan GF, Tsai SY, Tsai MJ, O'Malley BW. Mol. Endocrinol. 1993;7:1266–1274. doi: 10.1210/mend.7.10.8264659. [DOI] [PubMed] [Google Scholar]
- 65.Kraus WL, McInerney EM, Katzenellenbogen BS. Proc. Natl. Acad. Sci. U S A. 1995;92:12314–12318. doi: 10.1073/pnas.92.26.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, Chambon P. Cell. 1989;59:477–487. doi: 10.1016/0092-8674(89)90031-7. [DOI] [PubMed] [Google Scholar]
- 67.Tzukerman MT, Esty A, Santiso-Mere D, Danielian P, Parker MG, Stein RB, Pike JW, McDonnell DP. Mol. Endocrinol. 1994;8:21–30. doi: 10.1210/mend.8.1.8152428. [DOI] [PubMed] [Google Scholar]
- 68.McInerney EM, Tsai MJ, O'Malley BW, Katzenellenbogen BS. Proc. Natl. Acad. Sci. U S A. 1996;93:10069–10073. doi: 10.1073/pnas.93.19.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Webb P, Nguyen P, Shinsako J, Anderson C, Feng W, Nguyen MP, Chen D, Huang SM, Subramanian S, McKinerney E, Katzenellenbogen BS, Stallcup MR, Kushner PJ. Mol. Endocrinol. 1998;12:1605–1618. doi: 10.1210/mend.12.10.0185. [DOI] [PubMed] [Google Scholar]
- 70.Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld MG, Rose DW. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 71.Benecke A, Chambon P, Gronemeyer H. EMBO Rep. 2000;1:151–157. doi: 10.1093/embo-reports/kvd028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Uht RM, Webb P, Nguyen P, Price RH, Jr., Valentine C, Jr., Favre H, Kushner PJ. Nucl Recept. 2004;2:2. doi: 10.1186/1478-1336-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schultz JR, Petz LN, Nardulli AM. J. Biol. Chem. 2005;280:347–354. doi: 10.1074/jbc.M407879200. [DOI] [PubMed] [Google Scholar]
- 74.Saville B, Wormke M, Wang F, Nguyen T, Enmark E, Kuiper G, Gustafsson JA, Safe S. J. Biol. Chem. 2000;275:5379–5387. doi: 10.1074/jbc.275.8.5379. [DOI] [PubMed] [Google Scholar]
- 75.Kim K, Thu N, Saville B, Safe S. Mol. Endocrinol. 2003;17:804–817. doi: 10.1210/me.2002-0406. [DOI] [PubMed] [Google Scholar]
- 76.Muyan M, Yi P, Sathya G, Willmert LJ, Driscoll MD, Hilf R, Bambara RA. Mol. Cell. Endocrinol. 2001;182:249–263. doi: 10.1016/s0303-7207(01)00493-2. [DOI] [PubMed] [Google Scholar]
- 77.Livak KJ, Schmittgen TD. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]