

Abstract
Adult neurogenesis is a life-long developmental process that occurs in two discrete regions in the adult mammalian brain: the subgranular zone of the dentate gyrus (DG) and the subventricular zone (SVZ) along the lateral ventricles. Despite immense interest in the therapeutic potential of adult neural stem cells (aNSCs) residing along these two neurogenic regions, molecular and cellular mechanisms regulating this process are not fully defined. Defining the regulatory mechanisms responsible for the genesis of new neurons in the adult brain is integral to understanding the basic biology of aNSCs. The techniques described here provide a basic blueprint to isolate, culture, and perform experiments using aNSCs in vitro as well as providing methods to perform immunohistochemistry on brain sections.
Keywords: neurogenesis, adult neural stem cells, aNSC, immunohistochemistry, primary culture, primary cell isolation, brain section
Introduction
This unit outlines methods that are commonly used to study adult neurogenesis in mice both in vitro and in vivo. Specifically, we provide detailed information to perform experiments in vitro after the successful isolation of primary adult neural stem cells (aNSCs). Additionally, we supplement this protocol by providing details on immunolabeling of proliferating cells using 5-bromo-2'-deoxyuridine (BrdU). We also describe how to perform BrdU labeling in adult mice followed by immunohistochemistry conditions to analyze cell-type specific markers present along both neurogenic regions.
Basic protocol 1 describes the isolation, culture, and expansion of primary subgranular zone (SGZ) and subventricular zone (SVZ) aNSCs. We add to this protocol by providing plating conditions of aNSCs for downstream experiments including pulsing cultures with BrdU to identify proliferating cells in culture. Basic protocol 2 describes how to perform in vivo BrdU labeling as well as how to perform immunohistochemical analysis of adult brain sections. Our goal for this unit is to provide details for commonly used techniques to study adult neurogenesis in mice.
Basic Protocol 1: Isolation, culture, and expansion of primary adult neural stem cells from both neurogenic regions
The main agenda of this protocol is to define conditions to culture aNSCs for downstream experiments where the investigator can examine molecular and cellular mechanisms regulating aNSC proliferation, cell-fate specification, and/or differentiation. We start with a detailed description of the microdissection technique to isolate both the SVZ and SGZ and follow that up with dissociation conditions of the tissue and finish with ideal culture conditions to permit proliferation and expansion of neurospheres.
Labeling cells with BrdU allows identification of actively proliferating cells during the S-phase of the cell cycle. We provide details in pulsing cells with BrdU as well as immunocytochemistry techniques to identify these cells by fluorescence microscopy. Additionally, we list cell-type specific markers that are commonly used for in vitro identification of different cell types.
Materials
Fresh, whole adult (8–10 week old) mouse brain
Small dissecting scissors
Forceps
Fine-tip forceps
Scalpel
Autoclaved TC water
Hank's Balanced Salt Solution (HBSS, Invitrogen, Cat. 14025-092)
Advanced DMEM/F-12 (Invitrogen, Cat. 12634-010)
Fetal Bovine Serum (FBS, Gibco, Cat. 16140)
Cell strainer (40 μm, Fisher, Cat. 22-363-547)
Brain matrix (Kent Scientific, Cat. RBMA-200C)
Dissecting microscope
100 mm petri dish
60 mm petri dish
35 mm petri dish
37°C TC incubator with 6.5% CO2 setting
Neurosphere dissociation and monolayer culture for downstream in vitro experiments
Solution A (See Recipes)
Enzyme Mix 1
Enzyme Mix 2
Percoll Solution
Initial Proliferation Medium (IPM)
Serum-Free Media (SFM)
0.125% Trypsin-EDTA (Invitrogen, diluted from 0.25% in HBSS, Cat. 25200-056)
0.014% w/v Soybean Trypsin Inhibitor (Invitrogen, diluted in HBSS, Cat. 17075-029)
Poly-L-Ornithine (Sigma, Cat. P2533, diluted with Milli-Q water)
Fibronectin (Invitrogen, Cat. 33016-015, diluted with PBS)
Aclar film coverslips (Electron Microscopy Sciences, Cat. 50425)
BrdU labeling and immunocytochemistry
BrdU (Sigma, Cat. B9285)
Primary antibodies (See Table 1)
Alexa-Fluor Secondary Antibodies (Invitrogen) (See Table 2)
1 N Hydrochloric acid
2 N Hydrochloric acid
0.1 M Borate buffer, pH 8.5
5% BSA blocking buffer in PBST
2.5 μg/mL Hoechst 33342 (Invitrogen, diluted in PBS, Cat. H3570)
Table 1.
Primary antibodies for immunolabeling different cell types in vitro
| Antibody | Host | Dilution | Source |
|---|---|---|---|
| BrdU | Rat | 1:500 | (AbD Serotec, Cat. MCA2060) |
| PCNA | Mouse | 1:1,000 | (Millipore, Cat. MAB424) |
| Ki67 | Rabbit | 1:2,000 | (Leica, Cat. NCL-Ki67p) |
| Nestin | Mouse | 1:500 | (DSHB, Cat. Rat-401) |
| GFAP | Mouse | 1:1,000 | (Millipore, Cat. MAB360) |
| Sox2 | Mouse | 1:500 | (Millipore, Cat. MAB4343) |
| DCX | Goat | 1:500 | (Santa Cruz, Cat. sc-8067) |
| PSA-NCAM | Mouse | 1:500 | (DSHB, Cat. 5A5) |
| (β-III Tubulin | Mouse | 1:1,000 | (Promega, Cat. G7121) |
| NeuN | Mouse | 1:500 | (Millipore, Cat. MAB377) |
Table 2.
Primary antibodies for immunolabeling different cell types in vivo
| Antibody | Host | Dilution | Source |
|---|---|---|---|
| BrdU | Rat | 1:500 | (AbD Serotec, Cat. MCA2060) |
| PCNA | Mouse | 1:500 | (Millipore, Cat. MAB424) |
| Ki67 | Rabbit | 1:1,000 | (Leica, Cat. NCL-Ki67p) |
| GFAP | Mouse | 1:1,000 | (Millipore, Cat. MAB360) |
| Sox2 | Mouse | 1:500 | (R&D Systems, MAB2018) |
| DCX | Goat | 1:200 | (Santa Cruz, Cat. sc-8067) |
| PSA-NCAM | Mouse | 1:500 | (DSHB, Cat. 5A5) |
| NeuroD | Goat | 1:200 | (Santa Cruz, Cat. sc-1084) |
| Calretinin | Mouse | 1:500 | (Santa Cruz, Cat. sc-135853) |
| NeuN | Mouse | 1:500 | (Millipore, Cat. MAB377) |
| Calbindin | Mouse | 1:500 | (Santa Cruz, Cat. sc-58699) |
Microdissection and isolation of primary adult neural stem cells
-
1
Soak dissecting instruments in 70% EtOH for at least 30 min and allow to air dry under the hood for at least 5 min prior to beginning this protocol.
-
2
Add a few mLs of HBSS to 3–4 60 mm petri dishes and place on ice.
-
3
Harvest fresh, whole adult mouse brain following cervical dislocation. Take care not to damage the brain when removing from the skull and place the brain in one of the petri dishes containing HBSS.
-
4
Repeat harvesting of 2–4 more mouse brains and place in same petri dish.
-
5
Make up Solution A and aliquot 10 mL into 2 15 mL conical tubes and place on ice (one for SVZ and one for SGZ).
-
6
Take a 100 mm petri dish, fill with ice and place under the dissecting microscope. This will be the cold stage to perform the microdissection. (Note: all dissection procedures are done outside of the TC hood).
-
7
Place one whole brain in the brain matrix and use a clean scalpel blade and slice the brain into 1 mm sections. Take each brain section and place them into one of the other 60 mm petri dishes with HBSS.
-
8
Place this 60 mm petri dish on the ice stage under the dissection scope and examine each section for the SVZ and SGZ.
-
9
Take fine-tipped forceps and pinch away SVZ and SGZ from each brain section and place tissue in corresponding 15 mL conical tubes.
-
10
Continue this process for all brain sections until all SVZ and SGZ have been removed from brain sections that contain these neurogenic regions.
-
11
Repeat process with remaining brains.
-
12
Take both 15 mL conical tubes containing SVZ and SGZ and spin down for 10 min at 1,000 rpm to pellet all tissue.
-
13Aspirate supernatant and add 2 mL Enzyme Mix 1 into each tube, resuspend tissue fragments with p1000 pipette tip (up/down 15–20 times, pre-wet tip to avoid sample loss) and rock tubes at room temperature for 20 min. Pipette tissues up/down 5 times, and the rock tubes for another 5 min.For steps 13–22, please also refer to: (Guo et al., 2012; Luo et al., 2010)
-
14
Add 30 μL of Enzyme Mix 2 (freshly mixed) to each tube and rock tubes at room temperature for 15 min.
-
15
Triturate tissue fragments using p1000 pipette tip (pre-wet) for 10–15 times until all tissue fragments are dissociated. Take care to be gentle when triturating and minimize bubbles. (Note: can use glass Pasteur pipettes to triturate tissue as well).
-
16
Add 5 mL DMEM/F-12 medium with 10% FBS into each tube to stop digestion, and pipette up and down for 5–10 times.
-
17
Filter samples through a cell strainer (40 μm) and spin down for 3 min at 1,000 rpm.
-
18
Wash cell pellet with 5 mL of DMEM/F-12 medium with 10% FBS, and spin down for 3 min at 1,000 rpm.
-
19
Resuspend cell pellet with 3.9 mL of DMEM/F-12 medium with 10% FBS and 1.1 mL of Percoll solution in each tube. Mix well and spin down for 15 min at 1,500 rpm.
-
20
Remove supernatant, and resuspend cell pellet with 10 mL Solution A. Spin down for 5 min at 1,500 rpm.
-
21
Remove supernantant, and resuspend cell pellet with 10 mL Initial Proliferation Medium (IPM) and spin down for 3 min at 1,500 rpm.
-
22
Resuspend cell pellet with 3 mL IPM and plate cells into 35 mm petri dishes to permit neurosphere formation. (Note: There will be a lot of cellular debris in the dish, this is OK).
-
23Place petri dishes into 37°C incubator with 6.5% CO2 and culture for 7–14 d until visible neurospheres form. Refresh growth factors (EGF and bFGF) every 3–4 d until neurosphere formation.Note: SVZ neurospheres should appear between 7–10 d while SGZ neurospheres should appear between 10–14 d. SGZ neurospheres may be smaller than SVZ neurospheres.
Passaging primary neurospheres
-
24
Following primary neurosphere formation, use p100 pipette and pipette up neurospheres under standard light microscope. (Note: this is done outside the TC hood). Only select true neurospheres and minimize other cellular debris and pipette neurospheres into new 15 mL conical tube containing SFM.
-
25
Continue selecting healthy, visible neurospheres until all primary neurospheres have been collected.
-
26
After all primary neurospheres have been collected, spin down for 10 min at 1,000 rpm.
-
27
Aspirate supernatant and resuspend by trituration in fresh SFM media.
-
28
Plate cells in 35 mm petri dishes at a low cell density (10,000–20,000 cells/cm2) and permit secondary neurosphere formation for 7–10 d.
-
29Following secondary neurosphere formation, can dissociate spheres by trituration and freeze to store or continue passaging and expanding for experiments.Note: Freeze in 10% DMSO final concentration in SFM.
-
30
Commonly, neurospheres will attach and expand as a monolayer even on petri dishes. When passaging these attached neurospheres, transfer free-floating neurospheres in the media into a 50 mL conical tube first.
-
31
Add pre-warmed 0.125% Trypsin-EDTA to the dish to aide in detaching attached neurospheres and monolayer cells for 5 min at 37°C.
-
32
Gently pipette to ensure detachment of all cells from the dish.
-
33
Combine trypsinized cells to 50 mL conical tube, add equal volume of pre-warmed 0.014% trypsin inhibitor for 5 min at room temperature, spin down the cell pellet, triturate and dissociate as above and plate for further passage or experiments.
Plating adult neural stem cells for in vitro experiments
-
34
Adult neural stem cells are plated as a monolayer culture for experimentation.
-
35
Dissociate and digest neurospheres with 0.125% trypsin-EDTA at RT for 5–7 min.
-
36
Incubate with equal volume of 0.014% trypsin inhibitor at RT for 5 min.
-
37
Spin down cells at 1,000 rpm for 3 min and resuspend cell pellet in SFM.
-
38
Spin down cells again and then mechanically triturate cell pellet using a p1000 pipette until the cell suspension takes on a smooth appearance. Plate cells as a monolayer culture on coated (see Support Protocol 1 for double coating protocol) culture dishes or Aclar coverslips for experiments.
-
39
Plate single-cell suspension of aNSCs O/N and then perform downstream manipulation to examine effects on aNSCs.
BrdU labeling and immunocytochemistry
-
40
For in vitro BrdU labeling, plate cells at a density of 40,000–50,000 cells per well on coated coverslips in 48-well culture plates.
-
41
Twenty-four to 48 h after plating, cells can be treated with vehicle or growth factors for desired time, followed by BrdU (10 μM) incorporation for 2–24 h at 37°C depending on experiment requirements.
-
42
Fix cells with 4% paraformaldehyde (PFA)/sucrose at RT for 30 min, followed by immunocytochemical analysis.
-
43
Wash fixed cells with PBS for 3× for 5 min per wash.
-
44
Incubate sections in distilled water for 5 min at RT.
-
45
Denature DNA by incubating sections in 1 N ice-cold HCl for 10 min at 4°C
-
46
Continue denaturation by incubating sections in 2 N HCl at 37°C for 30 min. (Note: can add HCl at RT, then place in 37°C incubator).
-
47
Neutralize acid by washing brain sections in 0.5M borate buffer 2× for 15 min per wash.
-
48
Wash cells with PBST for 3× for 5 min per wash.
-
49
Apply blocking buffer (5% BSA/PBST) and incubate at room temperature for 2 h.
-
50
Incubate with rat monoclonal anti-BrdU at 4°C overnight.
-
51
Wash cells with PBST for 3× for 10 min per wash.
-
52
Incubate with secondary antibodies at 1:5,000 dilution (Alexa-Fluor-488, Invitrogen) or 1:2,000 dilution (Alexa-Fluor-594, Invitrogen) in blocking buffer at room temperature for 2 h.
-
53
Wash cells with PBST for 3× for 10 min per wash.
-
54
Incubate cells for 30 min to 1h in 2.5 μg/mL Hoechst 33342 at RT for nuclei visualization
-
55
Wash cells 1× for 10 min in PBST.
-
56
Mount onto slides using anti-fade Aqua Poly/Mount solution.
Support Protocol 1: Double coating culture surfaces
This protocol briefly describes the process we use for making double-coated culture dishes or Aclar coverslips for plating adult neural stem cells, as described in Basic Protocol 1.
Materials
Tissue culture dishes or Aclar coverslips
Poly-L-ornithine (15 μg/mL)
Media for making poly-L-ornithine dilution
PBS
Fibronectin (1mg/mL)
Coat tissue culture dishes or Aclar coverslips with 15 μg/ml poly-L-ornithine using 1/2 volume of media normally used.
Aclar film can be hole punched and washed in 70% EtOH O/N followed by 3–4 washes in autoclaved TC water.
Incubate dishes/coverslips overnight at 37°C.
Aspirate solution and wash twice with PBS, allowing 20 min incubation at 37°C for each wash.
Add 1 mg/L fibronectin to the poly-L-ornithine coated dishes/coverslips, spread and incubate 1–24 h at 37°C.
Aspirate solution, wash once with PBS.
Dishes/coverslips may be stored with PBS for up to 4 days at 37°C.
Rinse with media before use.
Basic Protocol 2: In vivo BrdU labeling and immunohistochemical analysis of adult brain tissue
This protocol is to provide the experimenter a method to label proliferating cells in the adult mouse brain and identify those cells by immunohistochemistry techniques frequently used when assessing adult neurogenesis. The premise of these techniques is derived from previously published reports (Pan et al., 2012a; Pan et al.; Pan et al., 2012b; Zou et al., 2012), and is expanded upon in greater detail here. BrdU is a commonly used intercalating agent to identify proliferating cells progressing through S-phase of the cell cycle. This is a permanent marker to, in effect, birthdate cells as they proliferate and/or differentiate into their specific cell type. Because birthdating proliferating cells can be examined hours or months after BrdU administration, it provides a very flexible and convenient tool to study aNSCs and adult-born neurons (Ming and Song, 2005; Taupin, 2006).
Materials
BrdU
Cryostat, microtome, or other frozen tissue sectioning equipment
12-well or 24-well plates
12-well or 24-well Netwell inserts (Corning)
1X PBS
Block buffer
Primary antibodies
Alexa-Fluor Secondary Antibodies (Invitrogen)
1 N Hydrochloric acid
2 N Hydrochloric acid
0.1 M Borate buffer, pH 8.5
Superfrost plus microscope slides (Fisher, Cat. 12-550-15)
Pap-pen liquid blocker (Electron Microscopy, Cat. 71312)
Gelatin
Size 0 paintbrush
Aqua/polymount media (Polysciences, Cat. 18606-20)
In vivo BrdU labeling of proliferating cells via free-floating IHC method
-
1
Freshly prepare or thaw out a previously prepared aliquot of 20 mg/mL BrdU. Dissolve at 56°C by constant vortexing.
-
2
Weigh mice and administer 100 mg/kg BrdU by intraperitoneal injection. (Note: BrdU can be administered either 1× or up to 5× every 2 h for 1 d depending on experimental conditions).
-
3
Sacrifice mice by intracardial perfusion after desired interval following BrdU administration.
-
4
Brains frozen after intracardial perfusion should be sectioned at 15–40 μm in the coronal or sagittal orientation.
-
5
Brain sections can be either directly mounted on slides or placed in 48-well or 24-well plates in cryoprotectant media as free-floating sections and stored at −20°C (Don't store sections in −80°C as cryoprotectant will freeze at this temperature).
-
6
Brain sections to be stained will be placed in netwells containing PBS. (Note: brain sections can be stored in PBS at 4°C after sectioning for 1–2 d prior to beginning IHC protocol).
-
7
Using either 12-well or 24-well netwells, wash tissue sections 4× in PBS for 10 min per wash. (Note: all free-floating staining steps should be performed on a platform shaker at RT unless otherwise stated).
-
8
Incubate sections in distilled water for 5 min.
-
9
Denature DNA by incubating sections in 1 N ice-cold HCl for 10 min at 4°C
-
10
Continue denaturation by incubating sections in 2 N HCl at 37°C for 30 min. (Note: can add HCl at RT, then place in 37°C incubator).
-
11
Neutralize acid by washing brain sections in 0.1M borate buffer 2× for 5 min per wash.
-
12
Wash sections in PBST 4× for 10 min per wash.
-
13
Block by incubating brain sections in blocking buffer for ≥2 h.
-
14
Add appropriate primary antibodies and incubate at 4°C for 48–60 h.
-
15
Wash sections in PBST 4× for 10 min per wash.
-
16
Incubate sections with appropriate Alexa-fluor secondary antibodies for 2 h at RT in the dark, or O/N at 4°C in the dark.
-
17
Wash sections in PBST 4× for 10 min per wash.
-
18
Incubate sections in 2.5 μg/mL Hoechst 33342 (diluted in PBS) for 30–45 min.
-
19
Wash sections in PBST 3× for 10 min per wash.
-
20
Remove one of the netwells containing brain sections and place into a larger container containing PBS. Maneuver netwells in PBS until all brain sections are out of the netwell.
-
21
Mount sections to gelatinized slides (See Support Protocol 2) using paintbrush taking care not to tear or cause wrinkles in the tissue.
-
22
Allow slides to dry for 5 min at RT and then apply Aqua/polymount medium and coverslip the slides and allow to dry O/N in the dark.
Immumohistochemistry staining using mounted sections
-
23
Take slides containing freshly cut brain sections or retrieve slides containing previously cut and frozen brain sections and pre-warm to RT.
-
24
Use Pap-pen to outline sections to be stained.
-
25
Wash sections in PBS 3× for 5 min per wash.
-
26
Incubate sections in 1% SDS for 10 min.
-
27
Wash sections in PBS 3× for 5 min per wash.
-
28
Wash sections in PBST 3× for 10 min per wash.
-
29
Apply blocking buffer for 1–2 h at RT.
-
30
Incubate sections in primary antibodies O/N-48 h at 4°C.
-
31
Wash sections in PBST 3× for 10 min per wash.
-
32
Incubate sections in Alexa-fluor secondary antibodies in blocking buffer for 2 h at RT.
-
33
Wash sections in PBST 3× for 10 min per wash.
-
34
Incubate sections in 2.5 μg/mL Hoechst 33342 (diluted in PBS) for 30 min at RT.
-
35
Wash sections in PBST 3× for 10 min per wash.
-
36
Allow slides to dry for 5 min at RT and then apply Aqua/polymount medium and coverslip the slides and allow to dry O/N in the dark.
Support Protocol 2: Making gelatinized slides
This short protocol details the process for making gelatinized slides for mounting tissue sections for downstream analysis (e.g. for IHC, as described in Basic Protocol 2).
Materials
2% gelatin, dissolved in Milli-Q H2O
Slide dipper reservoir
Slides
Slide box for storage
Dissolve 2% gelatin in Milli-Q water on hotplate by constant stirring. Do not heat above 60°C.
Pour 2% gelatin into slide dipper reservoir and dip slides for 10 min.
Repeat for more slides.
After 10 min gelatin dip, remove slides and allow to air dry O/N.
Remove slides and either use immediately or store in general slide box.
REAGENTS AND SOLUTIONS
Use of Milli-Q water is highly recommended for the preparation of all reagents and throughout the protocol.
Solution A
1X HBSS with Glucose (30 mM)
HEPES (pH 7.4, 2 mM)
NaHCO3 (26 mM)
Filter Sterilize
Initial Proliferation Medium (IPM); to make 25 mL
24 mL Neurobasal medium (Invitrogen, Cat. 21103-049)
0.5 mL B27 (Invitrogen, Cat. 17504-044)
0.25 mL L-Glutamine (Invitrogen, Cat. 25030-081)
0.25 mL Penicillin-Streptomycin (Invitrogen, Cat. 15140-122)
20 ng/mL EGF (EMD Chemicals, Cat. 324831)
10 ng/mL bFGF (Millipore, Cat. GF003)
Enzyme Mix 1
MACS Neural Tissue Dissociation Kit (Miltenyi Biotec, Cat. 130-092-628, relative volumes per brain)
Mix 50 μL of MACS Solution 1
2 mL of MACS Solution 2
2.5 μL of diluted β-mercaptoethanol (34.7 μL of β-mercaptoethanol diluted in 10 mL Milli-Q water)
Enzyme Mix 2
MACS Neural Tissue Dissociation Kit (relative volumes per brain)
20 μL of MACS Solution 3
10 μL of MACS Solution 4
Percoll Solution
9 mL Percoll (Amersham, Cat. 17-0891-01)
1 mL 10X PBS
Filter Sterilize
Serum-Free Media (SFM)
Advanced DMEM/F-12 (Invitrogen, Cat. 12634-010)
1X N-2 Supplement (Invitrogen, Cat. 17502-048)
1X B-27 Supplement w/out Vitamin A (Invitrogen, Cat. 12587-010)
100 U/mL Penicillin/Streptomycin (Invitrogen, Cat. 15140-122)
2 mM L-Glutamine (Invitrogen, Cat. 25030-081)
2 μg/mL Heparin Sodium Salt (Sigma, Cat. 84020)
20 ng/mL EGF (EMD, Cat. 324831)
10 ng/mL bFGF (Millipore, Cat. GF003)
10X Phosphate Buffered Saline (PBS)
Can be stored for up to 6 months at RT.
PBST
Dilute 10X PBS for final working concentration at 1X PBS.
Add triton X-100 for final working concentration of 0.25%
Stir to dissolve
Can be stored for up to 6 months at RT.
Block Buffer
PBST
10% Normal Serum (use as appropriate for antibody)
1% BSA
Make fresh for each use.
2% Gelatin
Gelatin
Milli-Q water
Heat up to 60°C on hot plate and stir to dissolve
Following each use, filter sterilize and store at 4°C for up to 6 months.
0.1M Borate Buffer, pH 8.5
Prepare 0.1 M boric acid
Prepare 0.1 M sodium borate
Add boric acid to sodium borate until pH 8.5 is reached (Note: ratio is approximately 5:1 of acid:base)
1% SDS
SDS
Milli-Q water
Stir to dissolve
BrdU Stock Solution (20 mg/mL)
BrdU
0.9% Bacteriostatic saline
0.007N NaOH
Heat at 56°C and vortex constantly to dissolve
Protect from light
Filter sterilize and store in −20°C for up to 1 year
COMMENTARY
Background information
Although the identification of proliferating cells in the adult rat brain in the 1960s provoked some thought that adult neurogenesis may exist (Altman and Das, 1965), it was not widely accepted until more evidence substantiated the generation of adult-born neurons 30–40 years later. Discoveries in the past decade led to the identification of neural stem cells capable of generating adult-born neurons under normal physiological conditions in the adult mammalian brain (Gage, 2000; Ming and Song, 2005; Taupin, 2006). Adult neural stem cells reside in two discrete regions: the lateral wall of the lateral ventricles and in the dentate gyrus of the hippocampal formation. Neurons born from the lateral ventricles migrate along the rostral migratory stream and integrate into the olfactory bulb of rodents, while neurons born in the dentate gyrus integrate locally and may contribute to learning and memory.
Immense interest in understanding the molecular and cellular mechanisms regulating adult neurogenesis has led to the identification of many signaling molecules which may play a role in the regulation of this process (Mu et al., 2010; Suh et al., 2009). Among those signaling molecules currently known in the literature, ERK5 MAPK has been recently described as a unique candidate to regulate the process of neuronal differentiation in the SGZ of the dentate gryus (Pan et al., 2012b). In addition, the functional significance of adult-born neurons and their contribution to hippocampus-dependent learning and memory has been expanded to include their involvement in the more challenging forms of memory where an animal needs to actively forget a previously learned memory in order to acquire a new memory as well as remote memory (Pan et al., 2012a; Pan et al.). These results directly demonstrate the necessity of ERK5 MAPK to regulate adult neurogenesis and highlights the importance of adult-born neurons to hippocampus-dependent learning and memory tasks.
Molecular and cellular characterization of adult neural stem cells can be directly studied by the isolation of primary cells from the two neurogenic regions. Methods previously developed to isolate and characterize SGZ- and SVZ-derived aNSCs have been evolved and is now presented here in this unit (Bonaguidi et al., 2008; Bull and Bartlett, 2005; Guo et al.; Reynolds and Weiss, 1992). These methods have been directly used in our lab with success. Downstream characterization of aNSCs isolated by this method is at the sole discretion of the researcher since the goal of this unit is to provide isolation, expansion, and culture conditions of aNSCs.
Studying the effects of adult neurogenesis in vivo is more demanding of the researcher since inter-species as well as inter-animal variables exist, thus requiring the likely use of more animals per experiment. However, the benefit of using in vivo models is the ability to examine effects of cellular manipulations and its affects systemically. BrdU is a commonly used intercalating agent to birthdate proliferating cells (Gould et al., 1999; Ming and Song, 2005; van Praag et al., 1999). It has been used extensively in many studies examining the proliferative capabilities of aNSCs as well as the differentiation, terminal differentiation, and survival of adult-born neurons (Gage, 2000; Gage et al., 1998; Ming and Song, 2005; Pan et al., 2012b; Zou et al., 2012). We describe in detail, the protocol we used in BrdU preparation, administration and immunostaining techniques to visualize BrdU+ cells in the brain.
Advancements in the field of adult neurogenesis rely on the ability of the next researcher to adequately and effectively obtain reproducible results without having to painstakingly troubleshoot previously published protocols. Toward this goal, this unit aimed to provide a more detailed description of our successes in using these techniques to study adult neurogenesis (Pan et al., 2012a; Pan et al.; Pan et al., 2012b; Zou et al., 2012).
Critical Parameters
For the isolation of aNSCs from fresh mouse brain, the most important parameters to consider are the quality of the microdissection for SGZ and SVZ, over-exposure to enzymatic digestion, and the forcefulness of the manual trituration technique. Following cervical dislocation of conscious mice, the brain must be removed with caution to maintain the structure and integrity of the whole brain. Whole brains are then placed on ice in HBSS and then subsequently placed in the brain matrix to slice the brain into 1 mm thick coronal sections. Microdissection ensues for each 1 mm slab of brain tissue and ensuring the isolation of the DG and SVZ is critical. It is advisable to know the general anatomy of the brain before starting the microdissection procedure to be more familiar with where to expect the DG and SVZ in each coronal section. Next, enzymatic digestion times and forcefulness of manual trituration are other critical parameters of this protocol. Do not over expose tissue/cells to enzymes for longer than the indicated times and do not over triturate the tissue/cells as both enzymatic and mechanical stress to the cells will decrease the overall yield of healthy aNSCs.
One other critical parameter to be cautious of is the use of sterile technique when performing the microdissection and isolation of primary neurospheres for downstream passaging. Both of these techniques are performed outside of the TC hood and have an increased probability of becoming contaminated. All dissecting instruments should be soaked in a 70% EtOH bath for at least 30 min and allowed to air dry for at least 5 min prior to beginning this procedure to ensure no EtOH carryover on the dissecting instruments. In addition to using sterile techniques, it is advisable to wear a facemask during these procedures. Use of filter-tipped pipette tips is advised when isolating primary neurospheres for expansion. Despite the exposure of the tissue and neurosphere samples to the environment, contamination of the culture should be minimal if sterile technique and general caution is considered.
Adult neural stem cell cultures should be passaged in petri dishes since TC-treated dishes generally cause neurospheres to attach and expand as a monoloayer. Some of the neurospheres cultured in petri dishes will still attach and expand as a monolayer, but less so than if TC-treated dishes were used. Although some of the neurospheres will have attached, they can still be used for downstream experiments as long as they are maintained in proliferating media containing growth factors. When needing to passage cells, transfer the media (containing free-floating neurospheres) into a 50 mL conical tube. Add pre-warmed 0.125% Trypsin-EDTA to the culture dish to help detach any remaining neurospheres or monolayers of aNSCs. Gently pipette cells and combine with previously transferred neurospheres. Note: It is easier to triturate and dissociate cells into a single-cell suspension following centrifugation if they are in a 50 mL conical tube.
BrdU stock preparation and immunostaining for BrdU visualization in vitro and in vivo are also areas to pay attention to. BrdU stock is prepared using 0.9% bacteriostatic saline and 0.007N NaOH at a final concentration of 20 mg/mL. BrdU will not go into solution by vortexing alone. Heat is necessary, but take caution to not heat the solution above 60°C. Constant vortexing of the solution and incubation on a heat block is sufficient to dissolve BrdU. When retrieving a frozen aliquot of previously prepared BrdU stock, it will be necessary to thaw and heat up the solution to ensure full dissolution of the precipitate BrdU that is sure to form from storage at −20°C. During the immunostaining steps for denaturing DNA via HCl treatment, it is important to be mindful of the overall time cells or tissues are incubated in HCl. Overexposure to HCl could cause damage to the cells and tissue and severely affect the quality of the staining. HCl treatments tend result in weak Hoechst staining; longer neutralization with borate buffer (2× 15 to 30min) should help to solve this issue.
Troubleshooting
The quality of aNSC cultures could stem from any of the three critical parameters described above. Of the three parameters, the key to a healthy culture is most typically the isolation of SGZ and SVZ. If problems arise with your aNSC culture, it is advisable to practice the microdissection technique on a few brains prior to beginning the actual culture for experiments. Enzymatic digestion times should not be altered and strictly followed to ensure a quality culture. It is possible that the microdissection and enzymatic digestion were performed as described and the culture is still not as viable. This could likely be due to over trituration of the tissue/cells, thereby causing too much mechanical stress to aNSCs. Although it can be frustrating to dissociate tissue fragments and neurospheres into a single-cell suspension, forceful trituration is not the answer. Continued trituration (without causing bubbles) is preferred over forceful trituration. If difficulty still persists, the use of a sterile Pasteur pipette attached to a pipetteman can be substituted for using a p1000 pipette. When triturating, ensure contact of the tip of the Pasteur pipette tip or p1000 pipette tip to the bottom of the conical tube; this should aide in the dissociation process and minimize bubbles.
Visualization of BrdU via immunostaining should be rather straightforward, however, should problems arise, remake the BrdU stock and do not over heat the solution when dissolving BrdU. Double-check the concentration and incubation times of HCl and borate buffer. Check the concentration of the BrdU antibody used and optimize if necessary.
Anticipated Results
Using the protocols provided in this unit, successful isolation and expansion of aNSCs should be attainable. Neural stem cells isolated using this protocol should resemble examples from previously published reports (Bonaguidi et al., 2008; Bull and Bartlett, 2005; Guo et al.; Reynolds and Weiss, 1992). It is known in the adult mouse brain that aNSCs reside along the SVZ and SGZ, so the successful primary culture of cells is directly related to the quality of the microdissection procedure. Use of aNSCs in monolayer culture for downstream experiments and immunostaining should resemble those from previously published reports (Figure 1) (Amiri et al., 2012; Gao et al., 2011; Pan et al., 2012b).
Figure 1.
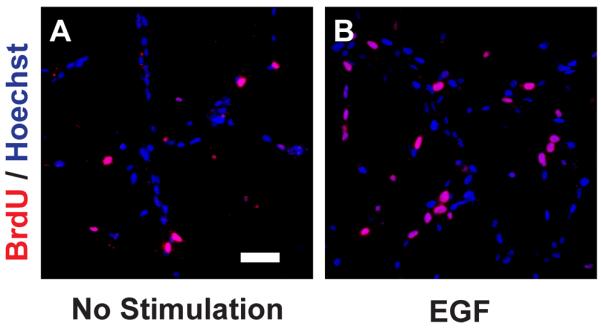
BrdU is incorporated into SVZ-derived aNSCs. Adult neural stem cells from the SVZ were pulsed with BrdU for 2 h and cells fixed and processed for immunostaining as described. A) Cells grown without growth factor stimulation. B) Cells grown in the presence of 100 ng/mL EGF for 18 h. Hoechst is used a s counterstain to identify all cell nuclei. Scale bar in A represents 50 μm and applies to both panels.
Immunostaining of BrdU in cells or in brain tissue samples should resemble previously published reports (Figure 2) (Brown et al., 2003; Gage et al., 1998; Hodge et al., 2008; Imayoshi et al., 2008; Pan et al., 2012b). The use of cell-type specific markers will be important to differentiate between the types of cells labeled with BrdU and those that are not. Depending on your research question, it may be necessary to birthdate aNSCs and examine their cellular phenotype within a couple hours of BrdU administration or months after BrdU administration. In either scenario, BrdU can be visualized using the protocol described in this unit.
Figure 2.

BrdU is incorporated in proliferating cells along the neurogenic regions. Mice were dosed with 100 mg/kg BrdU as described and sacrificed by transcardial perfusion 2 h after BrdU administration. Brains were harvested and processed for immunostaining as described. A–C) Proliferating cells at the time of BrdU administration is visualized along the SVZ. D–F) BrdU+ cells along the SGZ. Hoechst is used as a counterstain to identify all cell nuclei. Arrows point to the neurogenic regions the SVZ and SGZ of the lateral ventricle and dentate gyrus, respectively. Scale bar in A represents 100 μm and applies to all panels. Parts of this figure were originally published in The Journal of Biological Chemistry. Pan, YW; Zou, J; Wang, W; Sakagami, H; Garelick, MG; Abel, G; Kuo, CT; Storm, DR; and Xia, Z. Inducible and conditional deletion of extracellular signal-regulated kinase 5 disrupts adult hippocampal neurogenesis. The Journal of Biological Chemistry. 2012; Jul 6;287(28):23306–17. © the American Society for Biochemistry and Molecular Biology.
Time Considerations
During the aNSC isolation protocol, a total time of approximately 3–4 h should be budgeted for isolating aNSCs from 3–5 brains. Between 1–1.5 h should be allocated to the initial sacrificing of mice and the microdissection procedure. The remaining time will be used for the enzymatic digestion procedure, trituration of tissue samples, centrifugation steps, and plating.
Preparation of BrdU stock should take between 30–60 min. Administration of BrdU to in vitro cultures will depend on the time interval following BrdU that you are interested in. Likewise, the time interval between BrdU administration and examination of BrdU+ cells in vivo will depend upon the research question. The procedure of actually administering BrdU to mice takes approximately 1 min to weigh the mouse and inject the solution.
Brain sectioning for obtaining tissue sections will be largely dependent upon the instrument used to section the brain. Using a cryostat, we are able to process a brain capturing all coronal sections in 30–60 min. Immunohistochemistry for BrdU will take 4–5 d (free-floating method) and 3 d (mounted sections method) in total, beginning with the initial washing of brain sections to the application of mounting medium. The bulk of the time for the staining method is incubation of the brain sections in the primary and secondary antibodies. Washing brain sections, denaturing DNA, and blocking in the free-floating IHC method should take approximately 5 h. Following primary antibody incubation, the next wash step should take 40 min. After secondary antibody incubation, another 2–5 h should be allocated to wash, counterstain with Hoechst and mount brain sections to slides.
ACKNOWLEDGEMENT
The authors would like to thank all members of the Xia laboratory for critical input, evolution, and optimization of the protocols in this unit. In particular, we want to thank Dr. Junhui Zou and Mr. Tan Li for their insightful discussions and suggestions.
REFERENCES
- Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- Amiri A, Cho W, Zhou J, Birnbaum SG, Sinton CM, McKay RM, Parada LF. Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J Neurosci. 2012;32:5880–5890. doi: 10.1523/JNEUROSCI.5462-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, Peng CY, McGuire T, Falciglia G, Gobeske KT, Czeisler C, Kessler JA. Noggin expands neural stem cells in the adult hippocampus. J Neurosci. 2008;28:9194–9204. doi: 10.1523/JNEUROSCI.3314-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J, Cooper-Kuhn CM, Kempermann G, Van Praag H, Winkler J, Gage FH, Kuhn HG. Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur J Neurosci. 2003;17:2042–2046. doi: 10.1046/j.1460-9568.2003.02647.x. [DOI] [PubMed] [Google Scholar]
- Bull ND, Bartlett PF. The adult mouse hippocampal progenitor is neurogenic but not a stem cell. J Neurosci. 2005;25:10815–10821. doi: 10.1523/JNEUROSCI.3249-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Gage FH, Kempermann G, Palmer TD, Peterson DA, Ray J. Multipotent progenitor cells in the adult dentate gyrus. J Neurobiol. 1998;36:249–266. doi: 10.1002/(sici)1097-4695(199808)36:2<249::aid-neu11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Gao Z, Ure K, Ding P, Nashaat M, Yuan L, Ma J, Hammer RE, Hsieh J. The master negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J Neurosci. 2011;31:9772–9786. doi: 10.1523/JNEUROSCI.1604-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci. 1999;2:260–265. doi: 10.1038/6365. [DOI] [PubMed] [Google Scholar]
- Guo W, Patzlaff NE, Jobe EM, Zhao X. Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat Protoc. 7:2005–2012. doi: 10.1038/nprot.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Patzlaff NE, Jobe EM, Zhao X. Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat Protoc. 2012;7:2005–2012. doi: 10.1038/nprot.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, Kowalczyk TD, Wolf SA, Encinas JM, Rippey C, Enikolopov G, Kempermann G, Hevner RF. Intermediate progenitors in adult hippocampal neurogenesis: Tbr2 expression and coordinate regulation of neuronal output. J Neurosci. 2008;28:3707–3717. doi: 10.1523/JNEUROSCI.4280-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Ohtsuka T, Takao K, Miyakawa T, Yamaguchi M, Mori K, Ikeda T, Itohara S, Kageyama R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat Neurosci. 2008;11:1153–1161. doi: 10.1038/nn.2185. [DOI] [PubMed] [Google Scholar]
- Luo Y, Shan G, Guo W, Smrt RD, Johnson EB, Li X, Pfeiffer RL, Szulwach KE, Duan R, Barkho BZ, Li W, Liu C, Jin P, Zhao X. Fragile × mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 2010;6:e1000898. doi: 10.1371/journal.pgen.1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223–250. doi: 10.1146/annurev.neuro.28.051804.101459. [DOI] [PubMed] [Google Scholar]
- Mu Y, Lee SW, Gage FH. Signaling in adult neurogenesis. Curr Opin Neurobiol. 2010;20:416–423. doi: 10.1016/j.conb.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YW, Chan GCK, Kuo CT, Storm DR, Xia Z. Inhibition of Adult Neurogenesis by Inducible and Targeted Deletion of ERK5 Mitogen-Activated Protein Kinase Specifically in Adult Neurogenic Regions Impairs Contextual Fear Extinction and Remote Fear Memory. J Neurosci. 2012a;32:6444–6455. doi: 10.1523/JNEUROSCI.6076-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YW, Kuo CT, Storm DR, Xia Z. Inducible and Targeted Deletion of the ERK5 MAP Kinase in Adult Neurogenic Regions Impairs Adult Neurogenesis in the Olfactory Bulb and Several Forms of Olfactory Behavior. PLoS One. 7:e49622. doi: 10.1371/journal.pone.0049622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YW, Storm DR, Xia Z. The Maintenance of Established Remote Contextual Fear Memory Requires ERK5 MAP Kinase and Ongoing Adult Neurogenesis in the Hippocampus. PLoS One. 7:e50455. doi: 10.1371/journal.pone.0050455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YW, Zou J, Wang W, Sakagami H, Garelick MG, Abel G, Kuo CT, Storm DR, Xia Z. Inducible and conditional deletion of extracellular signal-regulated kinase 5 disrupts adult hippocampal neurogenesis. J Biol Chem. 2012b;287:23306. doi: 10.1074/jbc.M112.344762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Suh H, Deng W, Gage FH. Signaling in adult neurogenesis. Annu Rev Cell Dev Biol. 2009;25:253–275. doi: 10.1146/annurev.cellbio.042308.113256. [DOI] [PubMed] [Google Scholar]
- Taupin P. Neurogenesis in the adult central nervous system. C R Biol. 2006;329:465–475. doi: 10.1016/j.crvi.2006.04.001. [DOI] [PubMed] [Google Scholar]
- van Praag H, Kempermann G, Gage FH. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci. 1999;2:266–270. doi: 10.1038/6368. [DOI] [PubMed] [Google Scholar]
- Zou J, Pan YW, Wang Z, Chang SY, Wang W, Wang X, Tournier C, Storm DR, Xia Z. Targeted Deletion of ERK5 MAP Kinase in the Developing Nervous System Impairs Development of GABAergic Interneurons in the Main Olfactory Bulb and Behavioral Discrimination between Structurally Similar Odorants. J Neurosci. 2012;32:4118–4132. doi: 10.1523/JNEUROSCI.6260-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]