

Abstract
In the developing neocortex, brain-derived neurotrophic factor (BDNF) exerts a trophic activity to increase the expression and channel activity of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptor subunits. Here, we demonstrate that the epidermal growth factor (EGF) receptor (ErbB1) ligands exert the opposite biological activity in cultured neocortical neurons. Subchronic stimulation of ErbB1 with transforming growth factor α (TGFα), EGF, or heparin-binding EGF (HB-EGF) down-regulated protein expression of the GluR1 AMPA receptor subunit in cultured neocortical neurons. In agreement, TGFα treatment decreased the Bmax of [3H] AMPA binding and GluR1 mRNA levels. Immunocytochemistry revealed that the decrease in GluR1 was most pronounced in multipolar GABAergic neurons. To examine the physiological consequences, we recorded AMPA-evoked currents as well as miniature excitatory postsynaptic currents in morphologically identified putative GABAergic neurons in culture. Subchronic TGFα treatment decreased AMPA-triggered currents as well as the amplitude and frequency of miniature excitatory postsynaptic currents. An ErbB1 tyrosine kinase inhibitor, PD153035, inhibited the TGFα effect. Moreover, TGFα counteracted the neurotrophic activity of BDNF on AMPA receptor expression. Co-application of TGFα with BDNF blocked the BDNF-triggered up-regulation of AMPA receptor expression and currents. These observations reveal a negative regulatory activity of the ErbB1 ligand, TGFα, which reduces the input sensitivity of cortical GABAergic neurons to attenuate their inhibitory function.
Keywords: EGF, ErbB1, Her1, GluR1, BDNF, GABA, Neocortex
Introduction
Epidermal growth factor (EGF), transforming growth factor α (TGFα), and heparin-binding EGF (HB-EGF) all bind to one of EGF receptor subtypes, ErbB1 (Yamada et al., 1997; Harris et al., 2003; Xian and Zhou, 2004). These factors have neurotrophic actions on various types of neurons and glia (Maiese et al., 1993; Kornblum et al., 1999; Hanke et al., 2004). In the adult brain, brain injury, ischemic stress or seizure activity markedly induces TGFα and HB-EGF expression (Kawahara et al., 1999; Kornblum et al., 1994; Opanashuk et al., 1999). Accordingly, the neuroprotective activity of these factors is implicated in post-ischemic neurodegeneration as well as in counteracting glutamatergic neurotoxicity (Peng et al., 1998; Opanashuk et al., 1999; Petegnief et al., 2003).
During embryonic and postnatal development, TGFα and HB-EGF mRNAs are highly expressed in developing and mature neocortex (Kornblum et al., 1997, 1999; Assimacopoulos et al., 2003). Their receptor, ErbB1, is distributed in not only glial cells but also in neurons (Kornblum et al., 1995). In particular, ErbB1 is enriched in GABAergic neurons and dopaminergic neurons (Gómez-Pinilla et al., 1988; Kornblum et al., 1995, 1997). Mice lacking ErbB1 exhibit abnormal cortical development, including loss of astrocytes and impaired neuronal migration (Threadgill et al., 1995; Kornblum et al., 1998; Sibilia et al., 1998). The developmental effects of ErbB1 ligands on postmitotic neurons remain to be characterized, however. Our previous studies indicate that subchronic treatment with TGFα reduces voltage-gated currents in immature GABAergic neurons (Namba et al., 2003) and that repetitive EGF stimulation decreases the expression of AMPA-type glutamate receptors in neocortical cultures (Narisawa-Saito et al., 1999b). These results suggest the possibility that ErbB1 ligands may play a negative role in the development of GABAergic neurons or a protective role against excitotoxicity by reducing excitatory neurotransmission.
There are a large variety of neurotrophic factors and cytokines functioning in the central nervous system, most of which exert positive actions on neuronal differentiation or synaptic maturation and transmission. For example, BDNF promotes phenotypic maturation of neocortical GABAergic neurons (Ip et al., 1993; Nawa et al., 1993; Croll et al., 1994; Marty et al., 1996; Reibel et al., 2000; Nagano et al., 2003). Exogenous and endogenous BDNF acts on developing GABAergic neurons to enhance the expression of glutamic acid decarboxylase (GAD), presynaptic proteins, and AMPA-type glutamate receptors as well as morphological differentiation (Mizuno et al., 1994; Jones et al., 1994; Takei et al., 1997; Narisawa-Saito et al., 1999a; Rutherford et al., 1998; Bolton et al., 2000). Gene manipulation studies have confirmed the facilitatory effects of BDNF on the neocortical GABAergic inhibition as well (Jones et al., 1994; Huang et al., 1999; Gorski et al., 2003).
Like the immune and endocrine systems, the central nervous system presumably recruits both positive (facilitative) and negative (inhibitory) factors to precisely regulate the development of synaptic structures and function. Here, we show a novel inhibitory activity of the ErbB1 ligand, TGFα, that reduces AMPA receptor expression and function in cultured neocortical neurons, taking immunochemical approaches and the whole cell patch clamp method. The negative activity of TGFα is compared with the positive neurotrophic activity that BDNF exerts on AMPA receptor channel activity and expression.
Results
Subchronic effects of ErbB1 ligands on AMPA-type glutamate receptor expression
Using low-density neocortical cultures, we examined sub-chronic effects of EGF and other members of the EGF family, on AMPA receptor expression (Fig. 1A). Consistent with the results of Narisawa-Saito et al. (1999b), EGF treatment (20 ng/ml) significantly down-regulated protein levels of GluR1, but not those of GluR2/3, whereas BDNF (50 ng/ml) increased protein expression of GluR1 and GluR2/3. The other EGF family ligands, TGFα and HB-EGF (both 20 ng/ml), similarly decreased GluR1 protein levels without altering β-actin levels, which was used as an internal standard. The magnitude of GluR1 down-regulation by these ErbB1 ligands was similar: The ErbB1 ligands reduced GluR1 protein levels to approximately 50% of controls ( P < 0.05) (Fig. 1B). Although these factors are known to stimulate proliferation of progenitor and glial cells (Rabchevsky et al., 1998; Kornblum et al., 1999), the ErbB1 ligands had no effects on an astrocyte marker, glial fibrillary acidic protein (GFAP), and total protein recovery per dish under the given culture condition (see Experimental methods). The use of serum-free growth medium suppressed cell proliferation and reduced glial proportion to enable us to evaluate TGFα effects on cultured neurons (Yokomaku et al., 2005). As almost all ErbB1 ligands had the same impact on AMPA receptor expression, we focused on TGFα and analyzed its effects on AMPA-type glutamate receptor function.
Fig. 1.
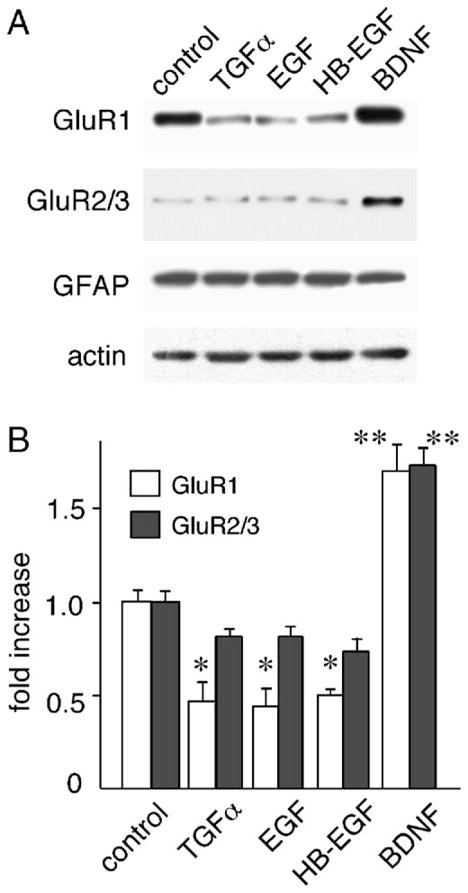
Effects of EGF family members on protein levels of AMPA-type glutamate receptor subunits. Neocortical neurons were cultured at a low density in serum-free medium in the presence or absence of EGF, HB-EGF or TGFα (20 ng/ml) for 5 days. Total cellular protein was collected and processed for immunoblotting. (A) Typical examples of immunoblotting are displayed. (B) The blotting results (from four cultures; total n = 4) were quantified by densitometry and plotted. *P < 0.05, **P < 0.01 with the unpaired t test.
TGFα decreases total AMPA binding in neocortical culture
[3H]AMPA binding assays were performed to test whether the down-regulation of GluR1 protein resulted in a change in AMPA receptor activity on the cell surface (Narisawa-Saito et al., 2002). After 4-day treatment with TGFα, various concentrations of [3H]AMPA were applied to living cultures, and the cell surface binding of radioactive AMPA was measured in each dish. The values of bound [3H]AMPA were subjected to Scatchard plot analysis, and the total number (Bmax) and dissociation constant (Kd) of functional AMPA receptors were determined for each culture (Fig. 2A). The Bmax of AMPA receptors significantly decreased from 4.16 ± 0.13 pmol/mg to 2.25 ± 0.15 pmol/mg. The dissociation constant of the AMPA receptors (Kd) decreased from 213.0 ± 12.4 nM to 60.7 ± 7.3 nM. A similar decrease in the maximal AMPA binding (with 300 nM [3H]AMPA) was observed on per-dish basis as well (from 1.74 ± 0.01 pmol/dish to 0.85 ± 0.02 pmol/dish; unpaired test, P = 0.044). The observed trend in AMPA binding per dish basis indicates that TGFα treatment also reduced the total number of surface AMPA receptors in culture.
Fig. 2.
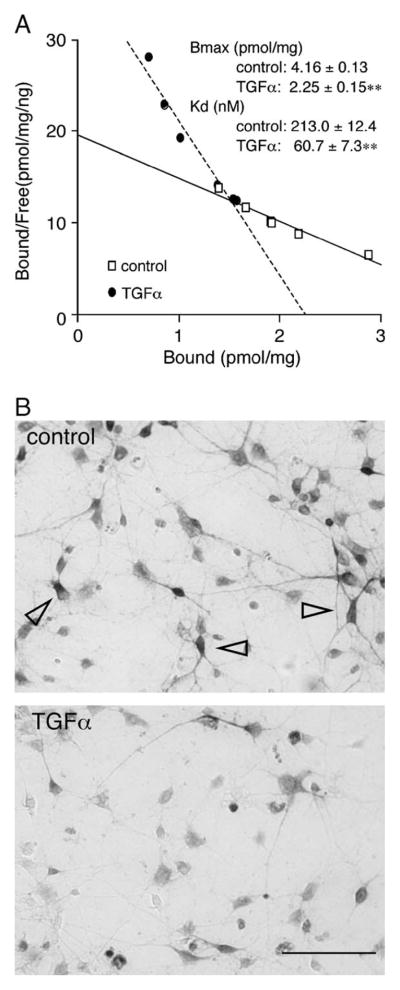
Decreased AMPA binding and GluR1-immunoreactivity in TGFα-treated neocortical cultures. (A) A typical Scatchard plot is shown for the radioactivity of [3H]AMPA bound to neocortical cultures treated with or without TGFα for 4 days. The set of binding assay was repeated four times, and the maximum amounts of bound [3H]AMPA (Bmax) and the dissociation constant (Kd) were calculated for each plot and averaged (n = 4). **P < 0.01. Non-specific background binding of [3H] AMPA to neurons (59 ± 8% of total [3H] AMPA binding) was estimated by replacing the [3H] AMPA with excess amounts of cold AMPA (100 μM) and subtracted from raw experimental values of [3H] AMPA binding. (B) GluR1-immunoreactivites were decreased in TGFα-treated cultures. Cortical cultures were treated with or without TGFα for 5 days. After fixation, cells were stained with the anti-GluR1 antibody and the immunoreactivity was visualized with the ABC kit. Arrowheads mark the typical multipolar neurons carrying higher levels of GluR1 immunoreactivity. Scale bar = 40 μm.
To examine how the decrease in total GluR1 protein levels was represented in individual neurons, cortical cultures were immunostained with the anti-GluR1 antibody (Fig. 2B). GluR1-immunoreactivity was detectable in almost all cells with different intensities, which is consistent with the previous report (Narisawa-Saito et al., 1999). The chronic treatment with TGFα appeared to decrease the number of the cells containing strong GluR1 immunoreactivity, which often exhibited multipolar dendritic morphology. The immunocytochemical data indicate that there are significant variations in the TGFα responses among individual cortical neurons.
Effect of TGFα on GluR1 immunoreactivity
In the neocortex, GABAergic neurons comprise approximately 20% of total neurons and some of them express high levels of the AMPA-type glutamate receptor subunit, GluR1 (Yin et al., 1994; Kharazia et al., 1996; Kondo et al., 2000). Therefore, we mainly focused on GABAergic neurons and investigated GluR1 expression in this population. The expression of EGF receptors (ErbB1) in cultured GABAergic neurons was confirmed using the anti-ErbB1 antibody (Gómez-Pinilla et al., 1988; Kornblum et al., 1995). In neocortical cultures, a majority of GAD67-positive cells (approximately 74%) were immunoreactive to the anti-ErbB1 antibody (Fig. 3A).
Fig. 3.
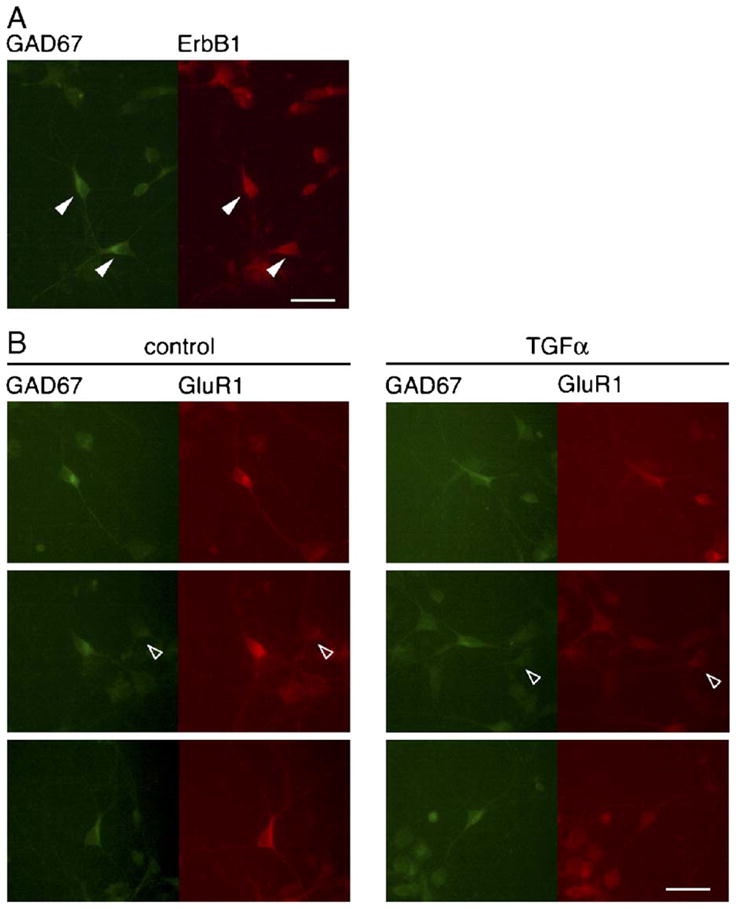
ErbB1- or GluR1-immunoreactivity of GAD67-positive neurons. (A) Colocalization of GAD67 with ErbB1 immunoreactivity. Control cultures were immunostained with anti-GAD67 and anti-ErbB1 antibodies followed by their secondary antibodies conjugated to Alexa Fluor® 488 (green) and Alexa Fluor® 546 (red), respectively. Arrowheads mark GAD67- and ErbB1-double positive cells. (B) Effects of TGFα on immunoreactivity of GluR1 in the GAD67 immunopositive neurons. Immunoreactivity for GAD67 and GluR1 proteins was visualized with Alexa Fluor® 488 (green) and Alexa Fluor® 546 (red), respectively. Stronger GluR1 immunoreactivity was detected in GAD67-positive multipolar neurons. Treatment with TGFα reduced GluR1 immunoreactivity of these neurons. Arrowheads indicate GAD67-negative cells. Scale bar = 20 μm.
Our previous study indicates that inhibitory neuronal populations expressing higher levels of GluR1 have a multipolar dendritic morphology in culture and retain developmental plasticity in response to BDNF (Nagano et al., 2003). The down-regulations in GluR1 protein observed in the present study might reflect negative reaction of the same GABAergic population. To distinguish GABAergic from non-GABAergic neurons, neocortical cultures were immunostained with an antibody raised against GAD67. The treatment with TGFα had no significant effect on the number of GAD67-immunopositive neurons at 5 DIV (control: 54.2 ± 3.2 cells/mm2, TGFα: 51.7 ± 1.5 cells/mm2, n = 4 each). In control cultures, almost all GAD67-positive cells expressed higher levels of GluR1 immunoreactivity and often had multipolar dendritic morphology (Figs. 3B and 4A). GluR1 immunoreactivity of these GABAergic cells appeared to decrease in TGFα-treated cultures: The intensity of GluR1 immunoreactivity in GABAergic neurons became comparable to that in non-GABAergic neurons (Fig. 3B).
Fig. 4.

Distributions of GluR1 immunofluorescence in GAD67-positive and negative cell populations. (A) The intensity of GluR1-immunoreactivity was compared between GAD67-positive and GAD67-negative cell populations in the control culture condition. Fluorescent intensities of somatic area were measured from four sister culture dishes and the frequency of each category of cells in total population was plotted. Mean Intensity of GluR1 immunoreactivity was 23.4 ± 0.4 and 9.7 ± 0.4 in GAD67-positive (closed bars) and GAD67-negative (open bars) cell populations, respectively. Note: The frequency of the cells that were markedly immunoreactive for the anti-GluR1 antibody (>20.0 intensity) was 69.5 ± 1.8 and 2.3 ± 0.9 in GAD67-positive and GAD67-negative cell populations, respectively. (B) The number of GluR1-immunoreactive cells was plotted over their GluR1 fluorescent intensities in the GAD67-positive multipolar population. Fluorescent intensities of somatic area were measured from four sister culture dishes. Mean intensity of GluR1 immunoreactivity in this population was 22.7 ± 0.6 and 17.6 ± 0.6 in control (open bars) and TGFα-treated (closed bars) cultures, respectively. (C) Fluorescent intensities in the GAD67-positive bipolar cells. Fluorescent intensities were measured from four sister culture dishes. Mean intensity of GluR1 immunoreactivity was 24.3 ± 0.8 and 21.7 ± 0.8 in control and TGFα-treated cultures, respectively. (D) Distribution of GluR1 fluorescent intensities in the GAD67-negative cell population. Mean intensity of GluR1 immunoreactivity was 8.2 ± 0.3 and 8.0 ± 0.3 in control and TGFα-treated cultures, respectively.
Neocortical neurons carrying GABA- or GAD-immunoreactivity display bipolar and multipolar dendritic morphology (de Lima and Voigt, 1997; Rutherford et al., 1998; Namba et al., 2003). In cortical cultures, a majority of GAD67-positive cells contained higher intensity of GluR1 immunoreactivity, compared with that of GAD67-negative cells (Fig. 4A). We grossly classified cultured neurons into three categories; GAD67-positive cells with multipolar dendrites, GAD67-positive cells with bipolar dendrites, and GAD67-negative cells. In each category, the intensity of GluR1 immunoreactivity and the cell number were plotted and subjected to statistical analysis to evaluate the effects of TGFα. In multipolar cells, the mean intensity of GluR1 immunofluorescence was significantly reduced by TGFα treatment, compared to control culture (control: n = 123, TGFα: n = 124, P < 0.001, unpaired t test) (Fig. 4B). In bipolar cells, similarly, TGFα treatment also decreased the frequency of immunoreactive neurons for GluR1, but the effect was modest (control: n = 84, TGFα: n = 82, P = 0.031, unpaired t test) (Fig. 4C). In contrast to the effects on these neuronal subpopulations, the distribution of GluR1 immunoreactivity in GAD67-negative cells was not significantly different between control and TGFα-treated cultures (control: n = 186, TGFα: n = 186, P = 0.68) (Fig. 4D). For instance, intensities of GluR1 immunoreactivity in non-GABAergic neurons (GAD67-negative cells) were rather lower and often indistinguishable in control and TGFα-treated cultures (Fig. 3B). Thus, it is likely that the GluR1 down-regulation at the total protein level mainly reflects the decrease observed in multipolar type GABAergic neurons.
Effects of TGFα on mRNA expression of AMPA receptors
To test whether the decrease in GluR1 protein is caused by a decline of its mRNA, we used quantitative real-time PCR analysis (Table 1) to measure the effects of TGFα on GluR1 and other AMPA receptor subunits. TGFα treatment significantly decreased GluR1 mRNA to ~63% of control level ( P < 0.001, unpaired t test) with no alteration in the levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as internal standard (Fig. 5). GluR2 mRNA was decreased to ~78% of control level ( P = 0.03, unpaired t test). In contrast, there were no significant alterations in mRNA levels of GluR3 ( P = 0.51) (Fig. 5B). These results suggest that TGFα treatment most markedly down-regulates GluR1 mRNA expression, presumably leading to the reduction in GluR1 protein levels as well as in surface AMPA binding activity.
Table 1.
Primer sequences for quantitative PCR reaction, anticipated amplicon length, and GenBank accession number
| Target | bp | GenBank | |
|---|---|---|---|
| GluR1 | |||
| Sense | TTACCACAGAGGAAGGCATGATC | 166bp | M36418 |
| Antisense | CCCTTGGGTGTCGCAATG | ||
| GluR2 | |||
| Sense | GTTTGTGAGGACTACCGCAGA | 176bp | M36419 |
| Antisense | ATCCTTTAGGTGTGGCGATG | ||
| GluR3 | |||
| Sense | GCAGAGCCATCTGTGTTTACCAA | 136bp | M36420 |
| Antisense | CACGGCTTTCTCTGCTCAATG | ||
| GAPDH | |||
| Sense | TGCACCACCAACTGCTAGC | 239bp | BC087743 |
| Antisense | GATGCAGGGATGATGTTCTG | ||
Fig. 5.
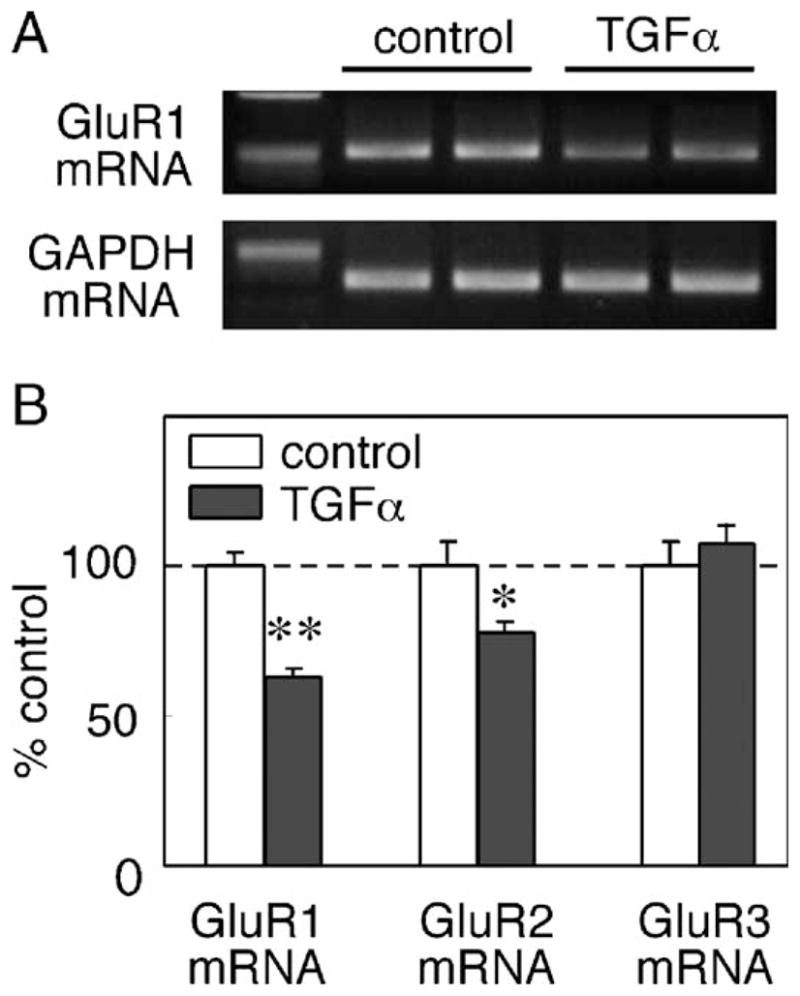
TGFα-triggered suppression of AMPA receptor mRNAs. Total RNA was extracted from untreated control and from cultures that were treated with TGFα for 3 days. (A) Typical examples of PCR products of GluR1 and GAPDH, which were amplified 29 and 28 cycles of PCR, respectively. PCR products were separated in 2% agarose gel electrophoresis and stained with ethidium bromide only for display. (B) mRNA levels for GluR1, GluR2 and GluR3 were independently determined by a quantitative real-time RT-PCR machine (Roche Diagnostics) using the specific primer sets and SYBR Green I (see Table 1). Relative mRNA levels for each glutamate receptor subunits were calculated by the Fit Point program and normalized to the amounts of amplified GAPDH cDNA in each sample (n = 5–8 each). *P < 0.05, **P < 0.01, unpaired t test.
Effects of TGFα on AMPA currents
Using the morphologic criteria, GABAergic neurons can be identified in cultures with an accuracy of approximately 80% (Nagano et al., 2003). To examine physiological significance of the GluR1 down-regulation, we performed whole-cell patch-clamp recording from the morphologically identified GABAergic neurons that display multipolar morphology. We measured the amplitude of inward currents triggered by local application of an agonist, AMPA (100 μM). Subchronic treatment of neocortical cultures with TGFα significantly decreased AMPA currents from 131.9 ± 8.6 to 68.0 ± 7.2 pA (Table 2, Fig. 6). TGFα treatment had no effect on passive-electrical properties of these cells, however (Table 2). Further, the suppressive effect on AMPA currents was blocked by co-application of the selective ErbB1 receptor inhibitor, PD153035 (100 nM). Application of PD153035 alone had no effect on AMPA current amplitudes. These results suggest that the down-regulation of AMPA currents is mediated by ErbB1 receptors.
Table 2.
AMPA currents and electrophysiological properties of multipolar-type neurons
| AMPA currents (pA) | Rm (MΩ) | Capacitance (pF) | Rs (MΩ) | |
|---|---|---|---|---|
| Control | 131.9 ± 8.6 | 2089 ± 376 | 39.6 ± 2.7 | 22.9 ± 1.3 |
| TGFα | 68.0 ± 7.2** | 2070 ± 266 | 36.1 ± 3.5 | 20.4 ± 0.9 |
| TGFα + PD | 130.4 ± 17.3a | 1900 ± 231 | 38.9 ± 3.2 | 23.7 ± 1.5 |
| PD alone | 120.3 ± 13.7 | 1972 ± 342 | 35.6 ± 3.9 | 21.2 ± 0.8 |
| BDNF | 357.0 ± 98.5* | 943 ± 89** | 65.2 ± 5.5** | 19.0 ± 1.0 |
| BDNF + TGFα | 121.3 ± 24.9b | 1445 ± 308 | 60.9 ± 4.9** | 22.0 ± 1.6 |
We analyzed electrophysiological properties of the cortical neurons (8–9 DIV) as described in Figs. 6 and 8. AMPA (100 μM) was locally applied to cell bodies of multipolar neurons by air pressure. Peak AMPA currents (pA) mean ± SE (n = 12 for each group). In parallel, passive properties of membrane resistance (Rm), capacitance and series resistance (Rs) were determined with hyperpolarizing voltage steps (see details in Experimental methods). PD: PD153035.
Comparisons were done: vs. TGFα (P < 0.05).
Comparisons were done: vs. BDNF (P < 0.05).
Comparisons were done: vs. control (P < 0.05).
Comparisons were done: vs. control (P < 0.01).
Fig. 6.
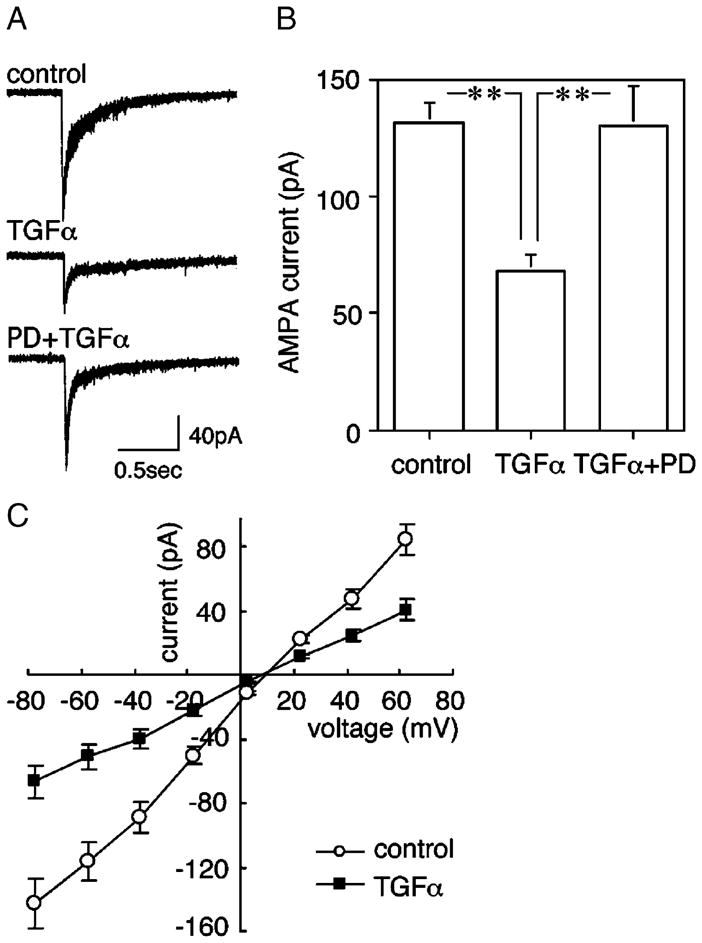
TGFα-triggered down-regulation of AMPA-evoked currents and effects of PD153035. Cultured cortical neurons were similarly prepared from embryonic rat neocortices and treated chronically with or without TGFα from 2 DIV to 6 DIV. Some cultures also contained a specific ErbB1 tyrosine kinase inhibitor, PD153035 (100 nM). AMPA (100 μM) was locally applied to cell bodies of multipolar neurons by air pressure. AMPA-evoked currents were recorded from multipolar neurons by a whole-cell patch-clamp technique (−78 mV). (A) Typical AMPA-evoked currents for display. (B) Peak AMPA currents (pA) mean ± SE (n = 12 for each group; ANOVA; F(2,33) = 9.42; P = 0.0006). post hoc: **P < 0.01. (C) I–V plots for AMPA-evoked currents are shown for control and TGFα-treated cultures.
Effects of TGFα on miniature excitatory postsynaptic currents
Miniature current analysis was performed to explore the effects of TGFα on AMPA receptor activity at synapses. To allow for synaptic maturation to produce a sufficient number of spontaneous synaptic events, the culture period was extended to 13–14 days in vitro (DIV). Whole-cell patch-clamp recording revealed that TGFα treatment decreased the miniature excitatory postsynaptic currents (mEPSC) in multipolar-type neurons from 24.4 ± 1.6 pA to 17.5 ± 1.4 pA (n = 12, P < 0.01) (Fig. 7). The treatment had no effect on the kinetics of mEPSC and there was no significant difference in current decay time (control: 2.0 ± 0.1 ms, TGFα: 2.3 ± 0.1 ms, P = 0.16). The frequency of mEPSCs decreased significantly from 0.89 ± 0.15 Hz to 0.45 ± 0.05 Hz ( P < 0.05). In contrast, TGFα treatment statistically had no apparent effect on the electrical properties of these multipolar GABAergic neurons in long-term culture, including input resistance (control: 917 ± 61.5 MΩ, TGFα: 715 ± 85.3 MΩ), membrane capacitance (control: 70.6 ± 3.40 pF, TGFα: 65.2 ± 6.32 pF), and series resistance (control: 15.6 ± 0.41 MΩ, TGFα: 16.1 ± 0.97 MΩ).
Fig. 7.

TGFα decreases mEPSC amplitudes in multipolar neurons. Cortical cultures were treated with or without TGFα for 5 days from 8 DIV and assayed at 13–14 DIV. (A) Spontaneous mEPSCs were recorded from multipolar neurons by a whole-cell patch-clamp technique. (B) Thirty mEPSC responses were calculated from multipolar neurons and averaged. (C) mEPSC frequencies were analyzed for multipolar neurons of control culture or TGFa-treated culture (n = 12 each, *P < 0.05, U = 32, Mann–Whitney U test). (D) mEPSC amplitudes of multipolar neurons were plotted (n = 12 each, **P < 0.01, U = 18, Mann–Whitney U test). Average quantal amplitudes of 100 events for the neurons that were grown with or without TGFa. (E) Cumulative amplitude histograms of mEPSCs were plotted for TGFa-treated and control multipolar neurons (P < 0.0001, Kormogorov–Smirnov test).
Antagonistic effects of TGFα against BDNF
Subchronic treatment with BDNF up-regulates the expression and activity of AMPA receptor channels in GABAergic neocortical neurons in culture (Narisawa-Saito et al., 2002; Nagano et al., 2003). The present results revealed an opposing activity of TGFα, reducing the expression and activity of AMPA receptor channels. Assuming that the target neurons of BDNF and TGFα are the same, do these factors counteract each other to regulate AMPA channel activity? We applied both factors to neocortical cultures and examined which activity predominates in the expression and channel function of AMPA receptors. Subchronic treatment with BDNF (50 ng/ml) increased GluR1 and GluR2/3 proteins to 150 ± 5% and 240 ± 13% of untreated culture, respectively (Figs. 8A and B), as reported previously (Narisawa-Saito et al., 1999a,b). Co-application of TGFα with BDNF, however, fully blocked the BDNF effects on GluR1 protein ( P < 0.01), but not on GluR2/3 protein ( P = 0.19).
Fig. 8.

TGFα antagonizes the upregulation of GluR1 protein and AMPA currents enhanced by chronic BDNF exposure. (A) Neocortical neurons were grown in serum-free condition for 5 days either with or without 50 ng/ml BDNF. In addition, 20 ng/ml TGFα was supplemented together with BDNF. GluR1 levels were determined in total protein lysate. Protein levels for GluR1 were significantly increased by chronic BDNF treatment (n = 4 each, ANOVA F(2,9) = 79.4, P < 0.001). (B) Protein levels for GluR2/3 were significantly increased by chronic BDNF treatment but the increase was not reversed by the coapplication of TGFα and BDNF (n = 4 each, ANOVA F(2,9) = 16.9, P < 0.001). (C) Typical AMPA-evoked currents when morphologically characterized neurons were clamped at −78 mV. Cultured cortical neurons were similarly prepared from embryonic rat neocortices and treated chronically with BDNF alone or with BDNF and TGFα. AMPA (100 μM) was locally applied to cell bodies by air pressure. Results are mean ± SE (n = 12 each; ANOVA F(2.33) = 5.12, P = 0.012). Note: there was no significant difference between AMPA-triggered currents in control and BDNF plus TGFα-treated cultures ( P = 0.90). post hoc: *P < 0.05, **P < 0.01, ***P < 0.001.
Physiological examination with whole-cell patch-clamp recording supported this immunoblot result (Fig. 8C, Table 2). BDNF elevated AMPA-evoked currents from 131.9 ± 8.6 to 357.0 ± 98.5 pA ( P < 0.05) in the multipolar-type neurons. This upregulation in AMPA currents was inhibited by co-application with TGFα. AMPA currents in TGFα/BDNF-treated culture were indistinguishable from those of untreated cultures. BDNF treatment caused an increase in membrane capacitance of the multipolar-type neurons, which presumably reflect an increase in somatic diameter and/or neurite length (McAllister et al., 1995; Jin et al., 2003; Nagano et al., 2003) (Table 2). Co-application with TGFα, however, failed to influence this activity of BDNF. These results indicate that TGFα specifically counteracts the neurotrophic action of BDNF on GluR1 receptor function.
Discussion
Growth factors and neurotrophic factors have long-lasting actions to promote the phenotypic development of neurons, including neurochemical maturation and synaptic formation (McAllister, 2002; Vicario-Abejon et al., 2002). For example, subchronic treatment with BDNF facilitates the expression of AMPA-type glutamate receptors, which lead to synaptic maturation in developing neocortex and hippocampus (Narisawa-Saito et al., 1999b; Rutherford et al., 1998; Sherwood and Lo, 1999; Bolton et al., 2000). An ErbB3/4 ligand, neuregulin-1, regulates the expression and function of NMDA-type glutamate receptors (Ozaki et al., 1997). The present study reveals an opposite activity of ErbB1 receptor ligands, which down-regulates the expression and function of the AMPA-type glutamate receptor, GluR1. Treatment with TGFα or other ErbB1 ligands (EGF and HB-EGF) decreased the expression of GluR1 protein in short-term neocortical cultures. The inhibitor for ErbB1 tyrosine receptor kinase fully blocked the action of these ErbB1 ligands. More prolonged treatment with TGFα decreased the amplitudes of spontaneous mEPSP, which presumably reflects a decrease in AMPA receptor expression at synapses. Other growth factors such as basic fibroblast growth factor (bFGF) and platelet-derived growth factor (PDGF), both of which are potent mitogens for astrocytes and fibroblast, have no ability to down-regulate AMPA receptor expression (Narisawa-Saito et al., 1999b; Jourdi and Nawa, 2002). These results suggest that peptide ligands for ErbB1 receptors have a unique biological activity to suppress the development of GluR1 receptor-containing synaptic AMPA receptors.
Competition between the positive and negative regulators, BDNF and TGFα
Previous studies suggest that subchronic stimulation of BDNF receptors increases and maintains the expression of both GluR1 and AMPA currents in multipolar GABAergic neurons both in vivo and in vitro (McLean Bolton et al., 2000; Nagano et al., 2003). The present investigation demonstrates that the BDNF-induced up-regulation of AMPA currents was reversed to control levels by co-application of TGFα. TGFα appeared to have a competitive role against conventional neurotrophic factor in the regulation of GluR1 subunit expression. BDNF also increases whole-cell capacitance in cultured neurons, which presumably represents an increase in soma size and/or neurite length (Jin et al., 2003; Widmer and Hefti, 1994; Wirth et al., 2003). Co-treatment with TGFα, however, failed to counteract this action of BDNF.
A large amount of BDNF is produced and released mainly from excitatory neurons, while TGFα is synthesized in both neurons and glial cells (Seroogy et al., 1993; Conner et al., 1997). These two different factors might converge onto the same population of GABAergic neurons to regulate GluR1 expression. These observations suggest that the counteraction of TGFα against BDNF is relatively specific for the regulation of AMPA receptor phenotype and might alter the composition of GluR1-containing AMPA receptor channel complexes at the synapse. What is the intracellular signal that plays a critical role in distinction of the biological reactions with TGFα and BDNF? This is an interesting question and remains to be characterized in future studies, as both factors activate the same set of signal transducers such as the Ras/MAP kinase cascade, the PI3 kinase/Akt cascade and the PLC gamma/ PKC cascade (Yamada et al., 1997; Harris et al., 2003; Xian and Zhou, 2004).
We have previously shown multiple effects of BDNF in the regulation of AMPA receptors, including their protein expression, interaction with cytoskeletal components and partner PDZ proteins, and channel activity and localizations (Narisawa-Saito et al., 2002; Jourdi et al., 2003). In addition, BDNF effects on AMPA receptors involve the activation of N-ethylmaleimide-sensitive factor (NSF) and the subsequent recruitment of the AMPA receptor-associated PDZ scaffolding molecules (Narisawa-Saito et al., 2002; Jourdi et al., 2003). Our recent results indicate that EGF and other ErbB1 ligands down-regulates the expression of the AMPA receptor-associating PDZ molecules such as SAP-97 and GRIP, which bind to AMPA receptors and stabilize them by providing a link to cytoskeleton of postsynaptic sites (Yokomaku et al., 2005). Therefore, it is possible that BDNF and TGFα also contribute to postsynaptic organization of the AMPA receptors by positively and negatively influencing the expression of PDZ scaffolding proteins as well.
Distinction between mitotic activity and AMPA receptor-reducing activity of TGFα
ErbB1 ligands are known to stimulate proliferation of various types of cells including astrocytes (Reynolds and Weiss, 1996; Rabchevsky et al., 1998). In the present study, however, the effects of TGFα on AMPA receptor, does not appear to involve astrocyte proliferation and was distinct from its mitotic action. The combination of serum-free medium with the anti-mitotic treatment attenuated glial proliferation. In agreement with the previous reports, a subpopulation of GABAergic cells expressed one of EGF receptor subtypes, ErbB1, and bound to fluorescent-labeled EGF (Gómez-Pinilla et al., 1988; Kornblum et al., 1995). Moreover, the GluR1-reducing activity of TGFα was apparent not only in AMPA binding activity per milligram protein but also on per culture dish basis: If the mitotic activity of TGFα contributed to the change in AMPA binding, then glial proliferation should either not influence or increase AMPA binding per dish, because astrocytes also express functional AMPA receptors (Seifert and Steinhauser, 2001; Iino et al., 2001). The marked decrease in AMPA binding by TGFα ruled out this explanation. In our physiological analysis, long-term culture of cortical neurons was required to obtain reasonable amounts of miniature currents in whole-cell patch-clamp recording, although the longer culture duration allowed astrocytes to proliferate. Even in the presence of a large glial population, however, there was a similar degree of decrease in AMPA receptors to that observed in the short-term cultures. Although we cannot rule out the possibility that non-neuronal population including glial cells might indirectly influence this process, it is less likely that glial cells played a central role on AMPA receptor down-regulation. The fact that other potent growth factors for glial cells (PDGF and bFGF) failed to mimic the AMPA receptor-reducing activity of TGFα may support this explanation (Narisawa-Saito et al., 1999b; Jourdi and Nawa, 2002).
Influences of TGFα on AMPA receptor binding kinetics
[3H]AMPA binding assays revealed that untreated control cultures exhibited similar Bmax and Kd values to those reported previously (Narisawa-Saito et al., 2002). Treatment of cultured cortical neurons with TGFα decreased the Bmax of the binding, which implies a decrease in the total number of surface AMPA receptors. This suggests that the TGFα-triggered reduction in total AMPA receptor content resulted in decreased surface AMPA receptors as well. In parallel, Kd values were decreased by TGFα treatment, suggesting an increase in AMPA receptor affinity. There are high and low affinity components of AMPA binding in brain preparations (Hall et al., 1992; Standley et al., 1994, 1998). It is possible that the observed alteration in the affinity of AMPA binding might be caused by post-translational modification of AMPA receptors following ErbB1 activation (Matsuda et al., 1999; Suzuki et al., 2005). Alternatively, the ErbB1 activation reduces the expression of AMPA receptor-associating PDZ proteins, SAP97 and GRIP1, and potentially influences subcellular distributions and affinity of the AMPA receptor subunits (Yokomaku et al., 2005). The real nature of the affinity increase awaits further investigation at molecular levels.
Biological implication of TGFα action
Both neural stem cells and their immediate postmitotic descendents express ErbB1 receptors in developing cortical structures (Eagleson et al., 1996; Fox and Kornblum, 2005). These in vivo observations on ErbB1 receptors agree with the present culture results. A significant proportion of cultured cortical neurons including GABAergic neurons, which were prepared from rat embryos at late gestation, were positive for ErbB1 and/or ErbB4 (Yau et al., 2003; Fox and Kornblum, 2005). Even at later postnatal stages, ErbB1 immunoreactivity or mRNA is detected in basket-like cells or GABAergic neurons in the neocortex (Gómez-Pinilla et al., 1988; Kornblum et al., 1995; Fox and Kornblum, 2005). Therefore, the down-regulation of AMPA-type glutamate receptor presumably represents the direct action of TGFα and any other ErbB1 ligands on developing GABAergic neurons. In vivo, however, the activation of the other ErbB receptors might mimic the action of TGFα. ErbB2–4 receptors are known to interact with ErbB1 and the heterodimerization of ErbB1 and the other ErbB receptor phosphorylates and transactivates ErbB1 receptors (Leahy, 2004; Xian and Zhou, 2004, both for review). Our preliminary result that in vivo challenge of another ErbB1 ligand, EGF, down-regulates AMPA receptor expression and function in the neocortex supports this argument (TN unpublished data).
At the adult stage, TGFα and HB-EGF are induced by seizures and suggested to protect neurons from glutamate-induced excitotoxic damage (Petegnief et al., 2003; Opanashuk et al., 1999). These previous results are consistent with the present finding. The ErbB1 ligands down-regulate the functional expression of the glutamate receptors and, therefore, exert a neuroprotective activity against excitotoxicity. Future studies will evaluate the pathological significance of the AMPA receptor reducing activity in ischemic and seizure paradigms as well. The present findings illustrate the presence of a negative regulatory factor(s) in the central nervous system, which might compete with conventional neurotrophic activities. Such negative regulatory factors, together with conventional positive neurotrophic factors, may play a role in local regulation of development or degeneration of inhibitory cortical neurons.
Experimental methods
Cell culture
The neocortex of embryonic day 19 (E19) rats (Sprague–Dawley; Nihon SLC, Shizuoka, Japan) was treated with papain and mechanically dissociated (Nagano et al., 2003). Dissociated neurons were suspended and plated onto poly-D-lysine-coated dishes at a density of 500–800 cells/mm2 in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% calf serum and grown in DMEM containing 0.5 mM glutamine, nutrient mixture N2, and 10 mM HEPES (pH 7.3) to minimize cell proliferation (serum-free N2 medium) (Narisawa-Saito et al., 1999a). In biochemical experiments, BDNF (Sumitomo Pharm. Ltd., Osaka, Japan, 50 ng/ml), human EGF (20 ng/ml; Sigma Chemical Co., St. Louis, MO), human TGFα (20 ng/ml; Sigma Chemical), or human HB-EGF (20 ng/ml; R&D Systems) was added daily from 1 DIV for 5 to 6 days. For electrophysiological analysis, cytosine beta-arabinofuranoside (AraC, 5 μM; Wako, Tokyo, Japan) was added to inhibit glial proliferation (see below). The use of serum-free N2 medium in combination with AraC reduced glial contamination to less than 5% as examined with glial fibrillary acidic protein immunostaining (Narisawa-Saito et al., 1999b).
Immunostaining
GluR1 immunoreactivity and its localizations in neuronal culture were examined by immunostaining. Neocortical culture was fixed with 4% paraformaldehyde in the presence or absence of 0.5% glutaraldehyde for 30 min and immunostained with the following primary antibodies; mouse anti-GAD67 (2 μg/ml; Chemicon Int., Temecra, CA), rabbit anti-GluR1 (2.5 μg/ml; Chemicon Int., Temecra, CA), rabbit anti-ErbB1 (5 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA), and rabbit anti-GABA antibodies (1:500, Immunotech, Plaque, Czech Republic). Their immunoreactivity was visualized with the incubation of the secondary antibodies conjugated with Alexa Fluor® 488 (1:200) or 546 (1:500) (both Molecular Probes, Eugene, OR) or the Vectastain ABC elite kit (1:100 for secondary antibody; Vector Laboratories, Burlingame, CA). Immunostained culture was observed with the aid of an Axioscop and pictured with a digital camera (1392 × 1040 pixels; Olympus, Tokyo, Japan). Captured neuron images were analyzed by imaging software (Adobe Photoshop® version 5.5, Adobe, San Jose, CA). The intensity of red fluorescence for GluR1 immunoreactivity was measured in somatic area and subtracted that in background area. Analysis was performed on 30–50 GAD67-immunoreactive multipolar and bipolar cells and GAD67-negative cells with or without pyramidal morphology chosen randomly from four sister culture dishes in each group.
Western blotting
Immunoblot analysis was performed according to standard procedures as previously described (Narisawa-Saito et al., 1999b; Xiong et al., 1999). Briefly, cultured cells were lysed in Laemmli sample buffer (2% sodium dodecyl sulfate (SDS), 62.5 mM Tris pH 6.8). After centrifugation, protein in supernatants was denatured at 95°C in the presence of 5% 2-mercaptoethanol and 10% glycerol. Protein (50 μg/lane) was separated by SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane by electrophoresis. Primary antibodies were diluted and incubated with the membrane at 4°C overnight. Immunoreactivity was detected with goat anti-rabbit or anti-mouse immunoglobulin conjugated to peroxidase (1: 10,000; Vector Laboratories, Burlingame, CA), followed by chemiluminescence reaction combined with film exposure (ECL kit, Amersham Biosciences, Tokyo, Japan). The following primary antibodies were used: affinity-purified rabbit anti-GluR1 (Chemicon International, Temecula, CA), anti-GluR2/3 (Chemicon International), rabbit anti-GFAP (DAKO, Denmark) and mouse anti-beta actin (Chemicon International) antibodies. The immunoreactivity of the bands was quantified by densitometric analysis.
[3H]AMPA binding assay
[3H]AMPA binding assays were performed by the procedures described previously (Narisawa-Saito et al., 2002). Briefly, cultures were rinsed with Tris buffer, pH 7.4, containing 100 μM sodium acetate, 2.5 mM CaCl2, and 5 g/L glucose. [3H]AMPA (Perkin Elmer Life Sciences) was adjusted to 50 nM with the Tris buffer and diluted with 0 to 800 nM cold AMPA (Sigma). Neocortical cultures were incubated with the AMPA solution on ice for 60 min. An excess amount of cold AMPA (100 μM) was applied to the culture to examine nonspecific [3H]AMPA binding to cells. After rapid washing with cold Tris buffer, cells were lysed with 0.5 N NaOH and the radioactivity of cell lysates was measured with an LSC-3050 liquid scintillation counter (Aloka, Tokyo, Japan).
Electrophysiological analysis
Electrophysiological data of AMPA currents were obtained from neocortical cultures of 8 and 9 DIV, which express membrane excitability and channel activity. Because TGFα has mitotic actions on putative progenitor cells (Kornblum et al., 1998, 1999; Reynolds and Weiss, 1992), chronic treatment for more than 5 days with TGFα markedly enhanced proliferation of these non-neuronal cells in neocortical cultures. To minimize the indirect effects of TGFα via proliferating non-neuronal cells, cultures were treated with AraC for 48 h from 4 DIV. A non-competitive ErbB1 tyrosine kinase inhibitor, PD 153035 (100 nM; Calbiochem, San Diego, CA) was applied 2 h before TGFα treatment (Bos et al., 1997). For observation of miniature excitatory postsynaptic currents (mEPSCs), TGFα (20 ng/ml) was applied daily from 8 DIV for 5 days. Tetrodotoxin (TTX; 1 μM; Wako) was also applied daily to eliminate the indirect influences of synaptic activity. AraC was applied for 24 h from 8 DIV. Electrophysiological results were obtained at 13 and 14 DIV.
GABAergic neurons were identified based on morphological criteria (de Lima and Voigt, 1997; Nagano et al., 2003). Typical multipolar-type GABAergic neurons were characterized by their triangular or rectangular somata with four to six thick dendrites (see Fig. 3). Cells were recorded using the whole-cell patch-clamp method. Recording medium contained 150 mM NaCl, 4 mM KCl, 2 mM CaCl2, 10 mM HEPES, 10 mM glucose, 1 mM MgSO4, and 10 mM sucrose (320 mosM; pH 7.4). Whole-cell patch-clamp recording was performed using borosilicate glass capillaries pulled to a tip resistance of 3 to 5 MΩ. The patch pipette contained 130 mM CsMeSO3, 10 mM CsCl, 10 mM HEPES, 0.5 mM EGTA, 5 mM QX-314Cl, 0.1 mM spermine–4HCl, and 3 mM Mg–ATP and sucrose was added to adjust the osmolarity to 300 mosM (pH 7.4). Input resistance and series resistance was determined by measuring the current change in response to a negative 5-mV pulse from a holding potential of −78 mV. Cell capacitance measurements were made by integration of capacitive transients. All data were filtered at 2 kHz and digitized at a sampling rate of 10 kHz. Recordings with a series resistance of more than 40 MΩ or in which the series resistance changed by more than 20% were excluded. Using a picospritzer (WPI, New Haven, CT) and a second recording pipette filled with 100 μM AMPA (Tocris Cookson Inc, Bristol, UK), we measured AMPA currents induced by local puff application (6 psi, 10 ms). For recording of miniature postsynaptic currents, 10 μM bicuculline (Sigma) and 1 μM TTX (Wako) were added to the external solution.
Real-time quantitative PCR
Total culture RNA from 4 DIV culture was extracted as described previously (Namba et al., 2003). Quantitative assessment of mRNA levels was carried out with a real time PCR machine (Light Cycler, Roche Diagnostics, Tokyo, Japan). Total RNA (20–200 ng) was subjected to RT-PCR amplification (60°C 10 min, 94°C 2 s, 35 cycles of 94°C, 55°C 5 s, 72°C 10 s and measuring fluorescent after 80–85°C incubation for 1 s) in a final volume of 25 μl using RT-PCR high plus (Toyobo, Osaka, Japan), containing of ×1/20,000 SYBR® Green I (Molecular Probes) and 5 pmol of primers listed in Table 1. PCR primers were characterized previously (Porter et al., 1998; Kamphuis et al., 2003), or designed with the computer program, Primer 3 (Rozen and Skaletsky, 2000). Relative amounts of mRNA of glutamate receptors were calculated by the differences of cycles during linear amplification, and normalized to the relative expression of GAPDH mRNA.
Statistics
Data are expressed as mean ± SE. Statistical analysis of molecular data was performed using one-way analysis of variance followed by the Bonferroni test. Alternatively, Student’s t test was applied to compare two experimental groups.
Acknowledgments
We thank Sumitomo Pharmaceuticals for recombinant BDNF. A preliminary form of the present study was published in the Society for Neuroscience Abstract (Namba et al., 2000). Supported by: Grant-in-Aid for Creative Scientific Research and for Young Investigators from the Ministry of Education, Culture, Sports, Science and Technology, and Grants for Promotion of Niigata University Research Projects.
References
- Assimacopoulos S, Grove EA, Ragsdale CW. Identification of a Pax6-dependent epidermal growth factor family signaling source at the lateral edge of the embryonic cerebral cortex. J Neurosci. 2003;23:6399–6403. doi: 10.1523/JNEUROSCI.23-16-06399.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton MM, Pittman AJ, Lo DC. Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci. 2000;20:3221–3232. doi: 10.1523/JNEUROSCI.20-09-03221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos M, Mendelsohn J, Kim YM, Albanell J, Fry DW, Baselga J. PD153035, a tyrosine kinase inhibitor, prevents epidermal growth factor receptor activation and inhibits growth of cancer cells in a receptor number-dependent manner. Clin Cancer Res. 1997;3:2099–2106. [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll SD, Wiegand SJ, Anderson KD, Lindsay RM, Nawa H. Regulation of neuropeptides in adult rat forebrain by the neurotrophins BDNF and NGF. Eur J Neurosci. 1994;6:1343–1353. doi: 10.1111/j.1460-9568.1994.tb00325.x. [DOI] [PubMed] [Google Scholar]
- de Lima AD, Voigt T. Identification of two distinct populations of gamma-aminobutyric acidergic neurons in cultures of the rat cerebral cortex. J Comp Neurol. 1997;388:526–540. [PubMed] [Google Scholar]
- Eagleson KL, Ferri RT, Levitt P. Complementary distribution of collagen type IV and the epidermal growth factor receptor in the rat embryonic telencephalon. Cereb Cortex. 1996;6:540–549. doi: 10.1093/cercor/6.3.540. [DOI] [PubMed] [Google Scholar]
- Fox IJ, Kornblum HI. Developmental profile of ErbB receptors in murine central nervous system: implications for functional interactions. J Neurosci Res. 2005;79:584–597. doi: 10.1002/jnr.20381. [DOI] [PubMed] [Google Scholar]
- Gómez-Pinilla F, Knauer DJ, Nieto-Sampedro M. Epidermal growth factor receptor immunoreactivity in rat brain. Development and cellular localization. Brain Res. 1988;438:385–390. doi: 10.1016/0006-8993(88)91369-8. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Zeiler SR, Tamowski S, Jones KR. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci. 2003;23:6856–6865. doi: 10.1523/JNEUROSCI.23-17-06856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RA, Kessler M, Lynch G. Evidence that high- and low-affinity DL-alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) binding sites reflect membrane-dependent states of a single receptor. J Neurochem. 1992;59:1997–2004. doi: 10.1111/j.1471-4159.1992.tb10086.x. [DOI] [PubMed] [Google Scholar]
- Hanke M, Farkas LM, Jakob M, Ries R, Pohl J, Sullivan AM. Heparin-binding epidermal growth factor-like growth factor: a component in chromaffin granules which promotes the survival of nigrostriatal dopaminergic neurones in vitro and in vivo. Neuroscience. 2004;124:757–766. doi: 10.1016/j.neuroscience.2003.12.033. [DOI] [PubMed] [Google Scholar]
- Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, Miwa A, Takayasu Y, Saito I, Tsuzuki K, Ozawa S. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292:926–929. doi: 10.1126/science.1058827. [DOI] [PubMed] [Google Scholar]
- Ip NY, Li Y, Yancopoulos GD, Lindsay RM. Cultured hippocampal neurons show responses to BDNF, NT-3, and NT-4, but not NGF. J Neurosci. 1993;13:3394–3405. doi: 10.1523/JNEUROSCI.13-08-03394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Hu H, Mathers PH, Agmon A. Brain-derived neurotrophic factor mediates activity-dependent dendritic growth in non-pyramidal neocortical interneurons in developing organotypic cultures. J Neurosci. 2003;23:5662–5673. doi: 10.1523/JNEUROSCI.23-13-05662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdi H, Nawa H. Basic fibroblast growth factor modulates the expression of PDZ domain-containing proteins in cultured cortical neurons. Acta Med Biol. 2002;50:107–115. [PMC free article] [PubMed] [Google Scholar]
- Jourdi H, Iwakura Y, Narisawa-Saito M, Ibaraki K, Xiong H, Watanabe M, Hayashi Y, Takei N, Nawa H. Brain-derived neurotrophic factor signal enhances and maintains the expression of AMPA receptor-associated PDZ proteins in developing cortical neurons. Dev Biol. 2003;263:216–230. doi: 10.1016/j.ydbio.2003.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Dijk F, O’Brien BJ. Gene expression of AMPA-type glutamate receptor subunits in rod-type ON bipolar cells of rat retina. Eur J Neurosci. 2003;18:1085–1092. doi: 10.1046/j.1460-9568.2003.02841.x. [DOI] [PubMed] [Google Scholar]
- Kawahara N, Mishima K, Higashiyama S, Taniguchi N, Tamura A, Kirino T. The gene for heparin-binding epidermal growth factor-like growth factor is stress-inducible: its role in cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:307–320. doi: 10.1097/00004647-199903000-00009. [DOI] [PubMed] [Google Scholar]
- Kharazia VN, Wenthold RJ, Weinberg RJ. GluR1-immunopositive interneurons in rat neocortex. J Comp Neurol. 1996;368:399–412. doi: 10.1002/(SICI)1096-9861(19960506)368:3<399::AID-CNE6>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Kondo M, Okabe S, Sumino R, Okado H. A high GluR1: GluR2 expression ratio is correlated with expression of Ca2+-binding proteins in rat forebrain neurons. Eur J Neurosci. 2000;12:2812–2822. doi: 10.1046/j.1460-9568.2000.00167.x. [DOI] [PubMed] [Google Scholar]
- Kornblum HI, Chugani HT, Tatsukawa K, Gall CM. Cerebral hemidecortication alters expression of transforming growth factor alpha mRNA in the neostriatum of developing rats. Mol Brain Res. 1994;21:107–114. doi: 10.1016/0169-328x(94)90383-2. [DOI] [PubMed] [Google Scholar]
- Kornblum HI, Gall CM, Seroogy KB, Lauterborn JC. A subpopulation of striatal GABAergic neurons expresses the epidermal growth factor receptor. Neuroscience. 1995;69:1025–1029. doi: 10.1016/0306-4522(95)00392-v. [DOI] [PubMed] [Google Scholar]
- Kornblum HI, Hussain RJ, Bronstein JM, Gall CM, Lee DC, Seroogy KB. Prenatal ontogeny of the epidermal growth factor receptor and its ligand, transforming growth factor alpha, in the rat brain. J Comp Neurol. 1997;380:243–261. doi: 10.1002/(sici)1096-9861(19970407)380:2<243::aid-cne7>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Kornblum HI, Hussain R, Wiesen J, Miettinen P, Zurcher SD, Chow K, Derynck R, Werb Z. Abnormal astrocyte development and neuronal death in mice lacking the epidermal growth factor receptor. J Neurosci Res. 1998;53:697–717. doi: 10.1002/(SICI)1097-4547(19980915)53:6<697::AID-JNR8>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Kornblum HI, Zurcher SD, Werb Z, Derynck R, Seroogy KB. Multiple trophic actions of heparin-binding epidermal growth factor (HB-EGF) in the central nervous system. Eur J Neurosci. 1999;11:3236–3246. doi: 10.1046/j.1460-9568.1999.00744.x. [DOI] [PubMed] [Google Scholar]
- Leahy DJ. Structure and function of the epidermal growth factor (EGF/ErbB) family of receptors. Adv Protein Chem. 2004;68:1–27. doi: 10.1016/S0065-3233(04)68001-6. [DOI] [PubMed] [Google Scholar]
- Maiese K, Boniece I, DeMeo D, Wagner JA. Peptide growth factors protect against ischemia in culture by preventing nitric oxide toxicity. J Neurosci. 1993;13:3034–3040. doi: 10.1523/JNEUROSCI.13-07-03034.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty S, Carroll P, Cellerino A, Castren E, Staiger V, Thoenen H, Lindholm D. Brain-derived neurotrophic factor promotes the differentiation of various hippocampal nonpyramidal neurons, including Cajal-Retzius cells, in organotypic slice cultures. J Neurosci. 1996;16:675–687. doi: 10.1523/JNEUROSCI.16-02-00675.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Mikawa S, Hirai H. Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J Neurochem. 1999;73:1765–1768. doi: 10.1046/j.1471-4159.1999.731765.x. [DOI] [PubMed] [Google Scholar]
- McAllister AK. Neurotrophins and cortical development. Results Probl Cell Differ. 2002;39:89–112. doi: 10.1007/978-3-540-46006-0_5. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- McLean Bolton M, Pittman AJ, Lo DC. Brain-derived neurotrophic factor differentially regulates excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci. 2000;20:3221–3232. doi: 10.1523/JNEUROSCI.20-09-03221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K, Carnahan J, Nawa H. Brain-derived neurotrophic factor promotes differentiation of striatal GABAergic neurons. Dev Biol. 1994;165:243–256. doi: 10.1006/dbio.1994.1250. [DOI] [PubMed] [Google Scholar]
- Nagano T, Yanagawa Y, Obata K, Narisawa-Saito M, Namba H, Otsu Y, Takei N, Nawa H. Brain-derived neurotrophic factor upregulates and maintains AMPA receptor currents in neocortical GABAergic neurons. Mol Cell Neurosci. 2003;24:340–356. doi: 10.1016/s1044-7431(03)00172-6. [DOI] [PubMed] [Google Scholar]
- Namba H, Xiong H, Futamura T, Otsu Y, Takei N, Nawa H. Factors in EGF family reduce spontaneous synaptic activities in developing rat neocortex by downregulating glutamate receptor expressions. Abstr-Soc Neurosci. 2000;26:223.15. [Google Scholar]
- Namba H, Takei N, Nawa H. Transforming growth factor α changes firing properties of developing neocortical GABAergic neurons by down-regulation of voltage-gated potassium currents. Neuroscience. 2003;122:637–646. doi: 10.1016/j.neuroscience.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Carnahan J, Araki K, Yamaguchi T, Nawa H. Brain-derived neurotrophic factor regulates the expression of AMPA receptor proteins in neocortical neurons. Neuroscience. 1999a;88:1009–1014. doi: 10.1016/s0306-4522(98)00496-5. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Silva AJ, Yamaguchi T, Hayashi T, Yamamoto T, Nawa H. Growth factor-mediated Fyn signaling regulates α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor expression in rodent neocortical neurons. Proc Natl Acad Sci U S A. 1999b;96:2461–2466. doi: 10.1073/pnas.96.5.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narisawa-Saito M, Iwakura Y, Kawamura M, Araki K, Kozaki S, Takei N, Nawa H. Brain-derived neurotrophic factor regulates surface expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors by enhancing the N-ethylmaleimide-sensitive factor/GluR2 interaction in developing neocortical neurons. J Biol Chem. 2002;277:40901–40910. doi: 10.1074/jbc.M202158200. [DOI] [PubMed] [Google Scholar]
- Nawa H, Bessho Y, Carnahan J, Nakanishi S, Mizuno K. Regulation of neuropeptide expression in cultured cerebral cortical neurons by brain-derived neurotrophic factor. J Neurochem. 1993;60:772–775. doi: 10.1111/j.1471-4159.1993.tb03216.x. [DOI] [PubMed] [Google Scholar]
- Opanashuk LA, Mark RJ, Porter J, Damm D, Mattson MP, Seroogy KB. Heparin-binding epidermal growth factor-like growth factor in hippocampus: modulation of expression by seizures and anti-excitotoxic action. J Neurosci. 1999;19:133–146. doi: 10.1523/JNEUROSCI.19-01-00133.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki M, Sasner M, Yano R, Lu HS, Buonanno A. Neuregulin-beta induces expression of an NMDA-receptor subunit. Nature. 1997;390:691–694. doi: 10.1038/37795. [DOI] [PubMed] [Google Scholar]
- Peng H, Wen TC, Tanaka J, Maeda N, Matsuda S, Desaki J, Sudo S, Zhang B, Sakanaka M. Epidermal growth factor protects neuronal cells in vivo and in vitro against transient forebrain ischemia-and free radical-induced injuries. J Cereb Blood Flow Metab. 1998;18:349–360. doi: 10.1097/00004647-199804000-00002. [DOI] [PubMed] [Google Scholar]
- Petegnief V, Friguls B, Sanfeliu C, Sunol C, Planas AM. Transforming growth factor-alpha attenuates N-methyl-D-aspartic acid toxicity in cortical cultures by preventing protein synthesis inhibition through an Erk1/2-dependent mechanism. J Biol Chem. 2003;278:29552–29559. doi: 10.1074/jbc.M300661200. [DOI] [PubMed] [Google Scholar]
- Porter JT, Cauli B, Staiger JF, Lambolez B, Rossier J, Audinat E. Properties of bipolar VIPergic interneurons and their excitation by pyramidal neurons in the rat neocortex. Eur J Neurosci. 1998;10:3617–3628. doi: 10.1046/j.1460-9568.1998.00367.x. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Weinitz JM, Coulpier M, Fages C, Tinel M, Junier MP. A role for transforming growth factor alpha as an inducer of astrogliosis. J Neurosci. 1998;18:10541–10552. doi: 10.1523/JNEUROSCI.18-24-10541.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reibel S, Vivien-Roels B, Le BT, Larmet Y, Carnahan J, Marescaux C, Depaulis A. Overexpression of neuropeptide Y induced by brain-derived neurotrophic factor in the rat hippocampus is long lasting. Eur J Neurosci. 2000;12:595–605. doi: 10.1046/j.1460-9568.2000.00941.x. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol. 1996;175:1–13. doi: 10.1006/dbio.1996.0090. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- Seifert G, Steinhauser C. Ionotropic glutamate receptors in astrocytes. Prog Brain Res. 2001;132:287–299. doi: 10.1016/S0079-6123(01)32083-6. [DOI] [PubMed] [Google Scholar]
- Seroogy KB, Lundgren KH, Lee DC, Guthrie KM, Gall CM. Cellular localization of transforming growth factor-alpha mRNA in rat forebrain. J Neurochem. 1993;60:1777–1782. doi: 10.1111/j.1471-4159.1993.tb13403.x. [DOI] [PubMed] [Google Scholar]
- Sherwood NT, Lo DC. Long-term enhancement of central synaptic transmission by chronic brain-derived neurotrophic factor treatment. J Neurosci. 1999;19:7025–7036. doi: 10.1523/JNEUROSCI.19-16-07025.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibilia M, Steinbach JP, Stingl L, Aguzzi A, Wagner EF. A strain-independent postnatal neurodegeneration in mice lacking the EGF receptor. EMBO J. 1998;17:719–731. doi: 10.1093/emboj/17.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley S, Irvin N, Baudry M. Differential subcellular localization of two populations of glutamate/AMPA receptors in the rat telencephalon. Neurochem Int. 1994;25:287–293. doi: 10.1016/0197-0186(94)90072-8. [DOI] [PubMed] [Google Scholar]
- Standley S, Tocco G, Wagle N, Baudry M. High- and low-affinity alpha-[3H]amino-3-hydroxy-5-methylisoxazole-4-propionic acid ([3H]AMPA) binding sites represent immature and mature forms of AMPA receptors and are composed of differentially glycosylated subunits. J Neurochem. 1998;70:2434–2445. doi: 10.1046/j.1471-4159.1998.70062434.x. [DOI] [PubMed] [Google Scholar]
- Suzuki E, Kessler M, Arai AC. C-terminal truncation affects kinetic properties of GluR1 receptors. Mol Cell Neurosci. 2005;29:1–10. doi: 10.1016/j.mcn.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Takei N, Sasaoka K, Inoue K, Takahashi M, Endo Y, Hatanaka H. Brain-derived neurotrophic factor increases the stimulation-evoked release of glutamate and the levels of exocytosis-associated proteins in cultured cortical neurons from embryonic rats. J Neurochem. 1997;68:370–375. doi: 10.1046/j.1471-4159.1997.68010370.x. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, Barnard JA, Yuspa SH, Coffey RJ, Magnuson T. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejon C, Owens D, McKay R, Segal M. Role of neurotrophins in central synapse formation and stabilization. Nat Rev, Neurosci. 2002;3:965–974. doi: 10.1038/nrn988. [DOI] [PubMed] [Google Scholar]
- Widmer HR, Hefti F. Stimulation of GABAergic neuron differentiation by NT-4/5 in cultures of rat cerebral cortex. Dev Brain Res. 1994;80:279–284. doi: 10.1016/0165-3806(94)90114-7. [DOI] [PubMed] [Google Scholar]
- Wirth MJ, Brun A, Grabert J, Patz S, Wahle P. Accelerated dendritic development of rat cortical pyramidal cells and interneurons after biolistic transfection with BDNF and NT4/5. Development. 2003;130:5827–5838. doi: 10.1242/dev.00826. [DOI] [PubMed] [Google Scholar]
- Xian CJ, Zhou XF. EGF family of growth factors: essential roles and functional redundancy in the nerve system. Front Biosci. 2004;9:85–92. doi: 10.2741/1210. [DOI] [PubMed] [Google Scholar]
- Xiong H, Narisawa-Saito M, Jourdi H, Nawa H. The glutamate receptor-mediated regulation of brain-derived neurotrophic factor production by cytokines in central nervous system. Abstr-Soc Neurosci. 1999;25:407.10. [Google Scholar]
- Yamada M, Ikeuchi T, Hatanaka H. The neurotrophic action and signalling of epidermal growth factor. Prog Neurobiol. 1997;51:19–37. doi: 10.1016/s0301-0082(96)00046-9. [DOI] [PubMed] [Google Scholar]
- Yau HJ, Wang HF, Lai C, Liu FC. Neural development of the neuregulin receptor ErbB4 in the cerebral cortex and the hippocampus: preferential expression by interneurons tangentially migrating from the ganglionic eminences. Cereb Cortex. 2003;13:252–264. doi: 10.1093/cercor/13.3.252. [DOI] [PubMed] [Google Scholar]
- Yin H, Turetsky D, Choi DW, Weiss JH. Cortical neurones with Ca2+ permeable AMPA/kainate channels display distinct receptor immunoreactivity and are GABAergic. Neurobiol Dis. 1994;1:43–49. doi: 10.1006/nbdi.1994.0006. [DOI] [PubMed] [Google Scholar]
- Yokomaku D, Jourdi H, Kakita A, Nagano T, Takahashi H, Takei N, Nawa H. ErbB1 receptor ligands attenuate the expression of synaptic scaffolding proteins, GRIP1 and SAP97, in developing neocortex. Neuroscience. 2005;136:1037–1047. doi: 10.1016/j.neuroscience.2005.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]