

Abstract
Dengue virus (DENV) is an enveloped flavivirus with a positive-sense RNA genome transmitted by Aedes mosquitoes, causing the most important arthropod-borne viral disease affecting humans. Relatively few cis-acting RNA regulatory elements have been described in the DENV coding-region. Here, by introducing silent mutations into a DENV-2 infectious clone, we identify the conserved capsid-coding region 1 (CCR1), an RNA sequence element that regulates viral replication in mammalian cells and to a greater extent in Ae. albopictus mosquito cells. These defects were confirmed in vivo, resulting in decreased replication in Ae. aegypti mosquito bodies and dissemination to the salivary glands. Furthermore, CCR1 does not regulate translation, RNA synthesis or virion retention but likely modulates assembly, as mutations resulted in the release of non-infectious viral particles from both cell types. Understanding the role of CCR1 could help characterize the poorly-defined stage of assembly in the DENV life cycle and uncover novel anti-viral targets.
Keywords: Dengue virus, Flavivirus, cis-acting RNA regulatory element, Viral replication, Aedes aegypti mosquitoes, Assembly
Introduction
Dengue is caused by four distinct dengue virus serotypes (DENV1–4), which are transmitted by the Aedes aegypti and Ae. albopictus mosquitoes, and is the most prevalent arthropod-borne viral illness affecting humans (Gubler, 2002b). DENV is a member of the family Flaviviridae, which includes other major public health concerns such as yellow fever virus, hepatitis C virus and West Nile virus (Burke and Monath, 2001; Gubler, 2002a). DENV causes a spectrum of clinical disease, with an estimated 40 million cases of dengue fever (DF) and 250,000–500,000 cases of the more severe forms of the illness, dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) annually (Kyle and Harris, 2008). Based on vector distribution, approximately 3 billion of the world’s population is at risk for DENV infection as a result of human population growth, increased urbanization, and greater ease in international shipping and travel (Gubler, 1998, 2002b; Kyle and Harris, 2008; Rosen, 1977). Currently, there are no effective antiviral therapies or vaccines available for dengue and treatment is largely supportive; thus, controlling the mosquito population in order to reduce transmission is our best defense.
DENV is a small (~40 nm), enveloped RNA virus with a positive-sense RNA genome of ~11 kilobases that encodes 10 viral proteins in a single open reading frame (ORF) (Clyde et al., 2006). The genome contains a 5′ type 1 7-methyl-G cap and an ORF that is flanked by highly structured 5′ and 3′ untranslated regions (UTRs), and it lacks a poly(A) tail (Chambers et al., 1990; Lindenbach et al., 2007). The ORF is translated as a single polyprotein that is cleaved co- and post-translationally by both viral and host proteases (Arias et al., 1993). The viral genome encodes three structural proteins (C, prM/M and E) and seven nonstructural (NS) proteins (NS1, NS2A/B, NS3, NS4A/B and NS5). As with other positive-sense RNA viruses, the 5′ and 3′ UTRs contain several conserved cis-acting RNA elements that play key roles in regulating viral translation and RNA synthesis (Lindenbach et al., 2007).
The fully assembled flavivirus virion consists of a nucleocapsid core containing the viral RNA (vRNA), surrounded by a lipid bilayer derived from the endoplasmic recticulum (ER), which includes the viral E and prM/M proteins (Lindenbach et al., 2007). Flavivirus virions are assembled in association with intracellular membranes (Murphy, 1980) and are first observed in the ER lumen in vitro (Hase et al., 1987a; Ishak et al., 1988; Ko et al., 1979; Leary and Blair, 1980; Matsumara et al., 1977; Ng, 1987; Ohyama et al., 1977; Sriurairatna and Bhamarapravati, 1977) and in vivo (Hase et al., 1987b; Sriurairatna et al., 1973). However, while virions accumulate within membrane-bound vesicles, clear budding intermediates and naked nucleocapsids have not been observed (Alvisi et al., 2011; Welsch et al., 2009), which suggests that viral assembly is rapid. Viral assembly is localized to the membranous sites of replication by the hydrophobic segments of the anchored C protein (Nowak et al., 1989), and nucleocapsids acquire an envelope by budding into the ER lumen. The viral NS3 protein has been implicated in intracellular trafficking of the progeny non-infectious virions from the ER through the Golgi compartment (Chua et al., 2004). During virion maturation in the trans Golgi compartment, the prM protein is processed to the mature M protein by the host protease furin, which exposes the E receptor-binding domain that confers viral infectivity (Stadler et al., 1997; Zybert et al., 2008). Intracellular M-containing virions have not been detected, suggesting that prM cleavage occurs just before the release of mature virions (Lindenbach et al., 2007; Nelson et al., 2008; Yu et al., 2009). Virion transport from the sites of replication to the cell surface involves vesicles derived from the ER (Deubel and Digoutte, 1981) and other components of the exocytic pathway (Hase et al., 1987a; Heinz et al., 1994; Sriurairatna and Bhamarapravati, 1977). Recently, it has been shown that the DEAD-box RNA helicase DDX6 interacts with the DENV2 vRNA in vitro and in vivo via binding to the dumbbell (DB) structures 1 and 2 in the 3′ UTR, modulating the production of infectious viral particles (Ward et al., 2011). It is likely that DDX6 and other stress granule (SG) proteins recruit the vRNA to the P body and SG sites of assembly through their interaction with the 3′ UTR (Ward et al., 2011). Despite the information available on viral assembly, the mechanism by which the vRNA is recognized by the capsid protein to form the nucleocapsid precursor is still unknown. Thus far, no candidate assembly signals have been identified within the DENV genome, and the ability of subgenomic replicons lacking the majority of the structural proteincoding sequence to be assembled in trans (Jones et al., 2005; Khromykh et al., 1998) suggests that the signal does not reside in the prM/M/E region of the genome.
Much of what is known about RNA elements located in the UTRs has been shown to be equally important for viral replication in mammalian cells and mosquito cells, with a few exceptions. Deleting the variable region (VR) within the 3′ UTR reduces RNA synthesis in the mammalian baby hamster kidney cell line (BHK) but slightly amplifies RNA synthesis in an Ae. albopictus mosquito cell line(C6/36) (Alvarez et al., 2005a). On the other hand, changes to the bottom stem structure of the 3′ stem-loop (SL), located at the 3′ end of the viral genome, were shown to have wild-type (WT) replication levels in BHK cells but not in C6/36 cells (Zeng et al., 1998). To date, these are the only regions within the DENV genome that display cell type-associated differential effects on the viral life cycle, likely due to interactions with host-dependent factors. Indeed, identifying host-specific protein interactions is vital for understanding how the virus differentially modulates its life cycle in the human host and the mosquito vector.
Little information is available with respect to cis-acting RNA regulatory elements located within the DENV coding-region, as compared to those located within the 5′ and 3′ UTRs. Previous studies have shown that the 5′ conserved sequence (CS), found within the capsid-coding sequence, regulates RNA synthesis through interaction with the complementary 3′ CS, located within the 3′ UTR (Chiu et al., 2005; Khromykh et al., 2001). Recently, another set of complementary sequences, termed the downstream AUG region (DAR) was identified, with the 5′ DAR located just downstream of the initiating AUG and the complementary 3′ DAR sequence found in the 3′ UTR (Friebe and Harris, 2010; Friebe et al., 2011). The 5′/3′ DAR acts together with the 5′/3′ CS and 5′/3′ upstream AUG region (UAR), another pair of complementary sequences, to circularize the viral genome, which is a requirement for efficient RNA synthesis but is not involved in viral translation (Alvarez et al., 2005b, 2008; Friebe and Harris, 2010; Friebe et al., 2011; Khromykh et al., 2001). Additionally, a cis-acting RNA secondary structure, termed the capsid-coding hairpin (cHP), was recently found to regulate both viral translation and RNA synthesis in mammalian and mosquito cells (Clyde et al., 2008; Clyde and Harris, 2006). Based on the structure prediction algorithms that have been published, the DENV coding sequence has been reported to be rather unstructured (Hofacker et al., 1994; Thurner et al., 2004), though no functional proof exists that the coding-region lacks conserved cis-acting RNA sequence elements.
While none have been identified yet in DENV, coding-region cis-acting RNA regulatory elements have been found in a number of animal and plant viruses, modulating various aspects of the viral life cycle besides viral translation and RNA synthesis. These include trafficking of the vRNA to the membranous sites of replication, viral assembly, and other less well-characterized steps in viral replication (Liu et al., 2009; Miller and White, 2006). Specifically, in the cucumber necrosis tombusvirus, recruitment of the positive- and negative-strand vRNA to peroxisomal membranes is mediated via binding of a viral protein to a coding-region cis-acting RNA element (Panavas et al., 2005). In flock house viruses, an RNA structural element in RNA1 is predicted to form two stem-loops with nearly identical sequences that are required both for the recruitment of RNA1 to the outer mitochondrial membranes and for positive- and negative-strand RNA synthesis (Van Wynsberghe and Ahlguist, 2009). In Sindbis virus, a 132 nucleotide-long segment of the nsp1-coding sequence is predicted to form four stem loop structures (Frolova et al., 1997; Linger et al., 2004; Weiss et al., 1994), two of which were found to be essential for vRNA encapsidation (Linger et al., 2004). In the ORF1b of group 2 corona mouse hepatitis virus, a 69-nucleotide bulged stem-loop is required for viral packaging (Fosmire et al., 1992), whereas in nodaviruses, the packaging signal for RNA2 is located within a 32-nucleotide region of RNA2 (Zhong et al., 1992).
This study sought to identify additional cis-acting RNA regulatory elements in the DENV coding region in order to expand our knowledge of how the viral life cycle is regulated. We describe a novel coding-region cis-acting RNA regulatory element termed the conserved capsid-coding region 1 (CCR1), which regulates the viral life cycle in both mammalian and mosquito cells, though it displays a more prominent role in mosquito cells. Consistent with our in vitro data, CCR1 mutant viruses do not replicate as well as WT DENV-2 in Ae. aegypti mosquitoes, resulting in a reduction of viral transmission to the salivary glands. We demonstrate that CCR1 plays a role in post-RNA replication events, possibly acting as an assembly signal for DENV in a sequence-dependent and cell type-specific manner. These studies are important for improving our understanding of cis-acting determinants that differentially regulate the viral life cycle in the human host and mosquito vector, potentially serving in the design of attenuated vaccine strains or as targets for novel antiviral agents.
Results
A novel RNA element is predicted to be conserved both in sequence and secondary structure in the DENV and TBE serogroups
Selection of conserved coding-region RNA elements was based on an alignment of sequences from the representative genomes of DENV1–4, generated using Clustal W2 (Thompson et al., 1997). Conserved RNA sequence elements were identified by comparing the level of homology within a given stretch to that of the surrounding sequences, which was defined as 30 nucleotides upstream and downstream of the uncharacterized element, requiring at least 70% homology in the region of interest. The sequence alignment for DENV1–4 revealed a short stretch of sequence in the capsid gene that contains a greater degree of sequence homology (~89%) than that of the surrounding sequences (~64%) (Fig. 1A). The same stretch of sequence was identified in the tick-borne encephalitis (TBE) serogroup alignment (100% homology vs. 65% background) (Fig. 1C); however, the sequence was not well-conserved in the Japanese encephalitis virus (JEV) serogroup (44% homology vs. 32% background, Fig. 1B). Based on this high level of sequence conservation in the DENV and TBE serogroups, the RNA sequence element was termed the conserved capsid-coding region 1 (CCR1).
Fig. 1.
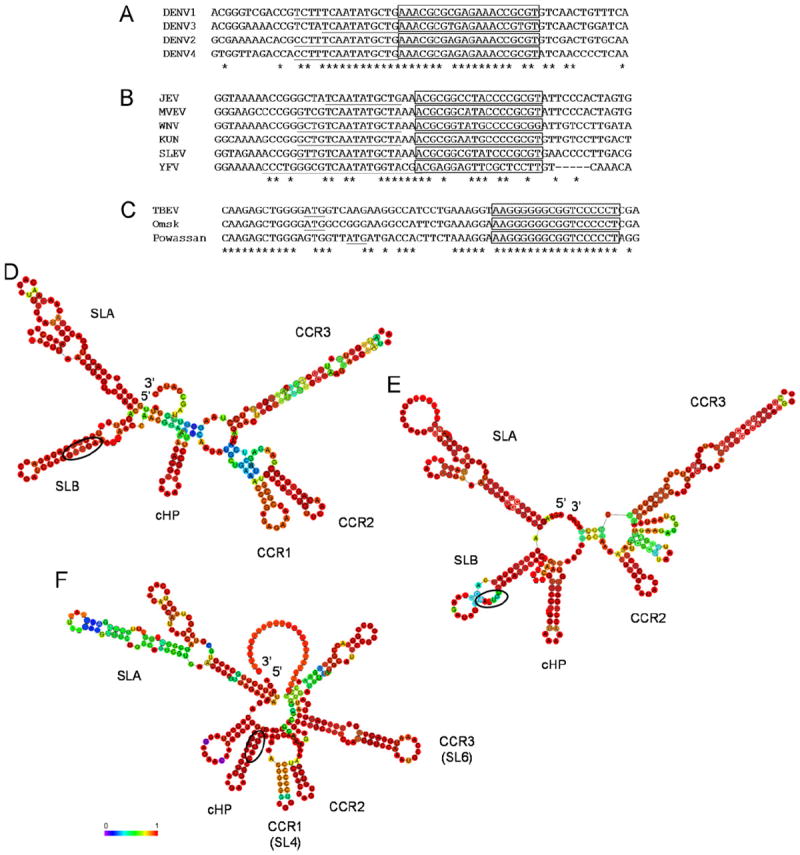
Conserved RNA elements are identified in the capsid coding-region of the DENV, JEV and TBE serogroups. Partial alignment of the representative genomes for the (A) DENV serogroup (nucleotides 118–177, numbering based on DENV2 Thai strain 16681; accession codes DENV1 U88535, DENV2 NC_001474, DENV3 AY099336 and DENV4 GU289913), (B) JEV serogroup (nucleotides 120–179, numbering based on JEV; accession codes JEV M18370, Murray Valley encephalitis virus AF161266, WNV NC_001563, Kunjin virus AY274505, St. Louis encephalitis virus DQ525916 and YFV U17067) and (C) TBE serogroup, including the tick-borne encephalitis, Powassan and Omsk hemorrhagic fever viruses (nucleotides 121–180, numbering based on TBEV; accession codes TBEV U27495, Powassan NC_003687 and Omsk AY193805), generated using Clustal W2 (Thompson et al., 1997). Positions of sequence homology are indicated by asterisks; the 5′ CS is underlined in the DENV and JEV serogroups; and the initiator AUG is underlined in the TBE serogroup. The conserved RNA sequence element CCR1 is boxed. Phylogenetic consensus structure based on the (D) DENV (nucleotides 1–274, numbering based on DENV2 Thai strain 16681), (E) JEV (nucleotides 1–273, numbering based on JEV) and (F) TBE serogroups (nucleotides 1–302, numbering based on TBEV) as computed by RNAalifold (Hofacker et al., 2002), where covariance is depicted as a colored spectrum ranging from purple (0) to red (1). CCR1, CCR2, CCR3 and other major known RNA elements, such as cHP and SLA, are indicated. The initiator AUG is circled.
The DENV, JEV and TBE serogroup alignments were modified so as to include the 5′ UTR as well as a large portion of the capsid-coding region to generate phylogenetically-conserved structure predictions using RNAalifold (Hofacker et al., 2002). The conserved RNA element CCR1 was found to be contained entirely within a small hairpin structure that is maintained by covariation in both the DENV and TBE serogroups (Fig. 1D and F) but not in the JEV serogroup (Fig. 1E). Though the CCR1 sequence and structure has not yet been identified in DENV, a homolog was recently characterized in TBE virus, termed stem-loop 4 (SL4), which was shown to be important for efficient viral translation, acting either directly or indirectly as a determinant of RNA replication in a manner proportional to its stability (Rouha et al., 2011). Our secondary structure predictions both confirm their results (Rouha et al., 2011) and demonstrate that CCR1 is a highly conserved nucleotide sequence and structural element in the TBE serogroup as well (Fig. 1C and F) though its role in the DENV life cycle has not yet been elucidated.
Since it is known that the DENV genome circularizes during viral replication, we created a modified DENV1–4 sequence alignment that included the 5′ and 3′ UTRs, the entire capsid-coding sequence and a large portion of the NS5-coding region, which was analyzed using RNalifold (Hofacker et al., 2002), demonstrating the predicted phylogenetic consensus structure for the DENV serogroup during viral replication where the ends of the genome basepair via the three sets of complementary sequences (5′/3′ CS, 5′/3′ UAR and 5′/3′ DAR) (Fig. 2). In addition to illustrating known cis-acting RNA structural elements such as SLA and cHP, CCR1 was easily identifiable (Fig. 2), confirming the results obtained in the previous analysis (Fig. 1D). However, while the predicted secondary structure of CCR1 shows a high degree of covariance, it is not as highly conserved as the predicted secondary structures of other known elements such as the SLA or cHP (Fig. 1D, F and Fig. 2), indicating that the predicted secondary structure of CCR1 may not be as important as the primary nucleotide sequence.
Fig. 2.

Conserved RNA structure elements in the context of a circularized DENV genome. Phylogenetic consensus structure based on the representative genomes for the DENV serogroup (position 1–417 and 9341–10723, numbering based on DENV2 Thai strain 16681; accession codes DENV1 U88535, DENV2 NC_001474, DENV3 AY099336 and DENV4 GU289913) as computed by RNAalifold (Hofacker et al., 2002), where covariance is depicted as circles. DENV serogrup alignment included the 5′ and 3′ UTRs, the entire capsid-coding sequence (nts 97–438) and a large portion of the NS5-coding region (nts 9341–10269). CCR1, CCR2, CCR3 and other major known RNA elements, such as cHP, SLA, 30 SL, 5′/3′ UAR, 5′/3′ DAR, and 5′/3′ CS are indicated.
Interestingly, both the larger circularized structure prediction and the smaller conserved structure predictions showed that there were two additional conserved RNA structural elements in the DENV capsid-coding region, termed conserved capsid-coding region 2 (CCR2) and CCR3 (Figs. 1D and 2), which were both found to be conserved among members of the JEV (Fig. 1E) and TBE (Fig. 1F) serogroups. Much like the cHP, the primary nucleotide sequence of CCR2 and CCR3 is not well-conserved in the DENV, JEV and TBE serogroups, despite the fact that the predicted secondary structures are conserved (data not shown). Recently, a homolog of CCR3 was identified in TBE virus, termed stem-loop 6 (SL6), which was shown to act as a structural element that is not essential for growth in tissue culture but functions as a replication enhancer (Tuplin et al., 2011). Taken together, it is likely that CCR1, CCR2 and CCR3 are conserved in sequence and/or secondary structure because they play a putative role in modulating the viral life cycle.
CCR1 modulates the viral life cycle in both BHK and C6/36 cells
To determine whether CCR1 regulates viral replication, silent point mutations were introduced into an infectious clone of the DENV2 Thai strain 16681, termed pD2/IC (Kinney et al., 1997). To disrupt the predicted secondary structure with minimal changes to the primary nucleotide sequence, two silent nucleotide changes were introduced into either side of the predicted stem structure (“5′/3′ flank mut,” Fig. 3A and Table 1), which linearized the stem-loop and eliminated competing structure predictions in the mfold prediction software (Zuker, 2003). Next, the primary nucleotide sequence was mutated without affecting the predicted secondary structure by introducing six silent nucleotide changes into CCR1 (“seq mut,” Fig. 3A and Table 1) in such a way that the predicted free energy is similar to that of the wild-type (WT). Thus, despite minor changes to the stem, the overall structure of CCR1 is maintained in this mutant construct, as predicted by mfold (Zuker, 2003). A third mutant, termed the “center mut” construct, contains only the nucleotide changes that were introduced in the predicted loop of the “seq mut” construct (Fig. 3A and Table 1).
Fig. 3.
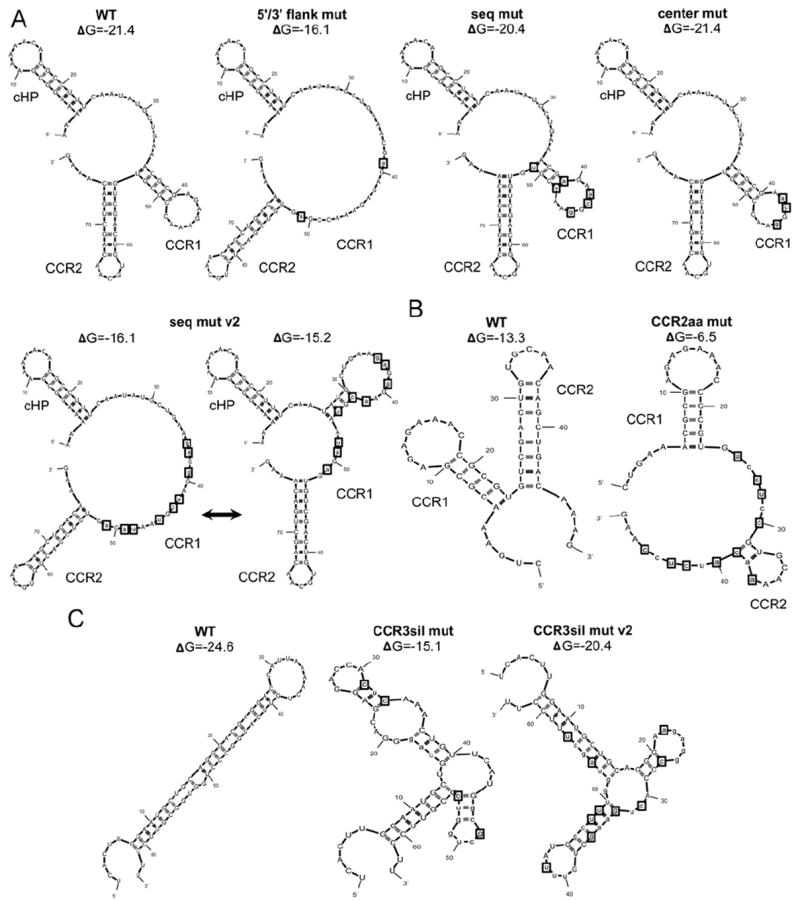
Mutational analysis of CCR1, CCR2 and CCR3 in order to examine their putative role as cis-acting RNA regulatory elements. (A) mfold predictions of the mutant constructs designed to disrupt the primary nucleotide sequence (“seq mut”) or the predicted secondary structure (“5′/3′ flank mut”). The sequence changes introduced into the predicted loop region of the “seq mut” construct were isolated in the “center mut” without altering the predicted secondary structure of CCR1 as predicted by mfold (Zuker, 2003). An additional sequence mutant was generated (“seq mut v2”), which oscillates between a linear conformation an alternative stem structure that is less stable than the WT. mfold predictions (Zuker, 2003) of the mutant constructs designed to disrupt the predicted secondary structures of conserved RNA structural elements (B) CCR2 and (C) CCR3. The predicted stem-loop of CCR2 was disrupted with conserved amino acid changes (“CCR2aa mut”). The predicted stem-loop structure of CCR3 was disrupted with silent point mutations (“CCR3sil mut” and “CCR3sil mut v2”). The positions of introduced mutations are indicated by boxes.
Table 1.
Sequence of WT, CCR1, CCR2 and CCR3 mutants.
| Construct | Nucleotide sequence | Amino acid sequence |
|---|---|---|
| WT | 5′–AAACGCGAGAGAAACCGCGTG–3′ | |
| 5′/3′ flank mut | 5′–AAACGaGAGAGAAACCGaGTG–3′ | |
| seq mut | 5′–AAACGaGAacGgAAtCGtGTG–3′ | |
| center mut | 5′–AAACGCGAacGgAACCGCGTG–3′ | |
| seq mut v2 | 5′–AAgaGgGAacGtAAtaGaGTG–3′ | |
| WT | 5′–GTGTCGACTGTGCAACAGCTGACAAAG-3′ | 5′–VSTVQQLTK-3′ |
| CCR2aa mut | 5′-GTGaCctCcGTGCAAaAcaTctCcAAG-3′ | 5′–VTSVQNISK-3′ |
| WT | 5′–CTTGGAATGCTGCAGGGACGAGGACCATTAAAACTGTTCATGGCCCTGGTGGCGTTCCTT–3′ | |
| CCR3sil mut | 5′-CTTGGAATGCTGCAGGGACGAGGACCAcTcAAACTGTTCATGGCgCTGGTcGCGTTCCTT-3′ | |
| CCR3sil mut v2 | 5′-CTTGGAATGCTGCAGGGAaGAGGcCCAcTgAAgCTGTTtATGGCttTGGTaGCtTTCCTT-3′ |
Amino acid changes are underlined where applicable.
Adjacent to CCR1 is the stem structure CCR2, which was predicted to act as a structural element given the lack of nucleotide sequence conservation. To disrupt the predicted hairpin structure of CCR2, ten mutations were introduced into the stem, though these sequence alterations also introduced conserved amino acid changes (“CCR2aa mut,” Fig. 3B and Table 1). Since CCR2 is directly downstream of CCR1, the mutant construct was designed so that the mutations would not affect neighboring RNA elements while still stably disrupting the CCR2 stem-structure as predicted by mfold (Zuker, 2003) (Fig. 3B). To disrupt the predicted secondary structure of CCR3 with minimal changes to the primary nucleotide sequence, a series of silent sequence changes were introduced into either side of the predicted stem structure (“CCR3sil mut” and “CCR3sil mut v2,” Fig. 3C and Table 1). For all mutant constructs designed, reading frame and codon usage bias in baby hamster kidney (BHK) and Ae. albopictus mosquito (C6/36) cells were maintained while preserving the surrounding secondary structures as predicted by the mfold (Zuker, 2003). Unless otherwise indicated, amino acid sequence was also maintained in all mutant constructs, and codon usage bias was taken into account to ensure that the mutated codon was equally available in both BHK and C6/36 cells.
RNA transcripts were transfected into BHK or C6/36 cell monolayers, and virus-containing supernatants were harvested at various timepoints (indicated as hours post-transfection, hpt) to be assayed for viral infectivity via plaque assays. As DENV replication kinetics are slower in C6/36 cells, virus-containing supernatants were assayed for viral infectivity at later times post-transfection than in BHK cells (BHK: 24, 48 and 72 hpt; C6/36: 72, 96, 120 and 144 hpt). Interestingly, all mutant CCR1 constructs (“5′/3′ flank mut,” “center mut,” and “seq mut”) demonstrated a significantly smaller plaque phenotype (see representative image from “seq mut” in Fig. 4A), but only the mutations that disrupted the primary nucleotide sequence (“seq mut”) rather than the predicted secondary structure (“5′/3′ flank mut”) significantly reduced the viral titer in BHK cells at all timepoints tested (Fig. 4B). Since the primary nucleotide sequence of CCR1 is important in BHK cells, an additional mutant construct was designed with nine silent sequence changes, termed “seq mut v2” (Fig. 3A and Table 1). Unlike the other CCR1 mutant constructs, the “seq mut v2” mfold structure prediction oscillates between a linearized form and a short hairpin structure, though the altered stem-structure is predicted to be less stable than the WT (Fig. 3A). When transfected into BHK cells, both sequence mutants (“seq mut” and “seq mut v2”) displayed a no-plaque phenotype at 24 hpt, and at later timepoints both showed a significantly lower viral titer in comparison to the WT (Fig. 4B). While the “seq mut” construct showed a small plaque phenotype, the “seq mut v2” construct demonstrated a significantly extra small plaque phenotype (see representative image from “seq mut v2” in Fig. 4A) at all timepoints yielding detectable virus. Altering the primary nucleotide sequence decreased viral titer in BHK cells, and this effect was influenced in part by the central sequences since the “center mut” construct had a significantly lower viral titer in comparison to the WT at all timepoints tested (Fig. 4B).
Fig. 4.
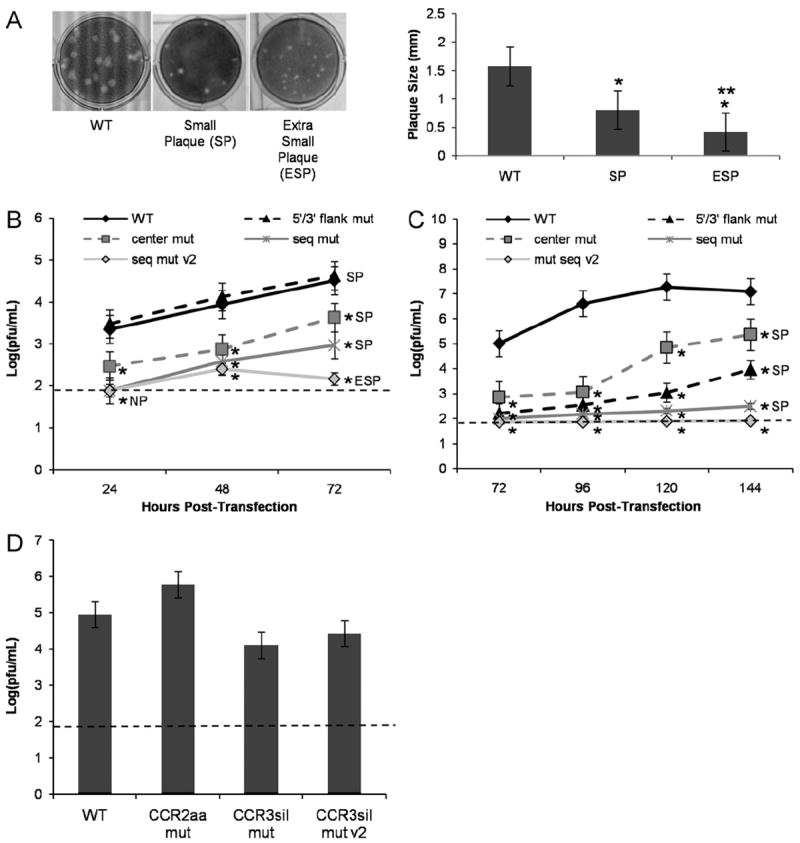
CCR1 acts as a cis-acting RNA sequence element in modulating the viral life cycle in BHK and C6/36 cells. (A) Plaque phenotype in BHK cells obtained by transfecting BHK or C6/36 cell monolayers with the described mutant constructs (SP, small plaque; ESP, extra small plaque). Left panel: A representative image for SP generated from “seq mut” construct in IC-transfected BHK cells and a representative image for ESP generated from “seq mut v2” construct in IC-transfected BHK cells. Right panel: Plaques were measured (millimeters, mm) for each described plaque phenotype using 60 plaques from 3 independent experiments. (B) BHK and (C) C6/36 cells were transfected with IC-based mutant CCR1 constructs and assayed for viral titer (pfu/mL) by plaque assay. Viral titers were normalized to the amount of vRNA present in the cells at the time of harvest as measured by qRT-PCR. Results were obtained from 5 independent experiments, conducted in duplicate. Plaque phenotypes are indicated for each construct. (D) BHK cells were transfected with IC-based mutant CCR2 and CCR3 constructs and assayed for viral titer (pfu/mL) by plaque assay at 72 hpt. Viral titers were normalized to the amount of vRNA present in the cells at the time of harvest as measured by qRT-PCR. Results were obtained from 3 independent experiments, conducted in duplicate. The limit of detection is shown by a dashed line (1.9 pfu/mL). *p<0.05 relative to the WT. **p<0.05 relative to the SP. Error bars indicate standard deviation.
In contrast to BHK cells, changes to the primary nucleotide sequence (“seq mut” and “seq mut v2”) as well as changes to the predicted secondary structure (“5′/3′ flank mut”) significantly decreased the viral titer in C6/36 cells at all timepoints tested (Fig. 4C). The “seq mut” construct did not result in any infectious virus at early timepoints, and viral titers barely exceeded the limit of detection at later times post-transfection, while the “seq mut v2” construct maintained a no-plaque phenotype at all timepoints tested (Fig. 4C). Unlike in BHK cells, the effect on viral titer in C6/36 cells appears to be primarily mediated by the 5′ and 3′ flanking sequences since the “center mut” construct had slightly higher titers at later timepoints than the “5′/3′ flank mut” construct, although the “center mut” construct displayed a significantly lower titer than the WT at all timepoints tested (Fig. 4C). In transfected C6/36 cells, all mutant constructs tested that produced detectable infectious virus displayed a small plaque phenotype (Fig. 4A). It is known that not all dengue viruses form plaques, and it is conceivable that the CCR1 mutant viruses may form diffuse plaques that are not easily detected using standard plaque assays. In order to confirm that plaque assays are a viable means of assessing infectivity of the mutant viruses, the virus-containing supernatants from IC-transfected BHK and C6/36 cells were also assessed for viral infectivity using an immunofocus assay as an alternative measure of infectivity that does not depend on plaque formation. The results from the immunofocus assays confirmed the plaque assay results (data not shown), demonstrating that mutations to CCR1 alter infectious particle production in both BHK and C6/36 cells.
Though the changes that were introduced into CCR1 are not predicted to have an effect on neighboring RNA elements, it was important to demonstrate that these sequence and structural changes would not disrupt the function of the cHP element, which has been shown to regulate start site selection in BHK and C6/36 cells (Clyde et al., 2008; Clyde and Harris, 2006). To this end, the mutations in the “5′/3′ flank mut,” the “seq mut” and the “seq mut v2” constructs were subcloned into the C-FLAG reporter that had been previously constructed to visualize the full-length capsid protein and the two truncated capsid proteins generated by alternate start codon usage (Clyde and Harris, 2006). As expected, the WT and mutant CCR1 C-FLAG reporter constructs continued to favor translation of full length capsid protein (Fig. S1), in contrast to a mutant cHP construct (HP0.2), which had been previously shown to favor selection of the second AUG start codon in BHK or C6/36 cells (Clyde and Harris, 2006). These data also demonstrate that the CCR1 element itself plays no role in start site selection and has a function distinct from the cHP. Taken together, these results indicate that CCR1 functions primarily as a cis-acting RNA sequence element in both BHK and C6/36 cells, though these effects are more dramatic in C6/36 cells. Furthermore, the structure of CCR1 may also be required for efficient replication in C6/36 cells since the “5′/3′ flank mut” construct did not replicate as well as the “center mut” construct, which contains a WT stem-structure (Fig. 4C).
Preliminary analysis of CCR2 and CCR3 indicated that the predicted secondary structures of CCR2 and CCR3 are not critical for the viral life cycle in BHK cells since the mutant constructs replicated as well as the WT at 72 hpt (Fig. 4D). The “CCR2aa mut” mutant replication levels also imply that this region is not sensitive to amino acid changes (Fig. 4D). Taken together, though their stem-structures are well conserved, neither CCR2 nor CCR3 appear to be critical cis-acting RNA regulatory elements in BHK cells. Because the effect of mutations in CCR2 and CCR3 were minimal compared to CCR1, these regions were not pursued further.
Altering CCR1 impairs viral replication in Ae. aegypti mosquitoes
Since changes to the CCR1 primary nucleotide sequence were shown to have more dramatic effects in mosquito cells (Fig. 4C), Ae. aegypti mosquitoes were infected with the various CCR1 mutant viruses to determine whether the results obtained in the in vitro experiments could be observed in an in vivo model of DENV infection. Biological transmission of flaviviruses by arthropods depends on ingesting a blood meal that contains sufficient virus to establish an infection in the epithelial cells lining the mesenteron (midgut) (Rosen and Gubler, 1974). However, since the mutant constructs were shown to have a delayed replication rate in transfected C6/36 cells when compared to WT DENV2, Ae. aegypti mosquitoes were inoculated intrathoracically (IT) so that all the viruses would begin their replication cycle at the same time in the parenteral tissues. Virus-containing supernatants were derived from BHK cells transfected with WT and CCR1 mutant constructs, titered via plaque assay, and used to infect Ae. aegypti mosquitoes IT with approximately 104 pfu/mL. All inoculated mosquitoes showed 75–100% infection by day 20 post-inoculation by plaque assay (Fig. 5A), indicating that all viruses were able to replicate in the parenteral tissues of Ae. aegypti mosquitoes after IT inoculation. The mosquito bodies were assessed for infectious virus at various timepoints (indicated as days post-inoculation, dpi; days 5, 10 and 20) by plaque assay (Table S1). All mutant viruses displayed a significantly lower viral titer than the WT at all timepoints tested, especially the “mut seq v2” and “5′/3′ flank mut” mutant viruses (Fig. 5B). Thus, the replication defects observed in the in vitro experiments are reflected in reduced replication in the mosquito vector for all mutant viruses at all timepoints tested. Overall, the in vivo infections in Ae. aegypti mosquitoes support the in vitro results that CCR1 functions predominately as a cis-acting RNA regulatory element, mediated in part by the 5′ and 3′ flanking sequences.
Fig. 5.
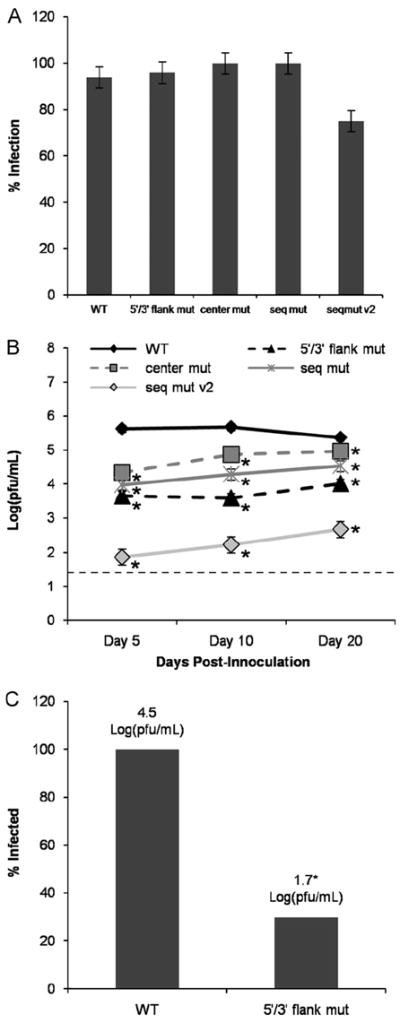
CCR1 modulates viral replication and dissemination to the salivary glands in Ae. aegypti mosquitoes. (A) the rates of infection at day 20 of colonized Panamanian Ae. aegypti mosquitoes inoculated IT with mutant viruses. (B) Viral titers harvested from the infected mosquitoes at the indicated timepoints, as assessed by plaque assay. (C) Salivary glands from mosquitoes inoculated with WT and “5′/3′ flank mut” mutant viruses harvested on day 10 were dissected and assayed for viral titer in Vero cells. Results were obtained from 3 independent mosquito infections. The limit of detection is shown by a dashed line (1.4 pfu/mL). *p<0.05 relative to the WT. Error bars indicate standard deviation.
During viral replication in the orally infected mosquito, the virus must escape from the midgut epithelium into the hemocele (body cavity) in order to disseminate to the brain, fat body and salivary glands of the infected mosquito (Rosen and Gubler, 1974). Once in the salivary glands, flaviviruses must undergo another round of replication to generate enough virus to be secreted in the saliva and transmitted to a susceptible host (Rosen and Gubler, 1974). To assess whether the replication defects observed in vivo could affect viral dissemination in the mosquito vector, the salivary glands were dissected from infected mosquitoes on day 10, and viral titer was assessed by plaque assay. Only 30% of the mosquitoes infected with the “5′/3′ flank mut” mutant viruses were found to have infected salivary glands, as compared to 100% of the WT-infected mosquitoes (Fig. 5C). In addition, there was a 2.6-fold decrease in the amount of virus present in the salivary glands of the “5′/3′ flank mut”-infected mosquitoes as compared to the mosquitoes infected with the WT virus (Fig. 5C). This implies that changes to the CCR1 sequence not only affect viral replication in the mosquito vector for up to 20 dpi, but that the effect on viral replication could have a major impact on viral transmission by day 10 post-inoculation, though additional experiments are necessary to determine if the replicating virus in the salivary glands is capable of being transmitted to susceptible Ae. aegypti mosquitoes. Thus, both the in vitro and in vivo experiments support the conclusion that CCR1 is an important cis-acting RNA sequence element; however, the question remains as to how it regulates the viral life cycle in both mammalian and mosquito cells.
Mutating CCR1 has no effect on viral translation or RNA synthesis in BHK and C6/36 cells
Most of the known RNA elements in DENV modulate the viral life cycle by affecting translation (Alvarez et al., 2005a; Chiu et al., 2005; Clyde and Harris, 2006) or RNA synthesis (Alvarez et al., 2005a, 2005b; Clyde et al., 2008; Filomatori et al., 2006; Friebe and Harris, 2010; Friebe et al., 2011). To determine whether the decreased viral titers in CCR1 mutants were due to effects on either viral translation or RNA synthesis, all mutations were subcloned into a Renilla luciferase reporter construct, termed pDRrep (Clyde et al., 2008). Since the first 72 nucleotides of the capsid-coding sequence present in the DENV2 reporter constructs contain the CCR1 sequence, it was not necessary to introduce additional changes into pDRrep for this study. The pDRrep-RdRpmut construct was used as a negative control (“GVD”) because this replicon cannot replicate due to a mutation in the active site of the RNA-dependent RNA polymerase (RdRp) (Clyde et al., 2008). As with other DENV and WNV replicons, the replication kinetics are slower compared to the infectious clone (Alvarez et al., 2005a; Holden et al., 2006; Lo et al., 2003; Tilgner and Shi, 2004).
RNA transcripts were transfected into BHK and C6/36 cell monolayers and assayed for luciferase activity at various timepoints post-transfection (BHK: 4, 8, 12, 24, 48 and 72 hpt; C6/36: 4, 8, 24, 72, 96, 120 and 144 hpt). None of the mutant constructs showed a significant difference in luciferase counts at early timepoints post-transfection when compared to the WT and GVD controls, indicating that CCR1 does not influence viral translation in BHK (Fig. 6A) or C6/36 (Fig. 6B, left panel) cells. In BHK cells, no significant difference in luciferase levels was observed at later timepoints either, indicating that mutations in CCR1 have no effect on RNA synthesis when compared to the WT (Fig. 6A). Transfection of C6/36 cells with replicon RNA proved particularly difficult at later timepoints; thus, only the 24 hpt data is shown for all mutant replicon constructs in C6/36 cells (Fig. 6B). However, the later timepoints were achieved for the GVD negative control and the “5′/3′ flank mut” construct, which is likely representative of the other mutant constructs used in this study (Fig. 6B, right panel). These data demonstrated that although the mutant IC construct displayed reduced and delayed growth in transfected C6/36 cells (Fig. 4C), the mutant replicon produces robust levels of luciferase activity at both early and late timepoints post-transfection when compared to the GVD control, which indicates that viral translation and RNA synthesis are not regulated by CCR1 in C6/36 cells (Fig. 6B, right panel). Deleting CCR1 from the Renilla luciferase replicon (“delta”) had no significant effect on luciferase counts at either early or late timepoints, implying that CCR1 is dispensable for viral translation and replication (Fig. 6A and B, left panel). These results imply that CCR1 influences a stage of the viral life cycle that occurs after viral translation and RNA synthesis in mammalian and mosquito cells, which has not yet been observed with the other known DENV cis-acting RNA regulatory elements identified thus far.
Fig. 6.

CCR1 does not affect viral translation, RNA synthesis or RNA stability in BHK cells. (A) mutations were subcloned into Renilla luciferase replicons, and the mutant replicons were transfected into BHK cell monolayers. Left panel: Cellular lysates were harvested at the indicated timepoints and assayed for luciferase activity. An additional mutant, termed “delta,” was created where the entire CCR1 sequence was removed from the replicon. Right panel: A separate graph illustrating the luciferase levels at 4 and 72 hpt for all the mutant constructs shown in the left panel. *p<0.05 relative to the WT replicon. (B) Renilla luciferase mutant replicons were transfected into C6/36 cell monolayers. Left panel: Graph illustrating the luciferase levels at 24 hpt for all the mutant constructs, demonstrating that CCR1 mutations have no significant effect on viral translation. Right panel: Cellular lysates were harvested at the indicated timepoints and assayed for luciferase activity, illustrating that the CCR1 “5′/3′ flank mut” mutations have no effect on viral translation and RNA synthesis, whereas the negative GVD control is defective in RNA synthesis. (C) Mutations were subcloned into a GVD variant of the IC and transfected into BHK cells. Cells were harvested at 4, 8, 24, 48 and 72 hpt, and the amount of vRNA present was quantified using qRT-PCR in order to calculate the RNA half-life in BHK cells. Results were obtained from 3 independent experiments, conducted in duplicate. *p<0.05 relative to the WT replicon. Error bars indicate standard deviation.
Observed effects of CCR1 mutant constructs on viral titer are not due to RNA instability
To determine whether the introduction of silent point mutations into the primary nucleotide sequence of CCR1 destabilized the vRNA, all mutations were subcloned into a GVD variant of the infectious clone, termed pD2/IC-RdRpmut (Clyde et al., 2008), which is unable to replicate due to a mutation in the active site of the RdRp. RNA transcripts derived from this backbone will persist within the cell after transfection, and the rate of RNA degradation for each construct can be measured using qRT-PCR to derive the RNA half-life, as compared to the GVD control. For all constructs tested, none of the introduced changes significantly altered the RNA half-life in comparison to the GVD control (Fig. 6C). The results derived from these stability assays are supported by the similar translation efficiencies observed early after transfection using the mutant and WT Renilla luciferase replicons in both BHK and C6/36 cells (Fig. 6A and B). This implies that the introduced mutations, including the deletion of the entire CCR1 sequence, do not alter the overall stability of the RNA, or else a drop in luciferase counts would be observed at early timepoints as a result of decreases in the availability of the replicon RNA. Furthermore, luciferase levels are maintained at the latest time-points tested (72 hpt in BHK cells, 144 hpt in C6/36 cells), also supporting the notion that the mutant replicon RNAs are as stable as the WT. Taken together, it is likely that RNA instability is not a cause for the observed effects of CCR1 on viral titer.
CCR1 regulates infectious particle production in BHK and C6/36 cells
Though infectious virus production can be measured by plaque assay, this does not account for non-infectious viral particles that are routinely released during infection or infectious virions that do not form plaques on the plaquing cell line. In order to determine the amount of assembled viral particles in the virus-containing supernatants from IC-transfected BHK and C6/36 cells, the amount of vRNA was quantified using qRT-PCR. For all the mutant constructs, there was no significant decrease in the amount of vRNA present in the supernatant as compared to the WT for all the timepoints tested in BHK cells (Fig. 7A). In C6/36 cells, only the “seq mut” and “seq mut v2” constructs displayed significantly lower amounts of vRNA as compared to the WT (Fig. 7B), though these levels (~106 G-Eq/mL) were still high considering that they had a no-plaque phenotype during transfections in C6/36 cells (Fig. 4C). However, the amount of vRNA released from transfected BHK (Fig. 7A) and C6/36 cells (Fig. 7B) was significantly higher than the GVD control at all timepoints tested. This result indicates that there are similar amounts of viral particles being released from WT and mutant IC-transfected BHK and C6/36 cells but they differ in their infectivity as determined by plaque assay or with an immunofocus assay.
Fig. 7.

CCR1 mediates infectious viral particle production. vRNA found in virus-containing supernatants from IC-transfected (A) BHK or (B) C6/36 cell monolayers was extracted at the indicated timepoints and measured by qRT-PCR. Particle:pfu ratios were generated by comparing vRNA concentrations (G-Eq/mL) in the supernatant to the viral titers (pfu/mL) obtained at the same timepoint in IC-transfected (C) BHK or (D) C6/36 cell monolayers. Results were obtained from 4 independent experiments, conducted in duplicate. *p<0.05 relative to the WT. Error bars indicate standard deviation.
To determine the efficiency of infectious particle production during transfections, the amount of vRNA present within the virus-containing supernatants was measured using qRT-PCR and compared to the viral titer, generating a particle-to-pfu ratio for each mutant construct. In BHK cells, the “5′/3′ flank mut” construct displayed the same particle:pfu ratio as the WT whereas the “seq mut,” “seq mut v2’‘ and “center mut” constructs displayed a significantly greater particle:pfu ratio at all time-points tested (Fig. 7C), indicating that more non-infectious viral particles are being released into the supernatant for these constructs. Interestingly, all the mutant constructs that showed lower replication kinetics in BHK cells (Fig. 4A) had a significantly higher particle:pfu ratio (Fig. 7C). When virus-containing supernatants derived from IC-transfected BHK cells were analyzed by capture ELISA, both the WT and the “seq mut” construct displayed equivalent amounts of viral E protein (Fig. S2, right panel). Considering that the same samples of virus-containing supernatants also contained similar amounts of vRNA (Fig. S2, left panel), it is likely that cells transfected with the “seq mut” construct release viral particles that do not, however, form plaques on the plaquing cell line. In C6/36 cells, both the “seq mut” and “seq mut v2” constructs have an increased particle:pfu ratio as compared to the WT, demonstrating that mutating the primary sequence of CCR1 impacts infectious particle production in both BHK and C6/36 cells (Fig. 7D), however, the amount of the “seq mut” mutant virus released into the supernatant was below the limit of detection of the capture ELISA (data not shown). In C6/36 cells, the “5′/3′ flank mut” construct also showed a significantly higher particle:pfu ratio than the WT, indicating that the 5′/3′ flanking sequences contribute substantially to the observed phenotype (Fig. 7D). In both cell types, altering the primary nucleotide sequence of CCR1 (“center mut” and “seq mut”) results in less infectious virus as compared to the WT, though there remains the possibility that CCR1 has a structural component in C6/36 cells. Taken together, this evidence suggests that specific portions of the CCR1 sequence have a cell-type dependent effect on a post-replication stage of the viral life cycle that modulates infectious particle production.
Thus, it is likely that mutating CCR1 interferes with post-replication events such as viral assembly, virion processing/maturation or virion release. To examine virion release, the amount of infectious particles outside the cell was compared to the amount of infectious particles within cells in both BHK and C6/36 cells. At various timepoints post-transfection (BHK: 24, 48 and 72 hpt; C6/36: 72, 96, 120 and 144 hpt), cell monolayers were stripped of extracellular virus using a high-pH, high-salt solution and subjected to a single freeze/thaw cycle in order to release intracellular infectious virus, which was then assayed via plaque assay alongside supernatants containing extracellular virus. None of the mutant constructs showed a significant effect on the ratio of extra- to intracellular virus in BHK cells, except for the “seq mut” and “seq mut v2” constructs at 24 hpt, though at this timepoint both constructs displayed a no-plaque phenotype (Fig. 8A). Similar observations were seen in C6/36 cells, where those constructs capable of producing detectable infectious virus showed no significant retention of intracellular infectious virus as compared to the WT (Fig. 8B). Since the decreases in viral titer are not due to accumulation of infectious virus within the cell, it is likely that mutating CCR1 results in a virion assembly or processing/maturation defect.
Fig. 8.

CCR1 has no effect on virion release or the amount of vRNA released by BHK or C6/36 cells. The amount of extracellular infectious virus was compared to the amount of intracellular infectious virus from IC-transfected (A) BHK and (B) C6/36 cell monolayers at the indicated timepoints. A ratio of extra- to intracellular virus greater than 1 was used to indicate that more virus was released from the cells whereas a ratio of less than 1 implies that more infectious virus is located within the cell. Results were obtained from 3 independent experiments, conducted in duplicate. A ratio of extra- to intracellular virus of 1 is indicated by a dashed line. *p<0.05 relative to the WT. Error bars indicate standard deviation.
Discussion
Sequence and structure conservation was used as a platform for identifying conserved RNA regulatory elements in the DENV coding-region, as RNA elements are more likely to perform a function in the viral life cycle if they are conserved. Several conserved RNA elements were selected based on their sequence or secondary structure conservation in the DENV, JEV and TBE serogroups. Although CCR2 and CCR3 were identified as RNA structural elements that were conserved in the three serogroups examined, initial screens in BHK cells showed that neither CCR2 nor CCR3 play a critical role in modulating the viral life cycle. We found CCR1 to be conserved both in primary nucleotide sequence and predicted secondary structure in both the DENV and TBE serogroups and while a TBEV homolog of CCR1 has been identified as a replication element (Rouha et al., 2011), CCR1 had not yet been characterized for its role in regulating the DENV life cycle. In this study, we demonstrate that DENV CCR1 functions as a cis-acting RNA sequence element in BHK and C6/36 cells, though its effects on viral titer and infectious virus production are more pronounced in mosquito cells. CCR1 functions primarily through the central sequences in BHK cells, whereas in C6/36 cells its effects are mediated by both the 5′ and 3′ flanking sequences as well as the central sequences, potentially indicating a role for the stem-loop structure of CCR1. Finally, CCR1 appears to modulate a post-RNA synthesis stage of the DENV life cycle, possibly regulating viral assembly.
RNA elements that act in a cell-type specific manner can help explain how viral replication is differentially regulated, which is of particular relevance to understanding differences in arboviral replication within the mammalian host and the mosquito vector. For example, within the SIN viral genome, there is an 51-nucleotide long conserved sequence element in the nsP1-coding sequence that is predicted to form two smaller stem-loop structures that act as replication enhancers whose effects are more important in mosquito cells than in mammalian cells (Fayzulin and Frolov, 2004; Frolov et al., 2001; Niesters and Strauss, 1990). The fact that different regions of the CCR1 sequence are implicated in mediating viral replication and infectious virus production in BHK and C6/36 cells implies the involvement of cell-type specific factors in its function. Though CCR1 has been shown to function as a cis-acting RNA sequence element, the fact that the “5′/3′ flank mut” construct had a more severe phenotype in C6/36 cells compared to the BHK cells could imply that the predicted secondary structure of CCR1 is more important to the viral life cycle in mosquito cells, possibly due to the lower temperature at which these cells are grown in vitro and in vivo. Alternatively, it is possible that CCR1 also functions predominantly as a cis-acting RNA sequence element in C6/36 cells and that the 5′ and 3′ flanking sequences are more important irrespective of the predicted secondary structure. While it has been shown that CCR2 and CCR3 are not critical cis-acting RNA regulatory elements in mammalian cells, preliminary evidence indicates that the CCR3 mutant constructs vary slightly in their ability to replicate in C6/36 cells, which could imply that its secondary structure is not as important as the primary nucleotide sequence (data not shown). Interestingly, a CCR3 homolog in TBE virus has recently been shown to act as a replication enhancer (Tuplin et al., 2011). Given its conservation in all three flavivirus serogroups examined and the initial phenotype in C6/36 cells, it would be of interest to introduce more specific sequence changes in CCR3, focusing on altering the primary nucleotide sequence rather than the predicted secondary structure and testing its effects on the DENV life cycle in C6/36 cells.
The ability to generate mutant viruses in BHK cells provided the opportunity to determine whether the defects observed in the in vitro experiments reflect the complexity of an in vivo infection by inoculating Ae. aegypti mosquitoes. Many flaviviruses exhibit a high degree of specificity in their ability to infect and be transmitted by arthropod species (Lindenbach et al., 2007; Rosen et al., 1985); however, infected mosquitoes remain infectious for life (1–2 weeks to ~174 day) (Gubler and Kuno, 1997). We show that mutating CCR1 not only affects viral replication in vitro but significantly lowers viral replication in the predominant DENV mosquito vector Ae. aegypti, providing an in vivo model to support our in vitro data. To sustain flavivirus transmission cycles in arthropods, a sufficient amount of virus must be ingested to establish an infection in the epithelial cells lining the mesenteron (midgut) and escape into the hemocele (body cavity) to spread to secondary amplification tissues (Rosen and Gubler, 1974). Changes to the 5′ and 3′ flanking sequences of CCR1 significantly decreased viral dissemination to the salivary glands, with a 2.6-fold decrease in the amount of virus that could be potentially transmitted by the mosquito vector as compared to the WT virus by day 10. This implies that altering CCR1 could have considerable effects on viral transmission by the arthropod vector, though more detailed studies examining the transmission rates for all the CCR1 mutant viruses is warranted. While the data indicate that the mutant viruses do not replicate at the same rate as the WT once within the body cavity, it would be of interest to investigate how well the mutant viruses can disseminate in Ae. aegypti mosquitoes when they are infected orally and virus must infect and escape from the midgut into the body cavity.
Unlike other known RNA elements in DENV, CCR1 was shown to have no effect on viral translation, RNA synthesis or stability in BHK cells. It is unlikely that RNA transcripts that are shown to be stable in one cell line would be unstable in another, so it is improbable that the effects on viral titer and infectious particle production observed in C6/36 cells are due to RNA instability. Altering either the primary nucleotide sequence or the predicted secondary structure of CCR1 had no effect on viral translation or RNA synthesis in BHK or C6/36 cells, indicating that CCR1 functions at a post-replication stage of the viral life cycle and supporting the notion that changes to CCR1 do not affect RNA stability.
It is possible that CCR1 could serve as a putative assembly signal for DENV; for instance, in retroviruses (Chadwick and Lever, 2000; Kuno, 1997) and SIN (Frolova et al., 1997; Linger et al., 2004; Weiss et al., 1994), the packaging signal is located close to or within the coding-region. CCR1 does not regulate early steps in the replication cycle such as viral translation and RNA synthesis, and altering CCR1 results in an increased particle:pfu ratio in BHK and C6/36 cells, demonstrating a defect in production of infectious virions. Additionally, when virus-containing supernatants were examined for infectivity using immunofocus assays, the mutant constructs produced less infected cells as compared to the WT in both BHK and C6/36 cells in a manner consistent with the observed viral titers. However, similar amounts of vRNA and viral E protein are released from cells transfected with WT or CCR1 mutant constructs, indicating that viral particles are indeed being formed but are not infectious as measured by plaque assay and immunofocus assay. Lastly, the amount of extracellular infectious virus is equivalent or greater to the amount of intracellular infectious virus in the WT and all CCR1 mutants, indicating that mutating CCR1 does not result in the retention of virus inside the cell. Together, these data suggest that CCR1 plays a role in viral assembly and/or virion processing/maturation and is more important in the mosquito vector. Our observations are consistent with the results reported recently by Samsa et al. (2012), which showed that mutations in this region of the capsid-coding sequence affect infectious particle production in mammalian and more significantly in mosquito cells but do not impact early replication stages of the viral life cycle. However, while the authors attribute these effects to amino acid changes, our mutations in CCR1 were silent and therefore point to the importance of the primary nucleotide sequence rather than the amino acid sequence of CCR1.
Flavivirus infectious particle production is achieved during maturation in the trans Golgi compartment, where the prM protein is cleaved into the mature M form. Although inhibiting prM cleavage does not impair virion release, studies on prM-containing particles suggest that furin cleavage (Stadler et al., 1997; Wengler and Wengler, 1989) or a major structural alteration in prM (Elshuber and Mandl, 2005) is required to generate highly infectious virus. It would be of interest to determine whether the observed effects of mutations to the CCR1 sequence on non-infectious particle production could result from a lack of prM cleavage due to decreased accessibility of prM to furin during virion maturation, which could explain the overproduction of non-infectious virions in both cell lines tested.
Many studies using electron microscopy have shown that unlike alphaviruses, neither mature (Kuhn et al., 2002; Zhang et al., 2003b) nor immature (Zhang et al., 2003b) flavivirus particles appear to have a capsid structure surrounding the vRNA, resulting from a lack of icosahedral symmetry in the nucleocapsid core (Zhang et al., 2007). The fact that there is little specific interaction between the glycoprotein ectodomain and the capsid protein molecules implies that flavivirus particles are assembled as they acquire their envelopes from the ER membrane (Zhang et al., 2003a, 2003b). If CCR1 acts as an assembly signal, CCR1 mutations may lead to disruptions in the nucleocapsid core, which nonetheless permit low levels of viral particle production since flaviviruses do not have a structured symmetrical nucleocapsid core (Zhang et al., 2007). Unfortunately, direct testing of CCR1 interaction with the DENV capsid protein by electrophoretic mobility shift assays was complicated by the fact that capsid associates nonspecifically with RNA and also spontaneously forms protein aggregates in vitro. To address viral RNA packaging, initial analyses included transfecting RNA replicon constructs from this study into a 293T cell line that stably expresses the DENV2 16681 structural genes (C-prM-E) in an attempt to produce DENV Reporter Virus Particles (RVPs) (Ansarah-Sobrinho et al., 2008; Mattia et al., 2011). However, we encountered technical difficulties in transfecting the replicon RNA, resulting in no Renilla luciferase signal being detected except for the control DNA replicon (Mattia et al., 2011). Currently, the role of CCR1 in viral assembly is under investigation, though it is still plausible that sequence changes to CCR1 may render the viral particles non-infectious but not necessarily halt the assembly of viral particles.
Here we have identified a novel cis-acting regulatory RNA sequence element located within the DENV capsid-coding sequence, which functions in a sequence-dependent, cell-type specific manner to regulate post-RNA replication production of infectious viral particles. Whether CCR1 serves as a putative assembly signal for DENV is still under investigation. Studies such as these provide evidence that RNA elements located within the coding-region can have diverse roles in regulating distinct stages within the viral life cycle in the mammalian host and mosquito vector. In addition, the identification of mutations affecting late steps in DENV replication should yield valuable information about the final stages in the viral life cycle, which are still poorly understood. The observation that CCR1 is more important in the mosquito vector both in vitro and in vivo allows for exploration into viral replication strategies in mosquito cells and the requirements for efficient viral transmission. Finally, conserved cis-acting RNA elements in the coding-region could serve as targets for novel therapeutics since development of resistance is constrained by both their RNA regulatory function and amino acid coding capacity.
Materials and methods
Sequence conservation and secondary structure prediction algorithms
For conserved sequence prediction, sequences were aligned using the Clustal W2 web software (Thompson et al., 1997) from the European Bioinformatics Institute. Portions of the Clustal W2 alignments were processed by the RNAalifold web server (Hofacker et al., 2002) from the Institute of Theoretical Chemistry at the University of Vienna to generate phylogenetically conserved secondary structure predictions. Individual RNA secondary structures were predicted using the mfold3 web server (Zuker, 2003) from the RNA Institute at the University at Albany. Reported ΔG values reflect enf refinement, the mfold free-energy computation incorporating coaxial stacking and the Jacobson–Stockmeyer theory for multibranched loops (Zuker, 2003).
Construction of mutant DNA constructs
Mutations were introduced into the capsid-coding region of an infectious clone (IC) of the DENV-2 Thai strain 16681 (pD2/IC-30P, hereafter pD2/IC; a gift from R. Kinney, Centers for Disease Control and Prevention, Fort Collins, CO) using splicing overlap extension-PCR (SOE-PCR), where the T7 promoter and the first 1391 nucleotides (nts) of DENV-2 were amplified. Resulting products were digested with SacI/SphI and ligated into a SacI/SphI-digested pD2/IC or a SacI/SphI-digested pD2/IC-RdRpmut (“GVD”), as previously described (Clyde et al., 2008; Clyde and Harris, 2006). For the Renilla luciferase reporter constructs, the T7 promoter and the first 167 nts from the IC constructs were amplified, and resulting products were digested with NotI/MluI and ligated into a NotI/MluI-digested pDRrep as previously described (Clyde et al., 2008). For the “delta” replicon, CCR1 was removed from pDRrep using SOE-PCR. Primer sequences are available upon request.
Generation of RNA templates
For all DNA templates (pD2/IC, pD2/IC-RdRpmut, pDRrep and pDRrep-RdRpmut), linearized templates were generated by digestion with XbaI and purified using a 25:24:1 phenol/chloroform/isoamyl alcohol extraction (pH 6.7 ± 0.2, Fischer Scientific). RNA templates were generated via in vitro transcription using the RiboMax Large Scale RNA Production System (T7, Promega) with the following modifications to the manufacturer’s protocol: 5 mM each GTP, CTP and UTP; 1 mM ATP; and 5 mM m7G(5′)ppp(5′)A cap analog (New England Biolabs) were incubated for 4 h at 30 °C with the addition of 2 mM ATP after 30 min. RNAs were subsequently treated with 80 U/mL TURBO DNase (Ambion) for 15 min at 37 °C, and unincorporated nucleotides were removed from DENV RNAs by size exclusion chromatography using Micro Bio-Spin P-30 Tris columns (BioRad Laboratories).
Cell culture
Baby hamster kidney cells (BHK-21, clone 15; hereafter BHK) were grown in minimal essential medium-alpha (MEMα, Gibco) with 5% fetal bovine serum (FBS; HyClone) at 37 °C in 5% CO2. A high temperature (HT) variant (gift from A. Garmanik, Fundación Instituto Leloir, Buenos Aires, Argentina) was utilized for transfection in Ae. albopictus mosquito cells (C6/36), which were grown in Leibovitz’s medium (L-15, Gibco) with 10% FBS at 33 °C without CO2. All media was supplemented with 10 mM HEPES (pH 7.5), 100 units/mL penicillin, and 100 μg/mL streptomycin.
RNA transfections
BHK cells were seeded in 24-well plates, grown to 50% confluence, and transfected with 0.5 μg IC or replicon RNA using 4 × Lipofectamine 2000 (Invitrogen), followed by a 2-h incubation at 37 °C in 5% CO2. BHK cells were washed 3 × in MEMα medium and incubated at 37 °C with 5% CO2 until harvest at indicated timepoints. C6/36 HT cells were seeded in 12-well plates, grown to 95% confluence, and transfected with 1 μg IC or replicon RNA using 6 × Lipofectamine 2000 (Invitrogen), followed by a 3-h incubation at 33 °C without CO2. C6/36 HT cells were washed 1 × in L-15 media and incubated at 33 °C without CO2 until harvest at indicated timepoints. Virus-containing supernatants and intracellular virus were collected, so that viral titer could be assessed by plaque assay. Cellular RNA was harvested from a duplicate well and purified using the Mini RNA Isolation II Kit (Zymo Research), and viral RNA (vRNA) concentrations were determined using quantitative reverse transcriptase (qRT)-PCR. vRNA in the virus-containing supernatants was isolated using the ZR Viral RNA kit (Zymo Research) and quantified using qRT-PCR.
Virus titration by plaque assay
Plaque assays using BHK cells for virus titration were conducted as described previously (Clyde et al., 2008; Clyde and Harris, 2006; Diamond et al., 2000). The viral titers in Ae. aegypti mosquito bodies and salivary glands were determined by plaque assay on African green monkey kidney cells (Vero, ATCC #CCL-81) as previously described (Payne et al., 2006). Viral titer was calculated as plaque forming units (pfu) per mL, and the limit of detection was defined as the lowest titer of virus that could be accurately measured in BHK (1.9 pfu/mL) and Vero (1.4 pfu/mL) cells. For transfections, viral titers were normalized to the amount of vRNA in the cells at the time that the virus-containing supernatants were harvested, as determined by qRT-PCR. p-values relative to the WT control were derived by Wilcoxon rank sum test in Mstat.
Quantitative RT-PCR
qRT-PCR was performed using a modification to the method of Laue et al. (1999), which uses a FAM-TAMRA labeled probe and primer set that is directed to the viral NS5 gene. The TaqMan One-Step Master Mix (Applied Biosystems) system was used with the following modifications to the protocol: the RT step was conducted at 48 °C for 30 min, the initial denaturation step was performed at 95 °C for 10 min, the annealing temperature was set at 60 °C, and the reaction volume was fixed at 20 μL. For the purposes of determining transfection efficiency, target vRNA was first normalized to the cellular 18S RNA using the TaqMan VIC-MGB Primer Limited Eukaryotic 18S rRNA Endogenous Control (Applied Biosystems) in a parallel reaction. qRT-PCR was conducted on a Sequence Detection System 7300 (Applied Biosystems).
Ae. aegypti mosquitoes
The Ae. aegypti colony originated from eggs collected in Panama by J. Loaiza and were received at the Arbovirus Laboratory (Wadsworth Center) in 2007. Colonized mosquitoes were maintained on defibrinated rabbit blood with 10% sucrose (for egg laying) and given 10% sucrose ad libitum. Larvae were reared and adults maintained at 27 °C with 70% relative humidity (RH) and under a 16:8 light:dark diurnal cycle in 30.5-cm3 cages.
Infection of Ae. aegypti mosquitoes
Virus-containing supernatants were harvested at 72 h post-transfection (hpt) from IC-transfected BHK cells. Virus-containing supernatants were concentrated by ultracentrifugation at 89,500g for 2-h using a 5% cold sucrose column, and viral titer was assessed by plaque assay. Viruses were diluted to a titer of 104 pfu/mL in mosquito diluent (MD; 20% heat-inactivated FBS in PBS (Dulbecco) supplemented with 50 μg/mL penicillin/streptomycin, 50 μg/mL gentamicin, and 2.5 μg/mL fungizone). Two-to-five day-old mosquitoes were inoculated intrathoracically (IT) with 0.1–1 pfu under CO2 anesthesia using the FemtoJet micro-injection system (Eppendorf) and held at 30 °C with a 16:8 light:dark photoperiod for up to 20-day post-inoculation (dpi). Mosquito bodies were placed into 1 mL MD, and samples were stored at −70 °C until they were homogenized in a mixer mill (Qiagen) and clarified by centrifugation at 1200 rpm for 3 min. Body titer was evaluated at indicated timepoints, and the viral load in infected mosquitoes was determined by plaque assay. Salivary glands were dissected from infected mosquitoes on day 10 post-inoculation and placed into 0.3 mL MD, while the mosquito remnants were placed in 1 mL MD; samples were stored at −70 °C. Viral load in the salivary glands was determined by plaque assay.
Luciferase assay
BHK and C6/36 HT cells were transfected with the Renilla luciferase reporter constructs and harvested at the indicated timepoints. Samples were analyzed with the Renilla Luciferase Assay System (Promega) according to the manufacturer’s instructions on a GloMax-96 Microplate Luminometer (Promega) with the following modifications: 50 μL cell lysate and 40 μL of 1 × Renilla Luciferase Assay Substrate was used for each sample. Luciferase activity levels were normalized to the relative transfection efficiency at 2 hpt as determined by qRT-PCR. p-values relative to the WT control were derived by Wilcoxon rank sum test in Mstat.
Determination of vRNA half-life
Total cellular RNA was harvested from BHK cells transfected with pD2/IC-RdRp constructs (“GVD”) at 2, 4, 8, 24, 48 and 72 hpt, treated with DNase, and quantified by NS5 qRT-PCR as described above. Values were graphed, and an exponential trendline was fitted to the data in Microsoft Excel 2007. The half-life of each variant in each experiment was determined from the exponential trendline equation, averaged and shown in Fig. 5C. p-values relative to the GVD control were derived by Wilcoxon rank sum test in Mstat.
Intracellular virus extraction
Virus-containing supernatants were harvested from IC-trans-fected BHK and C6/36 HT cell monolayers at indicated timepoints and assessed by plaque assay to determine the extracellular viral titers. To obtain the intracellular viral titers, cells were washed 1 × in PBS and then incubated for 3 min at 4 °C with an alkaline/high-salt solution of 1 M NaCl plus 50 mM sodium bicarbonate, pH 9.5, to remove the surface-bound virus (Diamond and Harris, 2001; Helt and Harris, 2005). Cells were washed 2 × in PBS before the addition of 0.25% Trypsin-EDTA (Gibco) to detach cells from the plates. Cells were harvested in MEMα (BHK) or L-15 (C6/36) medium and lysed during a single freeze–thaw cycle. Cellular debris was pelleted by centrifugation at 3200g for 5 min, and the intracellular virus-containing supernatant was removed and assessed for viral titer by plaque assay. Extra- vs. intracellular viral concentrations were calculated as ratios, where a ratio greater than one indicates that there was no accumulation of infectious virus within the cell and ratios less than one indicates that there was more intracellular infectious virus than what was being released into the supernatant. p-values relative to the WT control were derived by Wilcoxon rank sum test in Mstat.
Statistical analysis
Wilcoxon rank sum test were performed in Mstat. Statistical significance was defined as p < 0.05.
Supplementary Material
Acknowledgments
We would like to thank Andrea Garmanik (Fundación Instituto Leloir, Buenos Aires, Argentina) for providing the C6/36 HT variant used in this paper and Kimberly-Anne Mattia (Integral Molecular, Philadelphia, PA) for her work on packaging CCR1 replicon RNAs. We are grateful to Karen Clyde and Sondra Schlesinger for technical and scientific advice throughout the project. We appreciate Molly OhAinle, Poornima Parameswaran and Sondra Schlesinger for their careful reading of this manuscript and helpful discussions. We thank Alex Ciota for his helpful suggestions on design and interpretation of mosquito studies. This work was supported by NIH grant R01AI52324 (to E.H.).
Footnotes
Appendix A. Supplementary Information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.virol.2012.06.028.
References
- Alvarez DE, De Lella Ezcurra AL, Fucito S, Gamarnik AV. Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology. 2005a;339:200–212. doi: 10.1016/j.virol.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Alvarez DE, Filomatori CV, Gamarnik AV. Functional analysis of dengue virus cyclization sequences located at the 5′ and 3′UTRs. Virology. 2008;375:223–235. doi: 10.1016/j.virol.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Alvarez DE, Lodeiro MF, Ludueña SJ, Pietrasanta LI, Gamarnik AG. Long-range RNA–RNA interactions circularize the dengue virus genome. J Virol. 2005b;79:6631–6643. doi: 10.1128/JVI.79.11.6631-6643.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvisi G, Madan V, Bartenshlager R. Hepatitis C virus and host cell lipids: an intimate connection. RNA Biol. 2011;8:258–269. doi: 10.4161/rna.8.2.15011. [DOI] [PubMed] [Google Scholar]
- Ansarah-Sobrinho C, Nelson S, Jost CA, Whitehead SS, Pierson TC. Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology. 2008;381:67–74. doi: 10.1016/j.virol.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias CF, Preugschat F, Strauss JH. Dengue 2 virus NS2B and NS3 form a stable complex that can cleave NS3 within the helicase domain. Virology. 1993;193:888–899. doi: 10.1006/viro.1993.1198. [DOI] [PubMed] [Google Scholar]
- Burke DS, Monath TP. Flaviviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Vol. 1. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 1043–1126. [Google Scholar]
- Chadwick DR, Lever AM. Antisense RNA sequences targeting the 5′ leader packaging signal region of human immunodeficiency virus type-1 inhibits viral replication at post-transcriptional stages of the life cycle. Gene Ther. 2000;7:1362–1368. doi: 10.1038/sj.gt.3301254. [DOI] [PubMed] [Google Scholar]
- Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- Chiu WW, Kinney RM, Dreher TW. Control of translation by the 5′- and 3′-terminal regions of the dengue virus genome. J Virol. 2005;79:8303–8315. doi: 10.1128/JVI.79.13.8303-8315.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua JJ, Ng MM, Chow VT. The non-structural 3 (NS3) protein of dengue virus type 2 interacts with human nuclear receptor binding protein and is associated with alterations in membrane structure. Virus Res. 2004;102:151–163. doi: 10.1016/j.virusres.2004.01.025. [DOI] [PubMed] [Google Scholar]
- Clyde K, Barrera J, Harris E. The capsid-coding region hairpin element (cHP) is a critical determinant of dengue virus and West Nile virus RNA synthesis. Virology. 2008;379:314–323. doi: 10.1016/j.virol.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyde K, Harris E. RNA secondary structure in the coding region of dengue virus type 2 directs translation start codon selection and is required for viral replication. J Virol. 2006;80:2170–2182. doi: 10.1128/JVI.80.5.2170-2182.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyde K, Kyle JL, Harris E. Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J Virol. 2006;80:11418–11431. doi: 10.1128/JVI.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deubel V, Digoutte JP. Morphogenesis of yellow fever virus in Aedes aegypti cultured cells. I. Isolation of different cellular clones and the study of their susceptibility to infection with the virus. Am J Trop Med Hyg. 1981;30:1060–1070. doi: 10.4269/ajtmh.1981.30.1060. [DOI] [PubMed] [Google Scholar]
- Diamond MS, Edgil D, Roberts TG, Lu B, Harris E. Infection of human cells by dengue virus is modulated by different cell types and viral strains. J Virol. 2000;74:7814–7823. doi: 10.1128/jvi.74.17.7814-7823.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Harris E. Interferon inhibits dengue virus infection by preventing translation of viral RNA through a PKR-independent mechanism. Virology. 2001;289:297–311. doi: 10.1006/viro.2001.1114. [DOI] [PubMed] [Google Scholar]
- Elshuber S, Mandl CW. Resuscitating mutations in a furin cleavage-deficient mutant of the flavivirus tick-borne encephalitis virus. J Virol. 2005;79:11813–11823. doi: 10.1128/JVI.79.18.11813-11823.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayzulin R, Frolov I. Changes of the secondary structure of the 5′ end of the Sindbis virus genome inhibit virus growth in mosquito cells and lead to accumulation of adaptive mutations. J Virol. 2004;78:4953–4964. doi: 10.1128/JVI.78.10.4953-4964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filomatori CV, Lodeiro MF, Alvarez DE, Samsa MM, Pietrasanta L, Gamarnik AV. A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Gene Dev. 2006;20:2238–2249. doi: 10.1101/gad.1444206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosmire JA, Hwang K, Makino S. Identification and characterization of a coronavirus packaging signal. J Virol. 1992;66:3522–3530. doi: 10.1128/jvi.66.6.3522-3530.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe P, Harris E. Interplay of RNA elements in the dengue virus 5′ and 3′ ends required for viral RNA replication. J Virol. 2010;84:6103–6118. doi: 10.1128/JVI.02042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe P, Shi PY, Harris E. The 5′ and 3′ downstream AUG region elements are required for mosquito-borne flavivirus RNA replication. J Virol. 2011;85:1900–1905. doi: 10.1128/JVI.02037-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov I, Hardy R, Rice CM. Cis-acting RNA elements at the 5′ end of Sindbis virus genome RNA regulate minus- and plus-strand RNA synthesis. RNA. 2001;7:1638–1651. doi: 10.1017/s135583820101010x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova E, Frolov I, Schlesinger S. Packaging signals in alphaviruses. J Virol. 1997;71:248–258. doi: 10.1128/jvi.71.1.248-258.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler DJ. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev. 1998;11:480–496. doi: 10.1128/cmr.11.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubler DJ. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 2002a;10:100–103. doi: 10.1016/s0966-842x(01)02288-0. [DOI] [PubMed] [Google Scholar]
- Gubler DJ. The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res. 2002b;33:330–342. doi: 10.1016/s0188-4409(02)00378-8. [DOI] [PubMed] [Google Scholar]
- Gubler DJ, Kuno G. Dengue and Dengue Hemorrhagic Fever. CAB International; 1997. [Google Scholar]
- Hase T, Summers PL, Eckels KH, Baze WB. An electron and immunoelectron microscopic study of dengue-2 virus infection of cultured mosquito cells: maturation events. Arch Virol. 1987a;92:273–291. doi: 10.1007/BF01317484. [DOI] [PubMed] [Google Scholar]
- Hase T, Summers PL, Eckels KH, Baze WB. Maturation process of Japanese encephalitis virus in cultured mosquito cells in vitro and mouse brain cells in vivo. Arch Virol. 1987b;96:135–151. doi: 10.1007/BF01320956. [DOI] [PubMed] [Google Scholar]
- Heinz F, Auer G, Stiasny K, Holzmann H, Mandl C, Guirakhoo F, Kunz C. The interactions of the flavivirus envelope proteins: implications for virus entry and release. Arch Virol. 1994;9:339–348. doi: 10.1007/978-3-7091-9326-6_34. [DOI] [PubMed] [Google Scholar]
- Helt A-M, Harris E. S-phase-dependent enhancement of dengue virus 2 replication in mosquito cells, but not in human cells. J Virol. 2005;79:7291–7299. doi: 10.1128/JVI.79.21.13218-13230.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL, Fekete M, Stadler PF. Secondary structure prediction for aligned RNA sequences. J Mol Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- Hofacker IL, Fontana W, Stadler PF, Bonhoeffer S, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 1994;125:167–188. [Google Scholar]
- Holden KL, Stein D, Pierson TC, Ahmed A, Clyde K, Iverson P, Harris E. Inhibition of dengue virus translation and RNA synthesis by a morpholino oligomer to the top of the 3′ stem-loop structure. Virology. 2006;344:439–452. doi: 10.1016/j.virol.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Ishak R, Tovey DG, Howard CR. Morphogenesis of yellow fever virus 17D in infected cell cultures. J Gen Virol. 1988;69:325–335. doi: 10.1099/0022-1317-69-2-325. [DOI] [PubMed] [Google Scholar]
- Jones CT, Patkar CG, Kuhn RJ. Construction and application of yellow fever virus replicons. Virology. 2005;331:247–259. doi: 10.1016/j.virol.2004.10.034. [DOI] [PubMed] [Google Scholar]
- Khromykh AA, Meka H, Guyatt KJ, Westway EG. Essential role of cyclization domains in flavivirus RNA replication. J Virol. 2001;75:6719–6728. doi: 10.1128/JVI.75.14.6719-6728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khromykh AA, Varnavski AN, Westaway EG. Encapsidation of the flavivirus kunjin replicon RNA by using a complementation system providing Kunjin virus structural proteins in trans. J Virol. 1998;72:5967–5977. doi: 10.1128/jvi.72.7.5967-5977.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney RM, Butrapet S, Chang GJ, Tsuchiya KR, Roehrig JT, Bhamarapravati N, Gubler DJ. Construction of infectious cDNA clones for dengue 2 virus strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology. 1997;230:300–308. doi: 10.1006/viro.1997.8500. [DOI] [PubMed] [Google Scholar]
- Ko KK, Igarashi A, Fukai K. Electron microscopic observations on Aedes albopictus cells infected with dengue viruses. Arch Virol. 1979;62:41–52. doi: 10.1007/BF01314902. [DOI] [PubMed] [Google Scholar]
- Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 2002;108:717–725. doi: 10.1016/s0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno G. Factors influencing the tranmsission of dengue viruses. In: Gubler DJ, Kuno G, editors. Dengue and Dengue Hemorrhagic Fever. CAB International; New York: 1997. [Google Scholar]
- Kyle JL, Harris E. Global persistence and spread of dengue. Annu Rev Microbiol. 2008;62:71–92. doi: 10.1146/annurev.micro.62.081307.163005. [DOI] [PubMed] [Google Scholar]
- Laue T, Emmerich P, Schmitz H. Detection of dengue virus RNA in patients after primary or secondary dengue infection by using the TaqMan automated amplification system. J Clin Microbiol. 1999;37:2543–2547. doi: 10.1128/jcm.37.8.2543-2547.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary K, Blair CD. Sequential events in the morphogenesis of Japanese encephalitis virus. J Ultrastruct Res. 1980;72:123–129. doi: 10.1016/s0022-5320(80)90050-7. [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, Thiel HJ, Rice CM. Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Lippincott-Raven Publishers; Philadelphia: 2007. pp. 1101–1152. [Google Scholar]
- Linger BR, Kunovska L, Kuhn RJ, Golden BL. Sindbis virus nucleocapsid assembly: RNA folding promotes capsid protein dimerization. RNA. 2004;10:128–138. doi: 10.1261/rna.5127104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wimmer E, Paul AV. Cis-acting RNA elements in human and animal plus-strand RNA viruses. Biochim Biophys Acta. 2009;1789:495–517. doi: 10.1016/j.bbagrm.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M, Tilgner M, Bernard KA, Shi P-Y. Functional analysis of mosquitoborne flavivirus conserved sequence elements within 3′ untranslated region of West Nile Virus by use of a reporting replicon that differentiates between viral translation and RNA replication. J Virol. 2003;77:10004–10014. doi: 10.1128/JVI.77.18.10004-10014.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumara T, Shiraki K, Sashikata T, Hotta S. Morphogenesis of dengue-1 virus in cultures of a human leukemic leukocyte line (J-111) Microbiol Immunol. 1977;21:329–334. doi: 10.1111/j.1348-0421.1977.tb00295.x. [DOI] [PubMed] [Google Scholar]
- Mattia K, Puffer BA, Williams KL, Gonzalez R, Murray M, Sluzas E, Pagano D, Ajith A, Bower M, Berdougo Harris E, Doranz BJ. Dengue reporter virus particles for measuring neutralizing antibodies against each of the four dengue serotypes. PLoS One. 2011;6(1–9):e27252. doi: 10.1371/journal.pone.0027252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WA, White KA. Long-distance RNA-RNA interactions in plant virus gene expression and replication. Annu Rev Phytopathol. 2006;44:447–467. doi: 10.1146/annurev.phyto.44.070505.143353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy FA. Togavirus morphology and morphogenesis. In: Schlesinger RW, editor. The Togaviruses: Biology, Structure, Replication. Academic; New York: 1980. pp. 241–316. [Google Scholar]
- Nelson S, Jost CA, Xu Q, Ess J, Martin JE, Oliphant T, Whitehead SS, Durbin AP, Graham BS, Diamond MS, Pierson TC. Maturation of West Nile virus modulates sensitivity to antibody-mediated neutralization. PLoS Pathog. 2008;4:e1000060. doi: 10.1371/journal.ppat.1000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng ML. Ultrastructural studies of Kunjin virus-infected Aedes albopictus cells. J Gen Virol. 1987;68:577–582. doi: 10.1099/0022-1317-68-2-577. [DOI] [PubMed] [Google Scholar]
- Niesters HG, Strauss JH. Mutagenesis of the conserved 51-nucleotide region of Sindbis virus. J Virol. 1990;64:1639–1647. doi: 10.1128/jvi.64.4.1639-1647.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak T, Farber PM, Wengler G, Wengler G. Analyses of the terminal sequences of West Nile virus structural proteins and of the in vitro translation of these proteins allow the proposal of a complete scheme of the proteolytic cleavages involved in their synthesis. Virology. 1989;169:365–376. doi: 10.1016/0042-6822(89)90162-1. [DOI] [PubMed] [Google Scholar]
- Ohyama A, Ito T, Tanimura E, Huang SC, Hsue J. Electron microscopic observation of the budding maturation of group B arboviruses. Microbiol Immunol. 1977;21:535–538. doi: 10.1111/j.1348-0421.1977.tb00320.x. [DOI] [PubMed] [Google Scholar]
- Panavas T, Hawkins CM, Panaviene Z, Nagy PD. The role of the p33:p33/p92 interaction domain in RNA replication and intracellular localization of p33 and p92 proteins of Cucumber necrosis tombusvirus. Virology. 2005;338:81–95. doi: 10.1016/j.virol.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Payne AF, Binduga-Gajewska I, Kauffman EB, Kramer LD. Quantitation of flaviviruses by fluorescent focus assay. J Virol Methods. 2006;134:183–189. doi: 10.1016/j.jviromet.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Rosen L. The Emperor’s New Clothes revisited, or reflections on the pathogenesis of dengue hemorrhagic fever. Am J Trop Med Hyg. 1977;26:337–343. doi: 10.4269/ajtmh.1977.26.337. [DOI] [PubMed] [Google Scholar]
- Rosen L, Gubler D. The use of mosquitoes to detect and propagate dengue viruses. Am J Trop Med Hyg. 1974;23:1153–1160. doi: 10.4269/ajtmh.1974.23.1153. [DOI] [PubMed] [Google Scholar]
- Rosen L, Roseboom LE, Gubler DJ, Lien JC, Chaniotis BN. Comparative susceptibility of mosquito species and strains to oral and parenteral infection with dengue and Japanese encephalitis viruses. Am J Trop Med Hyg. 1985;34:603–615. doi: 10.4269/ajtmh.1985.34.603. [DOI] [PubMed] [Google Scholar]
- Rouha H, Hoenninger VM, Thurner C, Mandl CW. Mutational analysis of three predicted 5′-proximal stem-loop structures in the genome of tick-borne encephalitis virus indicates different roles in RNA replication and translation. Virology. 2011;417:79–86. doi: 10.1016/j.virol.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsa MM, Mondotte JA, Caramelo JJ, Gamarnik AV. Uncoupling cis-acting RNA elements from coding sequences revealed a requirement of the N-terminal region of dengue virus capsid protein in virus particle formation. J Virol. 2012;86:1046–1058. doi: 10.1128/JVI.05431-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriurairatna S, Bhamarapravati N. Replication of dengue-2 virus in Aedes albopictus mosquitoes. An electron microscopic study. Am J Trop Med Hyg. 1977;26:1199–1205. doi: 10.4269/ajtmh.1977.26.1199. [DOI] [PubMed] [Google Scholar]
- Sriurairatna S, Bhamarapravati N, Phalavadhtana O. Dengue virus infection of mice: morphology and morphogenesis of dengue type 2 virus in suckling mouse neurons. Infect Immun. 1973;8:1017–1029. doi: 10.1128/iai.8.6.1017-1028.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick-borne encephalitis virus by furin. J Virol. 1997;71:8475–8481. doi: 10.1128/jvi.71.11.8475-8481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurner C, Witwer C, Hofacker IL, Stadler PF. Conserved RNA secondary structures in Flaviviridae genomes. J Gen Virol. 2004;85:1113–1124. doi: 10.1099/vir.0.19462-0. [DOI] [PubMed] [Google Scholar]
- Tilgner M, Shi PY. Structure and function of the 3′ terminal six nucleotides of the West Nile virus genome in viral replication. J Virol. 2004;78:8159–8171. doi: 10.1128/JVI.78.15.8159-8171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuplin A, Evans DJ, Buckley A, Jones IM, Gould EA, Gritsun TS. Replication enhancer elements within the open reading frame of tick-borne encephalitis virus and their evolution within the Flavivirus genus. Nucleic Acids Res. 2011;39:7034–7048. doi: 10.1093/nar/gkr237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wynsberghe PM, Ahlguist P. 5′ cis elements direct nodavirus RNA1 recruitment to mitochondrial sites of replication complex formation. J Virol. 2009;83:2976–2988. doi: 10.1128/JVI.02040-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AM, Bidet K, Yinglin A, Ler SG, Hogue K, Blackstock W, Gunaratne J, Garcia-Blanco MA. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol. 2011;8:1–14. doi: 10.4161/rna.8.6.17836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B, Geigenmuller-Gnirke U, Schlesinger S. Interactions between Sindbis virus RNAs and a 68 amino acid derivative of the viral capsid protein further defines the capsid binding site. Nucleic Acids Res. 1994;22:780–786. doi: 10.1093/nar/22.5.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengler G, Wengler G. Cell-associated West Nile flavivirus is covered with E+pre-M protein heterodimers which are destroyed and reorganized by proteolytic cleavage during virus release. J Virol. 1989;63:2521–2526. doi: 10.1128/jvi.63.6.2521-2526.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu IM, Holdaway HA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. Association of the pr peptides with dengue virus at acidic pH blocks membrane fusion. J Virol. 2009;83:12101–12107. doi: 10.1128/JVI.01637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Falgout B, Markoff L. Identification of specific nucleotide sequences within conserved 3′-SL in the dengue type 2 virus genome required for replication. J Virol. 1998;72:7510–7522. doi: 10.1128/jvi.72.9.7510-7522.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Chipman PR, Corver J, Jonhnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 2003a;10:907–912. doi: 10.1038/nsb990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Corver J, Chipman PR, Zhang W, Pletnev SV, Sedlak D, Baker TS, Straus JH, Kuhn RJ, Rossmann MG. Structures of immature flavivirus particles. EMBO J. 2003b;22:2604–2613. doi: 10.1093/emboj/cdg270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kostyuchenko VA, Rossmann MG. Structural analysis of viral nucleocapsids by subtraction of partial projections. J Struct Biol. 2007;157:356–364. doi: 10.1016/j.jsb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Dasgupta R, Rueckert R. Evidence that the packaging signal for nodaviral RNA2 is a bulged stem-loop. Proc Natl Acad Sci USA. 1992;89:11146–11150. doi: 10.1073/pnas.89.23.11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybert IA, van der Ende-Metselaar H, Wilschut J, Smit JM. Functional importance of dengue virus maturation: infectious properties of immature virions. J Gen Virol. 2008;89:3047–3051. doi: 10.1099/vir.0.2008/002535-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.