

Abstract
The extracellular signal-related kinases (ERK1/2) are key proteins mediating mitogen-activated protein kinase signaling downstream of RAS: phosphorylation of ERK1/2 leads to nuclear uptake and modulation of multiple targets1. Here we show that reduced dosage of ERF, which encodes an inhibitory ETS transcription factor directly bound by ERK1/2 (refs 2-7), causes complex craniosynostosis (premature fusion of the cranial sutures) in humans and mice. Features of this newly recognized clinical disorder include multiple suture synostosis, craniofacial dysmorphism, Chiari malformation and language delay. Mice with functional Erf reduced to ~30% of normal exhibit postnatal multisuture synostosis; by contrast, embryonic calvarial development appears mildly delayed. Using chromatin immunoprecipitation in mouse embryonic fibroblasts and high-throughput sequencing, we find that ERF binds preferentially to distal regulatory elements containing RUNX or AP1 motifs. This work identifies ERF as a novel regulator of osteogenic stimulation by RAS-ERK signaling, potentially by competing with activating ETS factors in multifactor transcriptional complexes.
We used exome sequencing8 to analyze the DNA from a 15-year old boy with craniosynostosis affecting all sutures of the cranial vault. His 7-year old brother had metopic, sagittal and left coronal synostosis and their mother exhibited exorbitism and midface hypoplasia but did not have documented craniosynostosis (Fig. 1a-d). After excluding previously described variants and genomic regions for which the brothers did not share the maternal allele, 135 nonsynonymous sequence changes remained, including 5 nonsense mutations (Supplementary Table 1). One of the nonsense mutations (c.547C>T; p.Arg183*) was present in ERF, encoding an inhibitory ETS-family transcription factor located on chromosome 19q13.2. ERF was previously shown to be a prominent binding target of the paralogous kinases ERK1 and ERK2 (ERK1/2)7, key effectors of the RAS-MEK-ERK signal transduction cascade; the transcriptional activity of ERF is primarily regulated by ERK1/2-mediated phosphorylation, which leads to its export from the nucleus5,6. We considered ERF as a candidate because activation of ERK1/2 signaling was previously demonstrated in craniosynostosis9,10. We confirmed segregation of the mutation from the maternal grandmother to the two affected children (Family 1, Supplementary Fig. 1).
Figure 1.

Clinical features of subjects heterozygous for ERF mutations. (a-d) Family 1 showing subject IV-1 aged 10 yr, in whom exome sequencing was performed (a), his brother IV-2 aged 4 mo (b) and mother III-3 aged 37 yr (c). The computed tomographic head scan of IV-2 aged 5 mo (d) shows synostosis of the left coronal and sagittal sutures (arrowheads) associated with multiple craniolacunae; the lambdoid and squamosal sutures remain patent. (e-h) Subjects identified in follow-up sequencing had clinical diagnoses ranging from non-syndromic sagittal (e, III-1 in Family 5 aged 1.6 yr) or unilateral lambdoid synostosis (f, III-1 in Family 4 aged 1.2 yr) to FGFR2 mutation-negative Crouzon syndrome (g, II-1 in Family 10 aged 4 mo; h, III-1 in Family 7 aged 18 yr). (i) Magnetic resonance brain imaging (sagittal, T1 view) of III-1 in Family 4 aged 7.1 yr, showing Chiari malformation (12 mm herniation of cerebellar tonsils through the foramen magnum, arrow). (j) Comparison of average faces between ERF-mutant (n = 14) and control (n = 381) subjects. Red/blue denotes normalized displacement at over 1.5 SD, highlighting shared features of hypertelorism (left), vertical nasal displacement (centre) and prominent forehead with exorbitism (right). Written consent was provided for the publication of all photographs.
To analyze the possible contribution of ERF mutation to the phenotype, we sequenced the gene in 411 samples from unrelated subjects with craniosynostosis (Table 1) and 288 north European controls. Heterozygous loss-of-function mutations were present in a further 11 patient samples but in no normal controls (P = 0.004, Fisher’s exact test) (Fig. 2 and Table 2). No deletions were identified in 276 mutation-negative samples. We analyzed earlier generations, finding 26 mutation-positive individuals in total (Supplementary Fig. 1). In 4 families the mutation had arisen de novo from a parent (n = 2), grandparent (n = 1) or great-grandparent (n = 1) (Supplementary Fig. 2). The occurrence of mutations only in patient samples and the identification of multiple de novo cases, establish that ERF mutations are the cause of craniosynostosis in these families.
Table 1. Patients with craniosynostosis analyzed for ERF mutations by DNA sequencing.
| Non- syndromic |
Syndromic | Combined | ||||
|---|---|---|---|---|---|---|
| Total |
ERF mutation positive |
Total |
ERF mutation positive |
Total |
ERF mutation positive |
|
| Metopic | 46 | 0 | 13 | 0 | 59 | 0 |
| Sagittal | 70 | 1 | 16 | 1 | 86 | 2 |
| Unicoronal | 99 | 0 | 16 | 0 | 115 | 0 |
| Bicoronal | 25 | 0 | 24 | 0 | 49 | 0 |
| Uni- or bilambdoid |
13 | 1 | 0 | 0 | 13 | 1 |
| Multisuture | 26 | 1 | 40 | 7 | 66 | 8 |
| Sutures not specified |
6 | 0 | 18 | 1 | 24 | 1 |
| Combined | 285 | 3 | 127 | 9 | 412 | 12 |
Figure 2.

Exon and domain structure of ERF and mutations identified in craniosynostosis. ERF comprizes 4 exons (a) extending over 7.6 kb and encodes a 548 amino acid protein (b). The positions of serine (S) and threonine (T) sites phosphorylated by ERK4 are indicated. Two missense substitutions p.Arg65Gln and p.Arg86Cys localize to the ETS DNA-binding domain. The lineup in the bottom panel shows the ETS domain sequence in a representative member of each ETS subfamily from humans. Fully and partially conserved residues are filled dark and light grey, respectively.
Table 2. Mutations of ERF present in 12 families.
| Family # |
Proband ID |
Proband cranio- synostosis phenotypea |
Individuals with mutations (see Supplementary Figure 1) |
cDNA change | Predicted amino acid change |
De novo mutation |
|---|---|---|---|---|---|---|
| 1 | OX2158 | M | II-2, III-3, IV-1, IV-2 |
c.547C>T | p.Arg183* | |
| 2 | OX2729 | na | II-1 | c.1512delT | p.Phe504Leufs*27 | |
| 3 | OX2789 | P | II-1 | c.891_892delAG | p.Gly299Argfs*9 | |
| 4 | OX3247 | UL | II-2, III-1 | c.256C>T | p.Arg86Cys | Y |
| 5 | OX3248 | S | II-1, III-1 | c.194G>A | p.Arg65Gln | |
| 6 | OX3801 | M | II-1, III-1, III-2, IV-2 |
c.3G>A | p.0 | Y |
| 7 | OX3970 | M | II-2, III-1, III-2 | c.891_892delAG | p.Gly299Argfs*9 | |
| 8 | OX4097 | M | II-1 | c.891_892delAG | p.Gly299Argfs*9 | Y |
| 9 | OX4626 | P | II-2, III-1, III-2 | c.1270C>T | p.Gln424* | |
| 10 | OX4708 | M | II-1 | c.256C>T | p.Arg86Cys | Y |
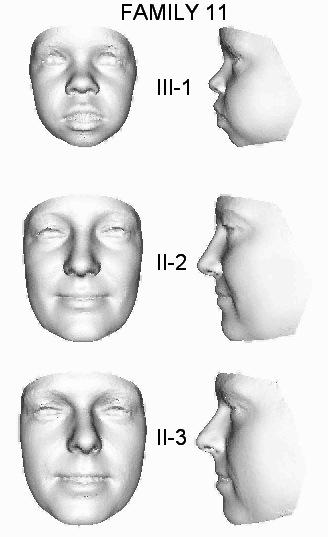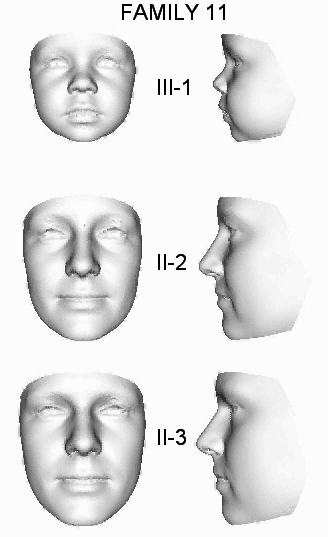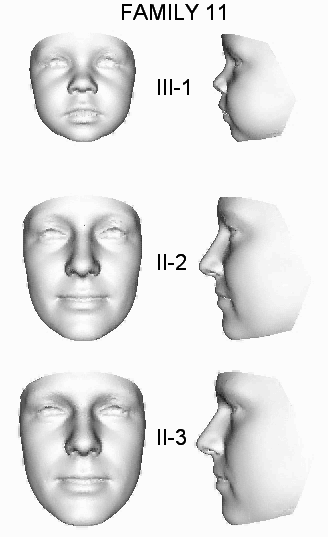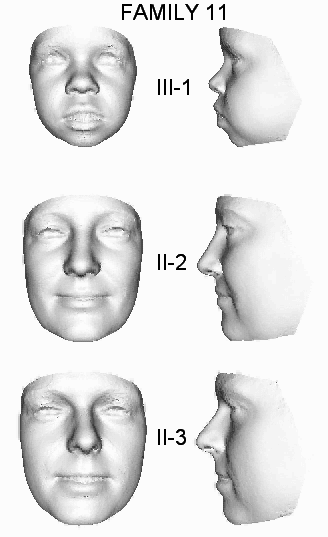
| 11 | OX4902 | S | II-2, II-3, III-1 | c.21A>T | p.Gly8_Phe9ins147 | |
| 12 | OX5072 | P | II-1 | c.1201_1202delAA | p.Lys401Glufs*10 |
M, multisuture synostosis; P, pansynostosis; S, sagittal synostosis; UL, unilateral lambdoid synostosis; na, not available.
ERF comprizes 4 exons (Fig. 2a) and encodes a ubiquitously expressed ETS transcription factor (numbering 28 members in humans)11, which acts as a negative regulator either by competing with other ETS-family members for DNA binding, or through unique targets2,3. Functionally characterized motifs in ERF comprize the N-terminal DNA-binding (ETS), central ERK interaction, and C-terminal repressor domains (Fig. 2b); DNA binding targets a core (5′-GGAA/T-3′) motif, with little sequence discrimination from other ETS-family members12. The mutations are diverse; the 3 missense changes (1 recurrent) are located in critical residues in the DNA-binding ETS domain or disrupt the initiation codon, whereas the remaining 8 mutations comprize a splice site mutation, two nonsense changes and 3 frameshifts (1 recurrent, present in 3 families) (Fig. 2, Table 2, Supplementary Fig. 3a). Immunoblotting of fibroblasts or lymphoblastoid cells from patients demonstrated reduced expression of full-length ERF associated with the initiation codon and nonsense mutations, but not the missense substitutions affecting the ETS domain (Supplementary Fig. 3b). DNA-binding domain mutants failed to repress Ets binding site-containing promoters (Supplementary Fig. 3c). These data suggest that the predominant pathophysiological mechanism is heterozygous loss-of-function (haploinsufficiency).
We analyzed the phenotype associated with this new syndrome in the 26 mutation-positive individuals (Supplementary Table 2). Of 14 pediatric cases, 13 had craniosynostosis; in the 8 with accurate assessment by 3-dimensional computed tomography (CT) of the skull, fusion affected the sagittal (n = 7), lambdoid (n = 5), coronal (n = 3) and metopic (n = 1) sutures (Fig. 1d, Supplementary Table 3), a pattern distinct from other monogenic types of craniosynostosis in which the coronal suture is most commonly affected13. Seven of 12 probands had syndromic multisuture synostosis (Table 1), representing a 13-fold enrichment compared to other diagnostic groups (P = 3 × 10−5, Fisher’s exact test), but 3 subjects presented with single synostosis of the sagittal (n = 2) or lambdoid (n = 1) sutures (Fig. 1e,f). In half of the families a diagnosis of Crouzon syndrome had been suggested because of exorbitism and midface hypoplasia (Fig. 1a,c,g,h, Supplementary Fig. 4); however, FGFR2 genetic testing14,15 was normal. Chiari type I malformations were diagnosed in 4 cases (Figure 1i); pathologically raised intracranial pressure was documented in 9 cases by invasive monitoring or skull radiology. Twelve (86%) pediatric cases had behavioural or learning problems, particularly affecting concentration and language acquisition (Supplementary Table 2). Notably, despite the multisuture involvement many affected individuals presented after infancy, and primary surgery was frequently delayed (Supplementary Fig. 5). The remainder of skeletal growth was normal except for occasional mild shortening of the digits; no health problems of later onset were consistently found in carrier adults, in many of whom mild craniofacial signs or macrocephaly were the only features. No genotype-phenotype correlation was evident. In 6 families, including 14 ERF mutation carriers, we used 3-dimensional scanning16 to document the facial phenotype; this showed that hypertelorism, shortening and/or vertical displacement of the nose, prominent orbits and forehead were consistently present but varied in severity (Fig. 1j, Supplementary Fig. 6, Supplementary Movies 1-6). This newly recognized disorder, which we term ERF-related craniosynostosis, was identified in 5/402 (1.2%) of patients requiring surgery for craniosynostosis in our extended Oxford cohort13 (children born between 1998 and 2006).
A specific role for ERF was not previously suspected either in the cranial sutures, or in osteogenesis more generally. In the mouse, heterozygous loss-of-function of the orthologous gene (Erf+/−) is not associated with any abnormality, whereas homozygous loss (Erf−/−) causes severe placental defects resulting in death by embryonic day (E) 10.5 (ref. 17). To explore the function of Erf during development, we engineered mice harboring a conditional allele (ErfloxP) containing a selectable marker, PGKneo, located within intron 1, together with tandem loxP sites to enable Cre-mediated excision (Supplementary Fig. 7a). Real-time reverse transcriptase (RT)-PCR analysis of Erf cDNA in dissected E16.5 mouse calvaria showed that in ErfloxP/− compound heterozygotes, Erf transcription was reduced to 29% of wild type (wt) (Fig. 3a). This indicates that the ErfloxP allele is hypomorphic, probably because the intronic PGKneo cassette reduces the efficiency of transcript production. Both heterozygous (ErfloxP/+) and homozygous (ErfloxP/loxP) conditional mice were grossly normal, but compound conditional/null heterozygotes (ErfloxP/−) exhibited domed heads that became apparent during the first 3-6 weeks of life. Micro-CT scanning showed craniosynostosis affecting multiple calvarial sutures (Fig. 3b-f, Supplementary Fig. 7b,c). No other specific skeletal abnormalities were evident on Alizarin red/Alcian blue staining (Supplementary Fig. 7d). As in humans, the cranial sutures appear particularly sensitive to reduced Erf dosage, but the threshold level required for phenotypic manifestation is lower in mice.
Figure 3.

Analysis of Erf in mouse mutants and embryonic fibroblasts. (a) Quantitative RT-PCR of Erf in E16.5 calvariae of different genotypes, showing reduced expression of ErfloxP relative to wild type Erf allele. Error bars indicate standard error of mean. (b-f), Micro-CT scanning of heads of mice aged 9 weeks. Note normal morphology and patent sagittal (s), coronal (c) and lambdoid (l) sutures in the Erf+/− mutant (b,e), whereas the ErfloxP/− littermates have craniosynostosis of the sagittal and coronal sutures (c) or sagittal, coronal and lambdoid sutures (d). Note dental malocclusion (arrow) on the side view (f) of skull shown in d. Scale bars: 1.12 mm (b,e), 1.01 mm (c,d,f). (g) Whole mount RNA in situ hybridization of Erf (left) and Runx2 (right) in wild type E16.5 mouse calvariae. Note similar expression patterns coinciding with osteogenic fronts of parietal (p) and interparietal (ip) bones. Scale bars: 1 mm. (h) Summary of ChIP-Seq analysis using antibody to Erf in mouse embryonic fibroblasts. In the upper panel, box shows number of peaks identified according to whether they were located within 1 kb of a transcription start site (TSS) and whether they showed loss of binding in the presence of FCS (−FCS/+FCS >3). In the lower panel, MEME analysis of the 2033 non-TSS dynamically bound peaks (>3) identifies enrichment for motifs corresponding to binding sites for AP-1 (#1: 5′-TGANTCA-3′), RUNX (#3: 5′-TGTGG-3′) and ETS (#4: 5′-TTCCT-3′). Motif #2 was also observed in TSS peaks (Supplementary Fig. 9a).
To explore the developmental origins of craniosynostosis in the mice, we examined Erf expression and calvarial osteogenesis in E16.5 calvariae by whole mount RNA in situ hybridization and real-time RT-PCR. In the wt, Erf is expressed along the osteogenic margins of the developing calvarial bones in a similar distribution to the master osteogenic regulator Runx2 (Fig. 3g); in humans haploinsufficiency of orthologous RUNX2 causes cleidocranial dysplasia associated with defective calvarial ossification, whereas complete duplications occur in craniosynostosis18-22. Comparison between wt and ErfloxP/− mutants for transcripts of Spp1 (osteopontin) or Bglap2 (osteocalcin; not shown), markers of late osteogenic differentiation23 revealed similar sutural gaps (Supplementary Fig. 8a). However quantitation of transcripts in E16.5 calvariae showed modest (up to 2-fold) downregulation of multiple osteogenic markers in ErfloxP/− mutants compared to wt littermates, significantly so (P < 0.05; t-test) in the case of Prkg2 and Serinc5 (Supplementary Fig. 8b)24. At this stage therefore, ossification appears mildly delayed in ErfloxP/− embryos. However by postnatal day (P) 14, the coronal sutures of the ErfloxP/−, but not Erf+/+ pups were variably fused (Supplementary Fig. 8c). Further detailed analysis of intermediate time points will be necessary to document the timing of craniosynostosis in the ErfloxP/− mutants and determine the relative contributions to this pathology of altered proliferation, differentiation, and apoptosis25.
To gain insight into the nuclear targets of Erf, we employed chromatin immunoprecipitation (ChIP) in mouse embryonic fibroblasts using a previously characterized antibody specific to the C-terminal domain3,26, combined with high throughput sequencing (ChIP-Seq). By comparing the enriched sequences from fibroblasts maintained without fetal calf serum for 4 hr (“-FCS”; Erk1/2 inactive; Erf nuclear), to those from cells supplemented with FCS (“+FCS”; leading to phosphorylation of Erk1/2, nuclear entry, and consequent phosphorylation and nuclear export of Erf )4,5,26, we could identify the component of the ChIP-Seq signal attributable to dynamic Erf binding (defined as a −FCS/+FCS ratio >3). We divided signals of dynamic binding according to whether they occurred within 1 kb of transcription start sites (TSS; putative promoters) or at greater distances (non-TSS; putative enhancers) (Fig. 3h, Supplementary Tables 4,5). MEME analysis27 identified two major sequences enriched near TSS (Supplementary Fig. 9a), one corresponding to the ETS binding consensus12 and the other to the sequence bound by Ronin/Hcf-1 (ref. 28); these motifs are virtually identical to those in promoters bound by ETS1 (ref. 29). In non-TSS, which are believed to identify tissue-specific interactions of ETS factors29, the three most highly specific sequence motifs were 5′-TGANTCA-3′, 5′-TGTGG-3′ and 5′-TTCCT-3′, corresponding to consensus motifs for AP1, RUNX and ETS factors respectively (Fig. 3h and Supplementary Fig. 9a). This suggests that Erf binding sites frequently lie close to sites for other transcription factors; corroborating this, enrichment of both AP1 and RUNX sites was observed in ChIP-Seq studies of other ETS proteins29,30. The non-randomness of these associations was confirmed by demonstrating that closely adjacent AP1-ETS and RUNX-ETS sites exhibit polarity consistent with interactions between pairs of transcription factors when binding DNA in specific orientation and separation (Supplementary Figure 9b,c, Supplementary Tables 6,7)29-33. Metacore analysis of −FCS/+FCS ratio >3, non-TSS ChIP-Seq targets (n = 2033) revealed epithelial/mesenchymal transition (EMT) as the process showing the highest enrichment (Supplementary Fig. 10); the RAS-MEK-ERK cascade and downstream AP1 factors locate within the center of this network. Validating this analysis, ERF was recently identified experimentally as a negative regulator of EMT34.
The identification of frequent RUNX motifs within distal (non-TSS) Erf targets is of interest because Runx2 is a key dosage-sensitive regulator of calvarial osteogenesis18,35. The ChIP-Seq data reveal two sites of dynamic binding within introns of Runx2, which might reflect direct regulation of transcription (Supplementary Table 6). Alternatively, Erf may regulate osteogenesis by altering the balance of positive and negative regulatory co-complexes formed on DNA targets by Runx2 and other Ets proteins (such as Ets2)36,37. We explored this hypothesis in two ways. First, we compared our Erf ChIP-Seq data with a dataset of 1603 RUNX2-occupied regions in human prostate cancer cells38. There were 130 orthologous genes that were nearest to, and had signals within 40 kb of a ChIP-Seq peak in each of the datasets (Fig. 4a and Supplementary Table 8), a highly significant overlap (P = 4 × 10−11; hypergeometric test); moreover, Little et al.38 identified enrichment of a hybrid ETS-RUNX binding motif very similar to the sequence (5′-GGATGTGG-3′) in our dataset (Supplementary Fig. 9c). Second, we examined the effects of ERF and RUNX2 on transactivation using a DNA-binding target containing this core sequence. Whereas RUNX2 alone elicited 1.6-fold upregulation of expression, this was suppressed by ERF to a sub-basal level similar to the effect of ERF added alone (Fig. 4b), indicating antagonism by ERF on RUNX2 transactivation in this model situation. Given that the ChIP-Seq data show potential interactions of Erf and RUNX2 at numerous targets (Fig. 4a, Supplementary Table 8), further work will be required to identify critical factors in osteogenic dysregulation in the ERF or RUNX2 haploinsufficiency states.
Figure 4.

Overlapping transcriptional targets of Erf and RUNX2 identified by ChIP-Seq. (a) Comparison of −FCS/+FCS >3, non-TSS murine Erf targets identified in this work (n = 2033) with orthologous human RUNX2 targets identified by Little et al. (n = 1603)38. To improve the specificity of linkage to regulated genes, ChIP-Seq peaks more than 40 kb from the closest RefSeq gene were excluded. Peaks were assigned to the gene with the closest exon. (b) Transactivation analysis using the hybrid Erf-Runx2 binding target identified by ChIP-Seq (sequence at top with core target capitalized). HeLa cells were transfected with 1 μg plasmid containing empty vector, ERF or RUNX2 cDNAs, or combinations as indicated. Results were normalized relative to empty vector and expressed as the mean ± SEM of 4 independent experiments.
Our genetic observations in humans and mice will focus renewed attention on the role of Ets factors in regulating osteogenesis, which although documented, is not well defined36,37,39. Direct Erk1/2-mediated phosphorylation was previously linked to stabilization and/or activation of Runx2 (refs 40,41); we speculate that additional indirect control of Runx2 action by Erk1/2 (ref. 42) may be mediated by its control of Erf localization. Our data provide a pathway-based phenotypic link with FGFR2 mutations, since several patients with ERF-related craniosynostosis were previously diagnosed with Crouzon syndrome (Supplementary Table 2)14,15. The position of ERF downstream of the RAS-MEK-ERK cascade, and the delayed onset of suture ossification associated with ERF haploinsufficiency (Supplementary Fig. 5), make ERF an attractive target for therapeutic modulation.
METHODS
Patients
The clinical study was approved by Oxfordshire Research Ethics Committee B (reference C02.143) and Riverside Research Ethics Committee (reference 09/H0706/20). Written informed consent to obtain samples for genetics research was obtained from each child’s parent/guardian. In most probands the clinical diagnosis of craniosynostosis was confirmed by computed tomographic scanning, although some individuals had skull radiography only. Venous blood was obtained for DNA extraction and preparation of lymphoblastoid cell lines. Fibroblast cultures were established from skin biopsies obtained from scalp incision during surgical intervention.
Exome sequencing and segregation analysis
We used an Agilent SureSelect Human All Exon Kit (v.1; 38 Mb) to capture exonic DNA from a library prepared from 3 μg of DNA extracted from whole blood (subject IV-1 in Family 1). Enriched DNA was sequenced on an Illumina GAIIx platform (51 bp paired-end reads) and the 3.1 Gb of sequence generated was mapped with Bowtie software43 (hg19 genome) which gave an average coverage of 43-fold. Variants were called using Samtools44 (Supplementary Table 9). The proband, his brother and parents were genotyped using an Illumina HumanCytoSNP-12 BeadChip (300k). Chromosomal regions where the two affected boys shared the same maternal allele were identified and used to filter the exome data.
Mutation screening
Mutation analysis was performed by sequencing genomic PCR amplification products (BigDye Terminator v3.1, Applied Biosystems). Copy number variation was analyzed by multiplex ligation-dependent probe amplification (MLPA). RNA was extracted from whole blood (PAXgene tubes, Qiagen), and lymphoblastoid cell lines (Trizol, Invitrogen), and cDNA synthesized with RevertAid first strand cDNA kit (Thermo). Primers for genomic and cDNA amplification, MLPA, and all experimental conditions are provided in Supplementary Table 10. cDNA numbering of ERF starts with A of the ATG (=1).
Three-dimensional facial imaging
Images were captured with a commercial photogrammetric device and manually landmarked, as were an additional 381 images of healthy controls for comparison. Dense surface model and signature analyses were undertaken as described16,45. Face signatures were visualized as color-coded heat maps, derived from lateral, vertical and depth differences of 24,000 surface points compared to corresponding positions on the mean face of the matched controls.
Western blots
Protein immunoblots were performed as described4. Cells or homogenized embryos were lysed in RIPA buffer containing protease and phosphatase inhibitors. Equal amounts of protein were separated by discontinuous SDS-electrophoresis and transferred onto nitrocellulose. ERF was detected with S17S anti-ERF rabbit polyclonal antibody (1:1000) and ERK1/2 with anti-ERK1/2 rabbit polyclonal antibody (Cell Signaling #9102; 1:1000) in TBS 0.1% Tween. Proteins were detected with anti-rabbit horseradish peroxidase antibody (Jackson Immunoresearch; 1:5000) and visualized by chemiluminescence. Autoradiographs were quantified using NIH Image software.
Promoter assays
Mutations identified in patients were introduced into wt ERF cDNA by site-directed mutagenesis (QuickChange™, Stratagene), cloned into pSG5-ERF and pSG424-ERF expression vectors3, and sequence verified. The ability of wt and mutated ERF to repress transcriptional activity was determined in HeLa cells. The pGL333 reporter (3 copies of the GATA1 ets-binding-site (ebs) and minimal TK1 promoter), generated by transferring the corresponding promoter fragment from pBLCAT333 (ref. 46) to pGL3-basic vector (Promega), was used to determine repression on ebs-containing promoters. The pGLGal4 reporter (gal4 DNA binding site and the SV40 promoter), generated by transferring the corresponding promoter fragment from SV40/GAL4 (ref. 3) to pGL3-basic vector (Promega), was used to assess ebs-independent repression.
For studies of RUNX2/ERF interaction, the SfiI restriction fragment from pENTR223.1-Runx2 plasmid (Open Biosystems), containing the RUNX2 cDNA was cloned into the EcoRI site of the pSG5 expression vector (Agilent Technologies) to generate pSG5-RUNX2. Double-strand oligonucleotides encompassing the core Ets/Runx binding site, (Supplementary Table 10) were cloned directionally between the NheI and BglII sites of the pGL3-promoter luciferase reporter vector (Promega) to generate pGL3prom-D1. HeLa cells were transfected with the calcium phosphate method using 0.5 μg pGL3prom-D1 reporter, 0.5 μg pRSV-Gal for transfection efficiency control, 1 μg pSG5-ERF and/or 1 μg pSG5-RUNX2, and/or 0-2 μg pSG5 to a total of 3 μg DNA. All pair-wise comparisons were significant (P < 0.01; t-test) with the exception of ERF vs ERF+RUNX2.
Generation of conditional ErfloxP mice
Mice were maintained in specific-pathogen-free facilities at the Institute of Molecular Biology and Biotechnology, Crete or Biomedical Services Unit, Oxford. Protocols were approved through the General Directorate of Veterinary Services, Region of Crete or the University of Oxford Local Ethical Review process. Experimental procedures were performed in accordance with the European Union DIRECTIVE 2010/63/EU and/or the UK Animals (Scientific Procedures) Act, 1986 (Project License 30/2660).
The Erf targeting vector (Supplementary Figure 7a) was prepared by inserting a loxP sequence (ApaI site 16 bp 3′ of the Erf stop codon) and a PGKneo-loxP cassette (BstZ17I site 350 bp 5′ of Erf exon 2) within the 7.3 kb Erf genomic fragment 59A. The loxP orientation was verified by sequencing and the targeting fragment inserted into pBSTK9 (ref. 47). R1 ES cells were electroporated and selected as described17 and clones screened by Southern blotting using a 2.2 kb BamHI-XbaI fragment (int-probe), for homologous recombination and absence of additional insertions. Positive clones were tested for the presence of the 3′ loxP site by PCR amplification using StopF2 and 5578R. Recombination after transfection with Cre recombinase-expressing plasmid was tested by PCR using Intr1-2 and 5578R (Supplementary Figure 7a and Supplementary Table 10).
Chimeric mice were generated by microinjection of ES clones as described17. Cells from clone no. 89 were injected into 3.5 days post conception C57BL/6 blastocysts and implanted into pseudopregnant CD1 females48. Male offspring with high levels of chimerism were mated to CBAxC57BL/6 females to produce ErfloxP/+ heterozygous mice. ErfloxP/− compound heterozygotes were obtained from a Erf+/− × ErfloxP/+ cross (Erf+/− animals were sourced either from a previously described knockout line17, or by crossing ErfloxP/loxP mice with Meox2tm1(cre)Sor/J mice49). At birth ErfloxP/− mice were on average ~18% lighter than their littermates, but they exhibited catch-up growth by 6 weeks.
Skeletal preparations and immunohistochemistry
Skeletons were fixed in 95% ethanol and stained with Alcian blue (0.03% w/v in 95% ethanol, 20% acetic acid) overnight. After several washes with 95% ethanol, skeletons were rehydrated, treated with 2% KOH (12 h) and stained in 1% KOH containing 75 μg/ml Alizarin red S (24 h). Excess stain was removed in 1% KOH/20% glycerol and, after washing in 0.2% KOH/20% glycerol, skeletons were stored in 50% glycerol.
The skulls of P14 mice were fixed overnight in 4% paraformaldehyde, decalcified using Calci-Clear (Raymond A Lamb, East Sussex, England) and paraffin-embedded. Immunohistochemistry was performed using a polyclonal RUNX2 antibody (M-70; Santa Cruz Biotechnology Inc.) on 8 μm sections that were counterstained with haematoxylin.
MicroCT analyses
Specimens for microCT were scanned using a General Electric Locus SP microCT scanner (GE Healthcare). Specimens were immobilized using cotton gauze, scanned to produce 14-28 μm voxel size volumes, and characterized further by making three-dimensional isosurfaces, generated and measured using Microview (GE).
Whole mount RNA in situ hybridization
Embryos were dissected and fixed in 4% paraformaldehyde, then dehydrated. In situ hybridization was performed as described50 using digoxigenin-incorporated riboprobes. The Erf probe (554 bp) was amplified from mouse cDNA with the primers mErfF2 and mErfR1 (Supplementary Table 10), and cloned into pGem-T Easy (Promega). The Spp1, Runx2 and Bglap2 probes were kind gifts from John Heath, Georg Schwabe and David Rice, respectively. For antibody detection, specimens were incubated with anti-digoxigenin antibody conjugated with alkaline phosphatase (1:1000, containing 2% fetal calf serum). Expression patterns were visualized with the BM Purple kit (Roche). Whole mounts were analyzed using a Leica MZFLIII microscope and LASAF software (Leica Microsystems, Milton Keynes, UK).
ChIP-Seq
For chromatin immunoprecipitation (ChIP), 20-25 ×106 mouse embryo fibroblasts from E13.5 wt embryos were grown in DMEM either supplemented with 10% fetal calf serum (+FCS), or in its absence (−FCS) for 4 h to induce Erf nuclear localization. ChIP was performed as described26 with S17S anti-ERF rabbit polyclonal antibody, which was previously demonstrated to be specific for ERF3. Cells were fixed with 1% formaldehyde in PBS for 10 min at room temperature, nuclei were isolated and sonicated in 50 mM Hepes pH 7.9, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate and 1% SDS. Antibody was added overnight at 4 °C followed by 2 h incubation with agarose-coupled protein G. The immunoprecipitated material was washed, de-cross-linked overnight at 65 °C and DNA purified by phenol extraction and ethanol precipitation. ChIP sequencing (ChIP-Seq) libraries were prepared and sequenced using the standard Illumina protocol.
ChIP-Seq analysis
Paired-end reads from ChIP-Seq and input samples were aligned to the mouse genome build using Bowtie (version 0.12.3)43. Peaks were called with SeqMonk (version 0.19), using the contig generator function (peak merge distance 50 bp, minimum peak size 50 bp, minimum fold enrichment relative to input 5-fold). The number of reads in a union set of peaks from the −FCS and +FCS samples were quantified and normalized for total aligned read count in each ChIP-Seq and input sample using the SeqMonk quantification function. The in-house PERL script Smonker.pl was used to normalize the peaks against input and to calculate the difference in enrichment for each peak in the −FCS and +FCS samples and to store these values in a GFF3 file. Peaks were annotated for overlap (within 1 kb) with transcription start site (TSS UCSC Known Gene mm9 build) and problematic copy number regions of the mm9 genome (ploidy peaks)51 using the in-house PERL script intersectandappend.pl. The resulting annotated and quantified peaks were stored in a Multi-image Genome browser (MIG) SQL database. Peaks were filtered in MIG on the basis of the calculated relative enrichments between the two samples and their overlap with TSSs to produce two datasets (TSS and non-TSS). Peaks associated with ploidy regions were excluded.
Motif analysis
The de novo identification of over-represented motifs around the centre of each peak (300 bp) was performed using MEME-chip tools27. The frequency of identified motifs in enriched and control peaks was calculated using the in-house program MotifQuant.pl. DNase1 hypersensitive regions from adult fibroblast cells (from the Encode project via UCSC table browser, file name: wgEncodeUwDnaseFibroblastC57bl6MAdult8wksPkRep1) that overlap with TSS or not, as appropriate, were used as control regions. MotifQuant.pl randomly sampled from these control sets the same number of peaks as in the test set, and repeated this sampling 1000 times to produce a mean frequency and a normal distribution for motif occurrence.
For the motif analyses presented in Supplementary Tables 6 and 7 and Supplementary Figures 9b and c, ETS-binding motifs were defined by the sequences 5′-GGAA-3′ or 5′-GGAT-3′; AP1 as 5′-TCANTGA-3′ and RUNX as 5′-TGTGG-3′ (of which 35% matched the extended 5′-TGTGGT-3′ consensus) (JASPAR database). Pathway analysis of gene lists from ChIP-Seq peaks was performed using GeneGo Metacore (Supplementary Figure 10).
Comparison with RUNX2 ChIP-Seq data
We extracted the list of 1603 ChIP-Seq peaks obtained for RUNX2 binding in a human prostate cancer cell line (Table S3 from ref. 38). From this we obtained the subset of 896 peaks located within 40 kb upstream or downstream of RefSeq genes, choosing the gene closest to the coordinates of the ChIP-Seq peaks. We converted the RefSeq ID for each annotated gene to the HomoloGene ID and compared this list to Erf ChIP-Seq data (Supplementary Table 4; non-TSS), also restricted to a 40 kb window either side of each gene. Significance of the overlap was tested using a hypergeometric distribution. The list of 130 shared genes (Fig. 4a) is provided in Supplementary Table 8.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
We thank R. Boehm, H. Care, C. Langman, J. Phipps and E. Sweeney for clinical assistance, L. Gregory, P. Piazza and staff at the High-Throughput Genomics facility at the Wellcome Trust Centre for Human Genetics (Oxford) for exome sequencing, C. Babbs, S. Butler, J. Frankland, C. Rode and T. Rostron for technical help, K. Kourouniotis for blastocyst injections and expert animal support, E. Giannoulatou for bioinformatics assistance and B. Graves and P. Hollenhorst for discussions. This work was funded by the Greek Ministry of Education grants PYTHAGORAS II KA2092, PENED 03ED626, HERAKLEITOS II KA 3396 and SYNERGASIA 09SYN-11-902 to GM., NIHR Biomedical Research Centre, Oxford, with funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme (S.J.L.K., A.O.M.W.), Oxford Craniofacial Unit Charitable Fund (V.P.S.), Department of Health QIDIS fund (V.P.S.) and Wellcome Trust (090532, S.J.L.K.; 093329, S.R.F.T., A.O.M.W.). The views expressed in this publication are those of the authors and not necessarily those of the Department of Health.
Footnotes
URLs. MLPA analysis, http://www.mrc-holland.com/pages/indexpag.html; MEME-chip, http://meme.sdsc.edu/meme/cgi-bin/meme-chip.cgi; UCSC genome browser, http://genome.ucsc.edu/; Polyphen, http://genetics.bwh.harvard.edu/pph2; SeqMonk, http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/; GeneGo Metacore, http://www.genego.com/metacore.php; JASPAR transcription factor binding profile database, http://jaspar.genereg.net/; Bowtie sequence alignment, http://bowtie-bio.sourceforge.net/index.shtm; Multi-image Genome browser (MIG), https://mig.molbiol.ox.ac.uk/mig/; HomoloGene (http://www.ncbi.nlm.nih.gov/homologene).
Accession codes. Human ERF mRNA and genomic sequence: NCBI reference sequences NM_006494.2 and NC_000019.9, respectively. Murine Erf mRNA sequence: NCBI reference sequence NM_010155.3. Human ETS DNA-binding proteins (Swissprot): ERF, P50548; SPI1, P17947; ETV6, P41212; ELF3; P78545; ELF1, P32519; SPDEF, O95238; ETV4, P43268; ELK1, P19419; ETS1, P14921; ETV2, O00321; ERG, P11308; GABPα, Q06546. Erf ChIP-Seq data: GSE42936 (GEO).
References
- 1.Plotnikov A, Zehorai E, Procaccia S, Seger R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta. 2011;1813:1619–1633. doi: 10.1016/j.bbamcr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Mavrothalassitis GJ, Papas TS. Positive and negative factors regulate the transcription of the ETS2 gene via an oncogene-responsive-like unit within the ETS2 promoter region. Cell Growth Differ. 1991;2:215–224. [PubMed] [Google Scholar]
- 3.Sgouras DN, et al. ERF: an ETS domain protein with strong transcriptional repressor activity, can suppress ets-associated tumorigenesis and is regulated by phosphorylation during cell cycle and mitogenic stimulation. EMBO J. 1995;14:4781–4793. doi: 10.1002/j.1460-2075.1995.tb00160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Gallic L, Sgouras D, Beal G, Mavrothalassitis G. Transcriptional repressor ERF is a Ras/mitogen-activated protein kinase target that regulates cellular proliferation. Mol. Cell. Biol. 1999;19:4121–4133. doi: 10.1128/mcb.19.6.4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le Gallic L, Virgilio L, Cohen P, Biteau B, Mavrothalassitis G. ERF nuclear shuttling, a continuous monitor of Erk activity that links it to cell cycle progression. Mol. Cell. Biol. 2004;24:1206–1218. doi: 10.1128/MCB.24.3.1206-1218.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polychronopoulos S, et al. The transcriptional ETS2 repressor factor associates with active and inactive Erks through distinct FXF motifs. J. Biol. Chem. 2006;281:25601–25611. doi: 10.1074/jbc.M605185200. [DOI] [PubMed] [Google Scholar]
- 7.von Kriegsheim A, et al. Cell fate decisions are specified by the dynamic ERK interactome. Nat. Cell Biol. 2009;11:1458–1464. doi: 10.1038/ncb1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng SB, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim H-J, et al. Erk pathway and activator protein 1 play crucial roles in FGF2-stimulated premature cranial suture closure. Dev. Dyn. 2003;227:335–346. doi: 10.1002/dvdy.10319. [DOI] [PubMed] [Google Scholar]
- 10.Shukla V, Coumoul X, Wang R-H, Kim H-S, Deng C-X. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat. Genet. 2007;39:1145–1150. doi: 10.1038/ng2096. [DOI] [PubMed] [Google Scholar]
- 11.Hollenhorst PC, McIntosh LP, Graves BJ. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011;80:437–471. doi: 10.1146/annurev.biochem.79.081507.103945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei G-H, et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010;29:2147–2160. doi: 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilkie AOM, et al. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics. 2010;126:e391–e400. doi: 10.1542/peds.2009-3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kan S-H, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am. J. Hum. Genet. 2002;70:472–486. doi: 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lajeunie E, et al. Mutation screening in patients with syndromic craniosynostoses indicates that a limited number of recurrent FGFR2 mutations accounts for severe forms of Pfeiffer syndrome. Eur. J. Hum. Genet. 2006;14:289–298. doi: 10.1038/sj.ejhg.5201558. [DOI] [PubMed] [Google Scholar]
- 16.Hammond P, Suttie MJ. Large-scale phenotyping of 3D facial morphology. Hum. Mutat. 2012;33:817–825. doi: 10.1002/humu.22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Papadaki C, et al. Transcriptional repressor Erf determines extraembryonic ectoderm differentiation. Mol. Cell. Biol. 2007;27:5201–5213. doi: 10.1128/MCB.02237-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mundlos S, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 19.Park M-H, et al. Differential expression patterns of Runx2 isoforms in cranial suture morphogenesis. J. Bone Miner. Res. 2001;16:885–892. doi: 10.1359/jbmr.2001.16.5.885. [DOI] [PubMed] [Google Scholar]
- 20.Komori T. Signaling networks in RUNX2-dependent bone development. J. Cell. Biochem. 2011;112:750–755. doi: 10.1002/jcb.22994. [DOI] [PubMed] [Google Scholar]
- 21.Mefford HC, et al. Copy number variation analysis in single-suture craniosynostosis: multiple rare variants including RUNX2 duplication in two cousins with metopic craniosynostosis. Am. J. Med. Genet. 2010;152A:2203–2210. doi: 10.1002/ajmg.a.33557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varvagiannis K, et al. Pure de novo partial trisomy 6p in a girl with craniosynostosis. Am. J. Med. Genet. A. 2013 doi: 10.1002/ajmg.a.35727. in press. [DOI] [PubMed] [Google Scholar]
- 23.Iseki S, Wilkie AOM, Morriss-Kay GM. Fgfr1 and Fgfr2 have distinct differentation- and proliferation-related roles in the developing mouse skull vault. Development. 1999;126:5611–5620. doi: 10.1242/dev.126.24.5611. [DOI] [PubMed] [Google Scholar]
- 24.Hecht J, et al. Detection of novel skeletogenesis target genes by comprehensive analysis of a Runx2−/− mouse model. Gene Expr. Patterns. 2007;7:102–112. doi: 10.1016/j.modgep.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 25.Morriss-Kay GM, Wilkie AOM. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J. Anat. 2005;207:637–653. doi: 10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verykokakis M, Papadaki C, Vorgia E, Le Gallic L, Mavrothalassitis G. The RAS-dependent ERF control of cell proliferation and differentiation is mediated by c-Myc repression. J. Biol. Chem. 2007;282:30285–30294. doi: 10.1074/jbc.M704428200. [DOI] [PubMed] [Google Scholar]
- 27.Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dejosez M, et al. Ronin/Hcf-1 binds to a hyperconserved enhancer element and regulates genes involved in the growth of embryonic stem cells. Genes Dev. 2010;24:1479–1484. doi: 10.1101/gad.1935210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollenhorst PC, et al. DNA specificity determinants associate with distinct transcription factor functions. PLoS Genetics. 2009;5:e1000778. doi: 10.1371/journal.pgen.1000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollenhorst PC, et al. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011;25:2147–2157. doi: 10.1101/gad.17546311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goetz TL, Gu TL, Speck NA, Graves BJ. Auto-inhibition of Ets-1 is counteracted by DNA binding cooperativity with core-binding factor alpha2. Mol. Cell. Biol. 2000;20:81–90. doi: 10.1128/mcb.20.1.81-90.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hess J, Porte D, Munz C, Angel P. AP-1 and Cbfa/runt physically interact and regulate parathyroid hormone-dependent MMP13 expression in osteoblasts through a new osteoblast-specific element 2/AP-1 composite element. J. Biol . Chem. 2001;276:20029–20038. doi: 10.1074/jbc.M010601200. [DOI] [PubMed] [Google Scholar]
- 33.D’Alonzo RC, Selvamurugan N, Karsenty G, Partridge NC. Physical interaction of the activator protein-1 factors c-Fos and c-Jun with Cbfa1 for collagenase-3 promoter activation. J. Biol. Chem. 2002;277:816–822. doi: 10.1074/jbc.M107082200. [DOI] [PubMed] [Google Scholar]
- 34.Allegra M, et al. Semaphorin-7a reverses the ERF-induced inhibition of EMT in Ras-dependent mouse mammary epithelial cells. Mol. Biol. Cell. 2012;23:3873–3881. doi: 10.1091/mbc.E12-04-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lou Y, et al. A Runx2 threshold for the cleidocranial dysplasia phenotype. Hum. Mol. Genet. 2009;18:556–568. doi: 10.1093/hmg/ddn383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sumarsono SH, et al. Down’s syndrome-like skeletal abnormalities in Ets2 transgenic mice. Nature. 1996;379:534–537. doi: 10.1038/379534a0. [DOI] [PubMed] [Google Scholar]
- 37.Vary CPH, et al. Involvement of Ets transcription factors and targets in osteoblast differentiation and matrix mineralization. Exp. Cell Res. 2000;257:213–222. doi: 10.1006/excr.2000.4879. [DOI] [PubMed] [Google Scholar]
- 38.Little GH, et al. Genome-wide Runx2 occupancy in prostate cancer cells suggests a role in regulating secretion. Nucleic Acids Res. 2012;40:3538–3547. doi: 10.1093/nar/gkr1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wai PY, et al. Ets-1 and runx2 regulate transcription of a metastatic gene, osteopontin, in murine colorectal cancer cells. J. Biol. Chem. 2006;281:18973–18982. doi: 10.1074/jbc.M511962200. [DOI] [PubMed] [Google Scholar]
- 40.Ge C, et al. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J. Biol. Chem. 2009;284:32533–32543. doi: 10.1074/jbc.M109.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park O-J, Kim H-J, Woo K-M, Baek J-H, Ryoo H-M. FGF2-activated ERK mitogen-activated protein kinase enhances Runx2 acetylation and stabilization. J. Biol. Chem. 2010;285:3568–3574. doi: 10.1074/jbc.M109.055053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsushita T, et al. Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol. Cell. Biol. 2009;29:5843–5857. doi: 10.1128/MCB.01549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hammond P. The use of 3D face shape modelling in dysmorphology. Arch. Dis. Child. 2007;92:1120–1126. doi: 10.1136/adc.2006.103507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watson DK, et al. The ERGB/Fli-1 gene: isolation and characterization of a new member of the family of human ETS transcription factors. Cell Growth Differ. 1992;3:705–713. [PubMed] [Google Scholar]
- 47.Spyropoulos DD, et al. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol. Cell. Biol. 2000;20:5643–5652. doi: 10.1128/mcb.20.15.5643-5652.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hogan B. Manipulating the Mouse Embryo: a Laboratory Manual. Cold Spring Harbor Laboratory Press; Plainview, New York, USA: 1994. [Google Scholar]
- 49.Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000;26:113–115. doi: 10.1002/(sici)1526-968x(200002)26:2<113::aid-gene3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 50.Wilkinson DG. In-situ Hybridization: a Practical Approach. Oxford University Press; Oxford, UK: 1992. [Google Scholar]
- 51.Kowalczyk MS, et al. Intragenic enhancers act as alternative promoters. Mol. Cell. 2012;45:447–458. doi: 10.1016/j.molcel.2011.12.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.