

Abstract
Formation, aggregation and transmission of abnormal proteins are common features in neurodegenerative disorders including Parkinson's disease, Alzheimer's disease, amyotrophic lateral sclerosis, and Huntington's disease. The mechanisms underlying protein alterations in neurodegenerative diseases remain controversial. Novel findings highlighted altered protein clearing systems as common biochemical pathways which generate protein misfolding, which in turn causes protein aggregation and protein spreading. In fact, proteinaceous aggregates are prone to cell-tocell propagation. This is reminiscent of what happens in prion disorders, where the prion protein misfolds thus forming aggregates which spread to neighbouring cells. For this reason, the term prionoids is currently used to emphasize how several misfolded proteins are transmitted in neurodegenerative diseases following this prion-like pattern. Histochemical techniques including the use of specific antibodies covering both light and electron microscopy offer a powerful tool to describe these phenomena and investigate specific molecular steps. These include: prion like protein alterations; glycation of prion-like altered proteins to form advanced glycation end-products (AGEs); mechanisms of extracellular secretion; interaction of AGEs with specific receptors placed on neighbouring cells (RAGEs). The present manuscript comments on these phenomena aimed to provide a consistent scenario of the available histochemical approaches to dissect each specific step.
Key words: advanced glycation end-products, exosome, synucleinopathies, neurodegenerative diseases, prion diseases, prionoids, RAGEs.
Introduction
The occurrence of protein aggregates
Neurodegenerative diseases (NDs) are chronic and progressive conditions which may affect, separately or in combination, autonomic, motor, sensory, mood and cognitive functions. Apart from juvenile cases, most of which are genetically determined, these disorders appear late in life, being characterized by marked neuronal loss which was traditionally described as trans-synaptic degeneration.1 At present, therapeutic strategies aimed at treating or preventing these disorders are under intense investigation.
Despite the occurrence of multiple mechanisms to generate cell pathology, the common feature which represents the starting point of disease progression is the occurrence of misfolded proteins. This point is key also for current histochemistry which allows detecting early in the disease process the occurrence of cell pathology. In fact, various NDs share the presence of altered protein aggregates within neuronal and non-neuronal cells. These proteins are expected to misfold, aggregate and propagate to sustain disease progression. For such a reason, these NDs are defined as protein misfolding diseases (PMDs) or conformational disorders. For instance, due to pathological conditions, normal proteins domain owing an α-helix may misfold into β-sheet structures whose conformation is further stabilized by pathobiochemical interactions, leading to the formation of oligomers that cannot be cleared.2–5
Misfolded proteins are pathological hallmarks which may be in common with different disorders or vary in specific diseases. For instance, α-synuclein aggregates characterize Parkinson's disease (PD)6 and other synucleinopathies, 7,8 whereas TDP43 and SOD1 are typically found as protein aggregates in amyotrophic lateral sclerosis (ALS)9. Huntingtin aggregates characterize Huntington's disease,10 whereas β-amyloid and tau are typical of degenerative dementia.11 In some cases, the same protein misfolds and accumulates in multiple disorders, such as TDP-43, which can be found both in ALS and fronto-temporal dementia.12 This concomitance indicates a continuum of disease progression9 and occurs more often for α-synuclein which may be found in a wide range of disorders. In fact, α-synuclein accumulates in Parkinsonism (including multiple system atrophy) but also in ALS13 in Huntington's disease10 and in specific forms of degenerative dementia.11 This poses the α-synuclein detection as a powerful marker to track neurodegeneration. In fact, the mechanisms by which α-synuclein misfolds, aggregates, propagates and leads to cell death are intensely investigated.14 The presence of altered α-synuclein in a variety of disorders is unlikely to be a coincidence but it rather witnesses for progressive involvement of different neuronal networks along the course of neurodegeneration.13,15–17
Histochemical detection of misfolded protein aggregates through the use of specific and sensitive antibodies allowed, in the last decades, enormous progress in the diagnosis and nosography of NDs. In particular, both light and electron microscopy, by profiting of sophisticated technologies to produce and characterize antibodies, provided a fairly disease-specific pattern of cell pathology. In this way, even in the presence of neuronal accumulation of α-synuclein, it is possible to distinguish whether this aggregation follows a Lewy body (LB)-like pattern (such as in Parkinsonism, where inclusions are formed by a negative α-synuclein core which is surrounded by α-synuclein-containing radiating filaments) or it is characterized by α-synuclein-positive neuronal vacuoles (as it occurs in Huntington's disease).18 The co-expression of α-synuclein with other protein aggregates is typical in specific diseases. This is the case of co-accumulation of α-synuclein with SOD1 in ALS,13 co-accumulation of α-synuclein with huntingtin in Huntington's disease18 or coexpression of α-synuclein, parkin and ubiquitin in PD.19 The cell type in α-synuclein aggregates is also useful in differential diagnosis. For instance, α-synuclein accumulates within neuronal cells in PD but also involves glial cells in multiple system atrophy.20
Protein aggregates are based upon protein misfolding
Why a given protein aggregates within neurons may depend on several mechanisms which in turn fall in two main classes: i) altered protein synthesis; ii) altered protein metabolism. In the first case, the gene coding for a certain protein is present in excess21 or it is altered by point mutations.22 In these cases, the nascent protein is prone to unfold or misfold.4,5In the case of altered protein metabolism, normal protein amounts and normal protein structure associate with altered protein clearing mechanisms which cannot remove the protein which accumulates making it likely to unfold or misfold.
Increased copies of the protein coding gene are detectable by RT-PCR measuring high amount of the gene which are paralleled by increased protein level (detected by SDS-PAGE immunoblotting). Again, if the gene is mutated, this can be detected by genetic analysis or the use of mutation specific antibodies. The occurrence of altered protein metabolism relies on two main protein clearing systems which are effective in eukaryotic cells, namely the ubiquitin proteasome system (UPS) and the autophagy pathway. There are now refined guidelines to detect the efficacy of these pathways and to establish the specific step which is impaired thus affecting protein metabolism. When a deficiency in either UPS or autophagy occurs, the excess of protein substrates leads to protein misfolding and aggregation. 23,24 Immunocytochemistry now provides the chance to localize the cell compartment triggering protein accumulation by staining the misfolded proteins in combination with compartment-specific antibodies. For instance, a defect in starting the autophagy pathway (critical for protein clearance) can be visualized in neuronal cells where misfolded protein is aggregated in the absence of effective autophagosomes where LC3-II particles are scarce. At the same time, a defect in the autophagy pathway can paradoxically associate with increased amount of LC3-II particles which are no longer metabolized if a defect in autophagy progression occurs. If this is the case, misfolded proteins are stagnant within big non-progressive autophagosomes. At present, the search for biochemical and immunohistochemical markers to measure protein clearance is under intense investigation.25,26 Deficiency in the clearance of misfolding-prone proteins may also derive from an excess of misfolded substrate, which even in the presence of normal nascent protein level (no genetic alterations), still produces misfolded protein aggregates. This may be due to specific environmental stimuli which alter protein conformation (oxidizing species, dopamine-derived quinones; chemical toxins and many others) beyond the compensatory increase in the activity of otherwise healthy cell protein clearing systems. This is the case of methamphetamine intoxication which increases the levels of misfolded α-synuclein beyond the clearing efficacy of the autophagy machinery thus leading to α-synuclein misfolding and aggregation.27
What brings to protein misfolding
When a protein is produced as an altered structure or it is accumulated at levels which overcome the ability of protein clearing systems, the cellular environment leads at variable rate to spontaneous misfolding, which is based on conformational changes where protein domains, structured as α-helix, are converted into β-sheet structures. Again, protein misfolding may derive from additional binding with specific compounds which promote the oxidation of SH groups into S-S disulphide bounds. Moreover, specific sugar residues may bind to proteins leading to a spontaneous reaction to form a Shiff's base leading to protein glycation. In detail, these steps occur as posttranslation modification which may apply both to normal and mutated proteins, thereby modifying their structure and conformation thus producing protein misfolding or unfolding.28 This applies both to prion protein and prionoids, such as SOD-1, β-amyoloid, tau, α-synuclein, huntingtin,29 and also to TDP43 and FUS which naturally own a prion-like domain.30,31 In the case of protein glycation, this occurs earlier though the formation of fructosamine which derives from the binding of a sugar residue, an aldehyde or a ketone to the amino-group of a given protein.32 Early steps in glycation follow an equilibrium which depends on the amount of protein substrates. Glycation proceeds though the final AGEs which derive from a cascade of reactions, all occurring spontaneously. AGEs may also derive directly from the binding of the proteins to methylglyoxal, glyoxal and 3-deoxyglucosone. All AGEs can be identified through immunohistochemical methods by using monoclonal antibodies against specific degradation product such as carboxymethyllysine, carboxyethyllysine, 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate, and argpyrimidine corresponding to the most common AGEs epitopes.
Final AGEs are stable thus making misfolded protein aggregates irreversible and protease-resistant.33 Therefore, a classic method used to detect misfolded proteins is based on pre-treating the sample with proteinase-K (PK) which destroys soluble proteins but cannot interact with misfolded protein aggregates, leaving intact their epitopes. This allows identifying authentic misfolded variants of prionoids.34 Therefore, the use of PK treatment, prior to immunocytochemistry, is routinely applied to remove folded proteins and detect their misfolded isoforms. The paradigm is represented by the immunoreactivity against cellular prion protein (PrPc) and PrPsc.35,36 Pretreatment with PK is also used to detect unsoluble α-synuclein modified as β-sheet oligomers as it occurs in dopamine (DA)-related neurodegenerative conditions or following the genesis of α-synuclein DA adducts.2,34
Apart from PK, which acts as a protease, the disruption of the secondary structure can also be carried out with the denaturing agent sarkosyl accordingly to Takahashi et al.37 Sarkosyl is sodium lauroyl sarcosinate, which reacts as an anionic detergent. This allows disrupting the strong binding within proteins which are critical to maintain the secondary structure.38
From an exquisite methodological perspective, it is worth mentioning potential discrepancies between pre-treatment with sarkosyl compared with PK. This is likely to depend on the loss of specific epitopes of the primary structure following the stronger effects of PK, whereas sarkosyl is expected to preserve entirely the amino acid chain of misfolded proteins. Therefore, it is not surprising that in some instances PK treatment leads to a total loss of staining, even in the presence of misfolded prionoids, whereas sarkosyl provides a fair antigen preservation. On the other hand, sarkosyl may lead to less specific results.
What happens to misfolded proteins
The status of misfolded protein in the form of AGEs confers to the prionoids the ability to spread from cell to cell. In fact AGEs can be expressed at the level of the plasma membrane from where they are released extracellularly. The combined histochemistry to detect glycated protein with the use of prionoid-specific antibodies allows detecting AGE on the plasma membranes. At the same time, AGEs contained in the endosomal compartment can merge with the plasma membrane and being released as exosomes in the extracellular space.39 The presence of specific receptor for AGEs, termed RAGEs, allows binding and entering prionoids at the level of neighbouring cells. Thus, misfolded proteins can either aggregate and persist in the original cell or move out to spread the disease. In the first case, protein aggregation may seed the formation of neuronal inclusions such as LB in PD. These inclusions should not be regarded as static milestones. In fact, if their detection by light and electron microscopy allows pathological diagnosis, they also represent dynamic structures which preserve antigen properties and enzymatic activity. In keeping with this, it is worth mentioning that protein clearing systems, such as UPS and autophagy, still persist within neuronal inclusions and own catalytic activity.40,41 By using advanced techniques of immuno-electron microscopy it is possible to purify misfolded protein aggregates (such as α-synuclein-containing aggregates) from neurons and assess the presence of specific enzyme belonging to the UPS or autophagy. This method is based on magnetic beads which bind to streptavidin and allow isolating α-synuclein containing structures. Lenzi et al.41 provided the description of this technique applied to the field of neurodegeneration. This procedure allows purifying in a prionoid-specific manner neuronal aggregates. An alternative technique based on differential centrifugation of neuronal pools allows localizing misfolded protein aggregates within specific cell compartments (including pathological compartments such as neuronal inclusions) where prionoids co-localize with protein clearing enzymes (see electron micrographs in Figure 1 for both procedures). The occurrence of enzymatic activity within misfolded prionoid aggregates suggests that these structures may represent the remnant of a compensatory mechanism which still engages misfolded substrates for their metabolism. In fact, it is a matter of debate whether prionoids aggregates are neuroprotective42 or rather their misfolded constituents exert neurotoxicity when available for various cell structures.43,44
Figure 1.
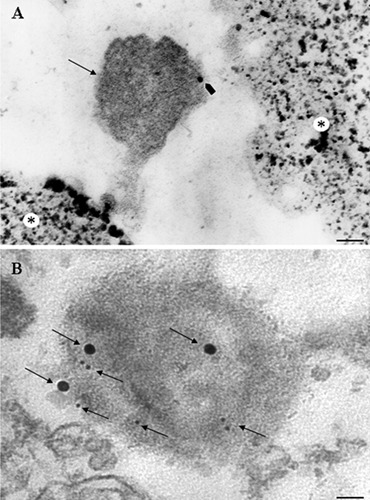
PC12 cells after methamphetamine treatment. A) Magnetic beads: an α-synuclein-positive aggregate (arrow) is attached to the magnetic bead (asterix); the α-synuclein prionoid co-localizes with the pro-autophagy protein cathepsin D, revealed by an immunogold particle (arrowhead); scale bar: 100 nm. B) Differential centrifugation; α-Synuclein-positive aggregate co-localizes with protein clearing marker cathepsin D for autophagy (20 nm and 10 nm immunogold particles, respectively; arrows); scale bar: 40 nm.
In spite of the faith of prionoid-containing neuronal inclusions for the original neuron, it is fascinating that in recent years these misfolded proteins were shown to spread from cell to cell.45 Prionoids spreading is supposed to occur via different mechanisms each including various steps described in the following paragraph.
How misfolded proteins spread from cell to cell
Recent evidence shows that self-propagation of the protein misfolding process is similar to prion diseases. In keeping with AGEs, their interaction with specific RAGEs represents a critical step. In fact, AGEs derived from specific prionoids including β-amyloid, tau, α-synuclein are implicated in cell-to-cell transmission.28,46–48 Namely, AGEs can interact with specific receptors, named RAGEs, placed on brain endothelium, microglia and neurons.49–51 The investigation of AGEs-RAGEs interaction opens new insights in the knowledge of cell-to-cell transmission of altered proteins52 or the implication of glia in the propagation of these altered proteins.53 At present, immunohistochemistry provided the chance to label and localize RAGEs in combination with major histocompatibility proteins bridging the spreading of neurodegeneration with altered immune system.54,55 This is further substantiated by the primary structure of RAGEs which belong to the superfamily of immunoglobulins. In fact, RAGEs possess three immunoglobulin domains, one extracellular V (variable) type and two C (constant) types which anchor the receptor to the cell membrane.56 The V domain binds AGEs and β-sheet chains. The binding of AGEs with RAGEs, apart from spreading prionoids, triggers a variety of transduction mechanisms which activate various biochemical cascade in neighbouring cells. These include RAGE-mediated activation of Protein Kinase C and NF-kB which leads to a further increase in RAGE expression by a positive feedback mechanism.28 Such a vicious circle may magnify the deleterious effects of prionoids spreading extracellularly. Apart from RAGEs, other mechanisms, including exosomes,57 tunnelling nanotubes,58 and trogocytosis,59 seem to be responsible for such a transmission.
Protein aggregates are often associated with the membrane trafficking machinery.60 This topography is critical for the faith of altered protein aggregates. In the case of prion disease, an endocytic recycling pathway has been demonstrated.60 Double immunostaining, with fluorescent probes (i.e., fluorescein and rodamine), allows detecting aggregates colocalised with different cell organelles, such as exosomes and microtubules.61 This immunohistochemical analysis demonstrated the association between exosomes and protein aggregates. 46,62 Morphological investigations of exosomes are critical considering that exosomes have been shown to secrete in the extracellular space prions,63 β-amyloid peptides,64 and α-synuclein.39
Cell-to-cell spreading of prionoids represents a hot research topic which is investigated in its specific steps. This process appears to be critical for the progression of neurodegenerative disorders and configures as a solid bridge between altered protein clearance, protein aggregates, post-translational protein alterations, and cell to cell transmission. This latter point applies both to altered neuronal transmission and altered immune networks which at this level possess a remarkable overlapping and enhance each other.
The chance to detect these specific steps by dedicated histochemical approaches was emphasized in the light of future research efforts.
References
- 1.Lach B, Grimes D, Benoit B, Minkiewicz-Janda A. Caudate nucleus pathology in Parkinson's disease: ultrastructural and biochemical findings in biopsy material. Acta Neuropathol. 1992;83:352–60. doi: 10.1007/BF00713525. [DOI] [PubMed] [Google Scholar]
- 2.Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopaminealpha-synuclein adduct. Science. 2001;294:1346–9. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 3.Soto C, Estrada LD. Protein misfolding and neurodegeneration. Arch Neurol. 2008;65:184–9. doi: 10.1001/archneurol.2007.56. [DOI] [PubMed] [Google Scholar]
- 4.Natale G, Pasquali L, Paparelli A, Fornai F. Parallel manifestations of neuropathologies in the enteric and central nervous systems. Neurogastroenterol Motil. 2011;23:1056–65. doi: 10.1111/j.1365-2982.2011.01794.x. [DOI] [PubMed] [Google Scholar]
- 5.Natale G, Ferrucci M, Lazzeri G, Paparelli A, Fornai F. Transmission of prions within the gut and towards the central nervous system. Prion. 2011;5:142–9. doi: 10.4161/pri.5.3.16328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 7.Galvin JE, Lee VM, Trojanowski JQ. Synucleinopathies: clinical and pathological implications. Arch Neurol. 2001;58:186–90. doi: 10.1001/archneur.58.2.186. [DOI] [PubMed] [Google Scholar]
- 8.Jellinger KA. Neuropathological spectrum of synucleinopathies. Mov Disord. 2003;18:S2–12. doi: 10.1002/mds.10557. [DOI] [PubMed] [Google Scholar]
- 9.Rusina R, Kovacs GG, Fiala J, Hort J, Ridzo P, Holmerová I, et al. FTLD-TDP with motor neuron disease, visuospatial impairment and a progressive supranuclear palsy-like syndrome: broadening the clinical phenotype of TDP-43 proteinopathies. A report of three cases. BMC Neurol. 2011;11:50–50. doi: 10.1186/1471-2377-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charles V, Mezey E, Reddy PH, Dehejia A, Young TA, Polymeropoulos MH, et al. Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington's disease patients and transgenic mouse models. Neurosci Lett. 2000;289:29–32. doi: 10.1016/s0304-3940(00)01247-7. [DOI] [PubMed] [Google Scholar]
- 11.Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and otherneurodegenerative disorders. Ann Neurol. 2011;70:532–40. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polymenidou M, Cleveland DW. The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 2011;147:498–508. doi: 10.1016/j.cell.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin LJ. Transgenic mice with human mutant genes causing Parkinson's disease and amyotrophic lateral sclerosis provide common insight into mechanisms of motor neuron selective vulnerability to degeneration. Rev Neurosci. 2007;18:115–36. doi: 10.1515/revneuro.2007.18.2.115. [DOI] [PubMed] [Google Scholar]
- 14.Steiner JA, Angot E, Brundin P. A deadly spread: cellular mechanisms of α-synuclein transfer. Cell Death Differ. 2011;18:1425–33. doi: 10.1038/cdd.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilbert RM, Fahn M, Mitsumoto H, Rowland LP. Parkinsonism and motor neuron diseases: twenty-seven patients with diverse overlap syndromes. Mov Disord. 2010;25:1868–5. doi: 10.1002/mds.23200. [DOI] [PubMed] [Google Scholar]
- 16.Vivacqua G, Casini A, Vaccaro R, Salvi EP, Pasquali L, Fornai F, et al. Spinal cord and parkinsonism: neuromorphological evidences in humans and experimental studies. J Chem Neuroanat. 2011;42:327–40. doi: 10.1016/j.jchemneu.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Vivacqua G, Biagioni F, Yu S, Casini A, Bucci D, D'Este L, et al. Loss of spinal motor neurons and alteration of alphasynuclein immunostaining in MPTP induced Parkinsonism in mice. J Chem Neuroanat. 2012;44:76–85. doi: 10.1016/j.jchemneu.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 18.Corrochano S, Renna M, Tomas-Zapico C, Brown SD, Lucas JJ, Rubinsztein DC, et al. α-Synuclein levels affect autophagosome numbers in vivo and modulate Huntington disease pathology. Autophagy. 2012;8:431–32. doi: 10.4161/auto.19259. [DOI] [PubMed] [Google Scholar]
- 19.Shults CW. Lewy bodies. Proc Natl Acad Sci USA. 2006;103:1661–8. doi: 10.1073/pnas.0509567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell BC, McLean CA, Culvenor JG, Gai WP, Blumbergs PC, Jäkälä P, et al. The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinson's disease. J Neurochem. 2001;76:87–96. doi: 10.1046/j.1471-4159.2001.00021.x. [DOI] [PubMed] [Google Scholar]
- 21.Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 22.Coppedè F. Genetics and epigenetics of Parkinson's disease. Scientific World Journal. 2012;2012:489830–489830. doi: 10.1100/2012/489830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Capobianco L, et al. Similarities between methamphetamine toxicity and proteasome inhibition. Ann N Y Acad Sci. 2004;1025:162–70. doi: 10.1196/annals.1316.021. [DOI] [PubMed] [Google Scholar]
- 24.Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, et al. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- 25.Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, et al. In search of an “autophagomometer”. Autophagy. 2009;5:585–9. doi: 10.4161/auto.5.5.8823. [DOI] [PubMed] [Google Scholar]
- 26.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubuleassociated Protein 1 Light Chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castino R, Lazzeri G, Lenzi P, Bellio N, Follo C, Ferrucci M, et al. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J Neurochem. 2008;106:1426–39. doi: 10.1111/j.1471-4159.2008.05488.x. [DOI] [PubMed] [Google Scholar]
- 28.Vicente MH, Outeiro TF. The sour side of neurodegenerative disorders: the effects of protein glycation. J Pathol. 2010;221:13–25. doi: 10.1002/path.2682. [DOI] [PubMed] [Google Scholar]
- 29.Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–90. doi: 10.1016/j.neuron.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 30.Ticozzi N, Silani V, LeClerc AL, Keagle P, Gellera C, Ratti A, et al. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology. 2009;73:1180–5. doi: 10.1212/WNL.0b013e3181bbff05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fornai F, Meininger V, Silani V. Future therapeutical strategies dictated by preclinical evidence in ALS. Arch Ital Biol. 2011;149:169–74. doi: 10.4449/aib.v149i1.1269. [DOI] [PubMed] [Google Scholar]
- 32.Grillo MA, Colombatto S. Advanced glycation end-products (AGEs): involvement in aging and in neurodegenerative diseases. Amino Acids. 2008;35:29–36. doi: 10.1007/s00726-007-0606-0. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Liu D, Sun L, Lu Y, Zhang Z. Advanced glycation end products and neurodegenerative diseases: mechanisms and perspective. J Neurol Sci. 2012;317:1–5. doi: 10.1016/j.jns.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 34.Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, et al. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse. 2007;61:991–1001. doi: 10.1002/syn.20456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haritani M, Spencer YI, Wells GA. Hydrated autoclave pretreatment enhancement of prion protein immunoreactivity in formalin-fixed bovine spongiform encephalopathy-affected brain. Acta Neuropathol. 1994;87:86–90. doi: 10.1007/BF00386258. [DOI] [PubMed] [Google Scholar]
- 36.Rosicarelli B, Serafini B, Sbriccoli M, Lu M, Cardone F, Pocchiari M, et al. Migration of dendritic cells into the brain in a mouse model of prion disease. J Neuroimmunol. 2005;165:114–20. doi: 10.1016/j.jneuroim.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi RH, Tobiume M, Sato Y, Sata T, Gouras GK, Takahashi H. Accumulation of cellular prion protein within dystrophic neurites of amyloid plaques in the Alzheimer's disease brain. Neuropathology. 2011;31:208–14. doi: 10.1111/j.1440-1789.2010.01158.x. [DOI] [PubMed] [Google Scholar]
- 38.Rahman S, Arai S, Saijo S, Yamato I, Murata T. Sarkosyl is a good regeneration reagent for studies on vacuolar-type ATPase subunit interactions in Biacore experiments. Anal Biochem. 2011;418:301–3. doi: 10.1016/j.ab.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 39.Emmanouilidou E, Melachroinou K, Roumeliotis, Garbis SD, Ntzouni M, Margaritis LH, et al. Cell produced alfasynuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010;30:6838–51. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fornai F, Lenzi P, Gesi M, Ferrucci M, Lazzeri G, Natale G, et al. Recent knowledge on molecular components of Lewy bodies discloses future therapeutic strategies in Parkinson's disease. Curr Drug Targets CNS Neurol Disord. 2003;2:149–52. doi: 10.2174/1568007033482904. [DOI] [PubMed] [Google Scholar]
- 41.Lenzi P, Fulceri F, Lazzeri G, Casini A, Ruggieri S, Paparelli A, et al. Analysis of single, purified inclusions as a novel approach to understand methamphetamine neurotoxicity. Ann N Y Acad Sci. 2008;1139:186–90. doi: 10.1196/annals.1432.014. [DOI] [PubMed] [Google Scholar]
- 42.Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, et al. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington's and Parkinson's diseases. Proc Natl Acad Sci USA. 2006;10:4246–51. doi: 10.1073/pnas.0511256103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mauceli G, Busceti CI, Pellegrini A, Soldani P, Lenzi P, Paparelli A, et al. Overexpression of alpha-synuclein following methamphetamine: is it good or bad? Ann N Y Acad Sci. 2006;1074:191–7. doi: 10.1196/annals.1369.019. [DOI] [PubMed] [Google Scholar]
- 44.Fornai F, Soldani P, Lazzeri G, di Poggio AB, Biagioni F, Fulceri F, et al. Neuronal inclusions in degenerative disorders Do they represent static features or a key to understand the dynamics of the disease? Brain Res Bull. 2005;65:275–90. doi: 10.1016/j.brainresbull.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 45.Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alphasynuclein. Proc Natl Acad Sci USA. 2009;106:13010–5. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Angot E, Steiner JA, Lema Tomé CM, Ekström P, Mattsson B, Björklund A, et al. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS One. 2012;7:e39465–e39465. doi: 10.1371/journal.pone.0039465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koch M, Chitayat S, Dattilo BM, Schiefner A, Diez J, Chazin WJ, et al. Structural basis for ligand recognition and activation of RAGE. Structure. 2010;18:1342–52. doi: 10.1016/j.str.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen L, Wei Y, Wang X, He R. Ribosylation rapidly induces alpha-synuclein to form highly cytotoxic molten globules of advanced glycation end products. PLoS One. 2010;5:e9052–e9052. doi: 10.1371/journal.pone.0009052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lue LF, Walker DG, Brachova L, Yan SD. Involvement of microglial receptor for advanced glycation endproducts (rage) in alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol. 2001;17:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- 50.Sasaki N, Toki S, Chowei H, Saito T, Nakano N, Hayashi Y, et al. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer's disease. Brain Res. 2001;888:256–262. doi: 10.1016/s0006-8993(00)03075-4. [DOI] [PubMed] [Google Scholar]
- 51.Zlokovic B.V. New therapeutic targets in the neurovascular pathway in Alzheimer's disease. Neurotherapeutics. 2008;5:409–41. doi: 10.1016/j.nurt.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma li ska D, Laure-Kamionowska M, Taraszewska A, Der gowski K, Ma li ski S. Immunodistribution of amyloid beta protein (Aβ) and advanced glycation end-product receptors (RAGE) in choroid plexus and ependyma of resuscitated patients. Folia Neuropathol. 2011;49:295–300. [PubMed] [Google Scholar]
- 53.Askarova S, Yang X, Sheng W, Sun GY, Lee JC. Role of Aβ-receptor for advanced glycation endproducts interaction in oxidative stress and cytosolic phospholipase Aβ activation in astrocytes and cerebral endothelial cells. Neuroscience. 2011;199:375–85. doi: 10.1016/j.neuroscience.2011.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–55. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casula M, Iyer AM, Spliet WG, Anink JJ, Steentjes K, Sta M, et al. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. 2011;179:233–43. doi: 10.1016/j.neuroscience.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 56.Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–5004. [PubMed] [Google Scholar]
- 57.Chivet M, Hemming F, Pernet-Gallay K, Fraboulet S, Sadoul R. Emerging role of neuronal exosomes in the central nervous system. Front Physiol. 2012;3:145–145. doi: 10.3389/fphys.2012.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gousset K, Zurzolo C. Tunnelling nanotubes: a highway for prion spreading? Prion. 2009;3:94–8. doi: 10.4161/pri.3.2.8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahmed KA, Xiang J. Mechanisms of cellular communication through intercellular protein transfer. J Cell Mol Med. 2011;15:1458–73. doi: 10.1111/j.1582-4934.2010.01008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamasaki T, Suzuki A, Shimizu T, Watarai M, Hasebe R, Horiuchi M. Characte-rization of intracellular localization of PrP(Sc) in prion-infected cells using a mAb that recognizes the region consisting of aa 119–127 of mouse PrP. J Gen Virol. 2012;93:668–80. doi: 10.1099/vir.0.037101-0. [DOI] [PubMed] [Google Scholar]
- 61.Santa-Maria I, Varghese M, Ksiezak-Reding H, Dzhun A, Wang J, Pasinetti GM. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of tau protein in aggresomes. J Biol Chem. 2012;287:20522–33. doi: 10.1074/jbc.M111.323279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vella LJ, Sharples RA, Nisbet RM, Cappai R, Hill AF. The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur Biophys J. 2008;37:323–32. doi: 10.1007/s00249-007-0246-z. [DOI] [PubMed] [Google Scholar]
- 63.Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci USA. 2004;101:9683–8. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, et al. Alzheimer's disease amyloid peptides are released in association with exosomes. Proc Natl Acad Sci USA. 2006;103:11172–77. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]