

Abstract
The Kaposi’s sarcoma-associated herpesvirus is the causative agent of primary effusion lymphoma (PEL), for which cytotoxic chemotherapy represents the standard of care. The high mortality associated with PEL may be explained in part by resistance of these tumors to chemotherapy. The membrane-bound glycoprotein emmprin (CD147) enhances chemoresistance in tumors through effects on transporter expression, trafficking and interactions. Interactions between hyaluronan and hyaluronan receptors on the cell surface also facilitate emmprin-mediated chemoresistance. Whether emmprin or hyaluronan-receptor interactions regulate chemotherapeutic resistance for virus-associated malignancies is unknown. Using human PEL tumor cells, we found that PEL sensitivity to chemotherapy is directly proportional to expression of emmprin, the lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1) and a drug transporter known as the breast cancer resistance protein/ABCG2 (BCRP), and that emmprin, LYVE-1 and BCRP interact with each other and colocalize on the PEL cell surface. In addition, we found that emmprin induces chemoresistance in PEL cells through upregulation of BCRP expression, and RNA interference targeting of emmprin, LYVE-1 or BCRP enhances PEL cell apoptosis induced by chemotherapy. Finally, disruption of hyaluronan-receptor interactions using small hyaluronan oligosaccharides reduces expression of emmprin and BCRP while sensitizing PEL cells to chemotherapy. Collectively, these data support interdependent roles for emmprin, LYVE-1 and BCRP in chemotherapeutic resistance for PEL.
Keywords: KSHV, lymphoma, drug resistance, emmprin, hyaluronan, LYVE-1
Introduction
The Kaposi’s sarcoma-associated herpesvirus (KSHV) is the etiologic agent of primary effusion lymphoma (PEL),1 multicentric Castleman’s disease2 and Kaposi’s sarcoma.3 PEL represents a rapidly progressive illness arising primarily in patients infected with the human immunodeficiency virus (HIV), although cases have also been documented in organ transplant recipients.4 Administration of cytotoxic chemotherapeutic agents represents the current standard of care for the treatment of PEL.5-7 However, the myelosuppressive effects of cytotoxic chemotherapy synergize with those caused by antiretroviral therapy or immune suppression.4,11 Furthermore, the prognosis for PEL remains poor with a median survival of ~6 months,5-7 dictating the need for safer and more effective therapeutic options. Therapies targeting the mammalian target of rapamycin or CD20 have proven helpful in select cases,8,9 although a lack of efficacy due to induction of alternative tumor-promoting signal transduction pathways or outgrowth of CD20-negative tumors limits the utility of these approaches.10 Many PEL tumors demonstrate resistance to chemotherapeutic agents used in the clinic.4 p53 mutagenesis and the KSHV-encoded latency-associated nuclear antigen-2 (LANA2) have been implicated in PEL resistance to chemotherapy,4,11 but a better understanding of mechanisms for PEL chemoresistance is needed in order to develop clinically applicable approaches for sensitizing PEL tumors to cytotoxic agents.
Emmprin (CD147; basigin) was originally identified as a membrane-bound inducer of matrix metalloproteinase (MMP) synthesis,12,13 enhanced tumor growth, and tumor cell invasion.14 More recent studies have demonstrated emmprin interactions with monocarboxylate and ATP-binding cassette (ABC)-family multidrug transporters to facilitate export of lactate or chemotherapeutic agents, respectively.15-19 Emmprin also stimulates production of hyaluronan,20 an extracellular polysaccharide that promotes tumor chemoresistance through interactions with the cell surface receptor CD44.21-25 Small hyaluronan oligosaccharides (oHAs) interact monovalently with CD44, competitively blocking polyvalent interactions between CD44 and endogenous hyaluronan,26,27 and oHAs sensitize murine lymphoma, malignant peripheral nerve sheath tumor, glioma and various carcinoma cell lines to chemotherapy in vitro and in vivo.18,21-24,28 The lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1), which has structural similarity to CD44, also serves as a receptor for hyaluronan.29 Interestingly, LYVE-1 is expressed by KSHV-infected cells and within KSHV-associated tumors,30-32 although a role for LYVE-1 in KSHV pathogenesis has not been established. Furthermore, surface expression of CD44 is negligible for PEL cells.33 It is unknown whether emmprin, hyaluronan receptors or other associated proteins regulate chemotherapeutic resistance for virus-mediated tumors.
Using patient-derived PEL tumors, we found recently that PEL cells express emmprin, LYVE-1 and the ABC family transporter known as the breast cancer resistance protein/ABCG2 (BCRP) on the cell surface. Therefore, we sought to determine whether emmprin, LYVE-1 and/or BCRP, either alone or through interdependent interactions, regulate PEL resistance to chemotherapeutic agents.
Materials and methods
Cell culture
KSHV-infected PEL cells, including BC-1, BC-3, BCP-1 and BCBL-1 cell lines, were kindly provided by the laboratories of Dr Dean H Kedes (University of Virginia) and Dr Dirk Dittmer (University of North Carolina, Chapel Hill). All PEL cells were maintained in RPMI-1640 media (Gibco, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum, 10 mm HEPES (pH 7.5), 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mm l-glutamine, 0.05 mm β-mercaptoethanol and 0.02% (wt/vol) sodium bicarbonate.
Preparation of oHAs
oHAs were prepared as described previously.17 Briefly, the oHA preparation comprises a mixed fraction of average molecular weight (MW) ~2.5 × 103 composed of 3 to 10 disaccharide units fractionated from testicular hyaluronidase (type 1-S) digests of hyaluronan polymer (Sigma-Aldrich (St Louis, MO, USA), sodium salt). Fractionation was performed using trichloroacetic acid precipitation followed by serial dialysis with 5000 MWCO (Amicon Ultra Ultracel, Millipore, Billerica, MA, USA) and 1000 MWCO (Spectra/Por Membrane, Spectrum Laboratories, Rancho Dominguez, CA, USA) membranes.
Cell viability assays
Cell viability was assessed using both MTT and Trypan blue exclusion assays as previously described.34 For MTT assays, a total of 5 × 103 PEL cells were incubated individual wells of a 96-well plate for 24 h. Serial dilutions of paclitaxel, doxorubicin or oHAs were added and subsequently incubated in 1 mg/ml MTT solution (Sigma-Aldrich) at 37°C for 3 h. Thereafter, cells were incubated in 50% dimethylsulfoxide overnight and optical densities determined at 570 nm using a spectrophotometer (Thermo Labsystems, West Palm Beach, FL, USA). For Trypan blue exclusion assays, cells were incubated with 0.4% Trypan blue (MP Biomedicals, Northbrook, IL, USA) and observed under light microscopy. Relative cell viability was determined after assessment of at least 1000 cells per condition for each experiment using the following formula: (no. of live cells/no. of total cells for experimental conditions)/ (no. of live cells/no. of total cells for vehicle-treated control cells).
Gene amplification
Total RNA was isolated using the RNeasy Mini kit according to the manufacturer’s instructions (QIAGEN, Valencia, CA, USA). Complementary DNA was synthesized from equivalent concentrations of total RNA using the SuperScript III First-Strand Synthesis SuperMix Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Coding sequences for hyaluronan synthases 1–3 (has1–3; and β-actin for internal controls) were amplified from 200 ng input complementary DNA using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). Custom primer sequences used for amplification experiments were as follows: has1 sense 5′-CAAGGCGCTCGGAGATTC-3′; has1 antisense 5′-GACCGCTGATGCAGGATACA-3′; has2 sense 5′-CATCATCCAAAGCCTGTT-3′; has2 antisense 5′-TCTTCTGAGTTCCCATCTA-3′; has3 sense 5′-TGGCTCAACCAGCAAACC-3′; has3 antisense 5′-CAGCAGGAAGAGGAGAATGT-3′; β-GGAAATCGTGCGTGACATT-3′; β-actin antisense 5′-GACTCGTCATACTCCTGCTTG-3′. Amplification was carried out using an iCycler IQ Real-Time PCR Detection System, and cycle threshold (Ct) values determined in duplicate for emmprin has transcripts and β-actin for each experiment. ‘No template’ (water) and ‘no-RT’ controls were used to ensure minimal background DNA contamination. Fold changes for experimental groups relative to assigned controls were calculated using automated iQ5 2.0 software (Bio-Rad).
RNA interference (RNAi)
Emmprin, LYVE-1, BCRP and non-target small interfering RNAs were purchased from the manufacturer (ON-TARGET plus SMART pool, Dharmacon, Lafayette, CO, USA). Cells were incubated with small interfering RNAs in 12-well plates using DharmaFECT Transfection Reagent (Dharmacon) according to the manufacturer’s instructions, and gene silencing assessed using immunoblots within 48 h.
Immunoprecipitation and immunoblot assays
Cells were lysed in buffer containing 20 mm Tris (pH 7.5), 150 mm NaCl, 1% NP40, 1 mm EDTA, 5 mm NaF and 5 mm Na3VO4. Total cell lysates (30 μg) were resolved by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and immunoblotted with 100–200 μg/ml antibodies recognizing the following proteins: BCRP, LYVE-1 (Santa Cruz, Santa Cruz, CA, USA), Bax, pro-/cleaved caspase-9, pro-/cleaved caspase-3, Bcl-2 (Cell Signaling, Boston, MA, USA) and emmprin (BD Pharmingen, San Jose, CA, USA). For loading controls, blots were reacted with antibodies detecting β-actin (Sigma-Aldrich). Immunoreactive bands were developed using an enhanced chemiluminescence reaction (Perkin-Elmer, San Jose, CA, USA), and visualized by autoradiography. Immunoprecipitation assays were performed using the Catch and Release v2.0 Reversible Immunoprecipitation System (Millipore) according to the manufacturer’s instructions (Invitrogen). Mouse or rabbit IgG were used as negative controls.
Flow cytometry
PEL cells were resuspended in 3% bovine serum albumin in 1 × phosphate-buffered saline, incubated on ice for 10 min, and then incubated with primary antibodies (diluted 1:50 for emmprin, and 1:20 for BCRP and LYVE-1) for an additional 30 min. Following two subsequent wash steps, cells were incubated for an additional 30 min with either goat anti-rabbit IgG Alexa-647 or goat anti-mouse IgG Alexa-647 (Invitrogen) diluted 1:200. Control cells were incubated with secondary antibodies only. Cells were resuspended in 1 × phosphate-buffered saline before analysis. For quantitative apoptosis assays, the fluorescein isothiocyanate Annexin V Apoptosis Detection Kit I (BD Pharmingen) and propidium iodide were used according to the manufacturer’s instructions to identify early apoptotic (annexin+ propidium iodide−) and late apoptotic (annexin+ propidium iodide+) cells for 10 000 cells in each experimental and control condition. Data were collected using a FACS Calibur four-color flow cytometer (Bio-Rad), and FlowJo software (TreeStar, San Carlos, CA, USA) was used to quantify cell surface localization of target proteins. The percentage of total apoptotic cells in each sample was calculated as follows: (early apoptotic + late apoptotic cells) / total cells analyzed.
Immunofluorescence assays
PEL cells were incubated in 3% paraformaldehyde at 4 °C for fixation, and then with a blocking reagent (3% bovine serum albumin in 1 × phosphate-buffered saline) for an additional 30 min. Cells were subsequently incubated for 1 h at 25 °C with primary antibodies (diluted 1:50 for emmprin, and 1:20 for BCRP and LYVE-1), followed by goat anti-rabbit IgG Texas Red or goat anti-mouse IgG Alexa-488 (Invitrogen) diluted 1:100 for an additional 1 h at 25 °C. To detect the presence of doxorubicin within individual cells, doxorubicin was excited using an argon laser (λex-488 nm) and detected using an emission filter set at 505–530 nm, as described previously.35 Images were captured using a Leica TCS SP5 AOBS confocal microscope (Leica Microsystems Inc., Buffalo Grove, IL, USA) equipped with a × 63/1.4 objective lens.
Transduction assays
PEL cells were transduced (multiplicity of infection ~20) using a recombinant adenoviral vector encoding emmprin or a control vector as previously described.36 After 24 h, cells were incubated with paclitaxel and doxorubicin (Sigma-Aldrich) with or without 100 μg/ml oHA before quantification of cell viability.
Hyaluronan quantification
Hyaluronan concentrations were determined within cell supernatants using an enzyme-linked immunosorbent-like assay described previously.37
Statistical analysis
Significance for differences between experimental and control groups was determined using the two-tailed Student’s t-test (Excel 8.0), and P-values <0.05 or <0.01 were considered significant or highly significant, respectively.
Results
PEL chemoresistance correlates directly with emmprin, LYVE-1 and BCRP expression
Using four representative human PEL cell lines, we sought to determine whether chemoresistance for PEL cells correlates with their expression of emmprin, LYVE-1 and BCRP. We chose to focus on BCRP as we observed its clear expression on the PEL cell surface (Figure 1b), whereas we did not observe appreciable PEL cell expression of the other ubiquitous, well-characterized ABC transporter, P-glycoprotein (not shown). For our experiments, we used four human PEL cell lines: two ‘chemoresistant’ cell lines (BCP-1 and BCBL-1 cells) and two ‘chemosensitive’ cell lines (BC-1 and BC-3 cells), previously characterized based on their relative sensitivity to the DNA synthesis inhibitor doxorubicin.4 Using immunoblotting and flow cytometry, respectively, we found that total protein expression and membrane localization of emmprin, LYVE-1 and BCRP were significantly greater for chemoresistant PEL cells (Figures 1a and b). Of note, chemoresistant PEL cells exhibited greater expression of both high-MW (~65 kDa) and low-MW (~35 kDa) emmprin glycoforms. Emmprin isoforms with high or low levels of glycosylation demonstrate biologic activity with respect to induction of MMP expression.38,39 Correlating with these results, we also noted greater expression of representative MMPs (MMP1, MMP2 and MMP9) in chemoresistant PEL cells (Supplementary Figure S1). In addition, chemoresistant PEL cells exhibited increased hyaluronan secretion and greater expression of hyaluronan synthase transcripts (has1–3 for BCP-1; has2/3 for BCBL-1) relative to chemosensitive PEL cells (Figures 1c and d). Protein complexes containing emmprin or CD44 and drug transporters have been previously identified on the surface of tumor cells.18,21 Using confocal microscopy, we observed colocalization of emmprin, LYVE-1 and BCRP on the PEL cell surface (Figure 2a). Moreover, BCRP and LYVE-1 co-immunoprecipitated with emmprin, and BCRP and emmprin co-immunoprecipitated with LYVE-1 (Figures 2b and c). These results support interactions between emmprin, LYVE-1 and BCRP on the surface of chemoresistant PEL cells.
Figure 1.
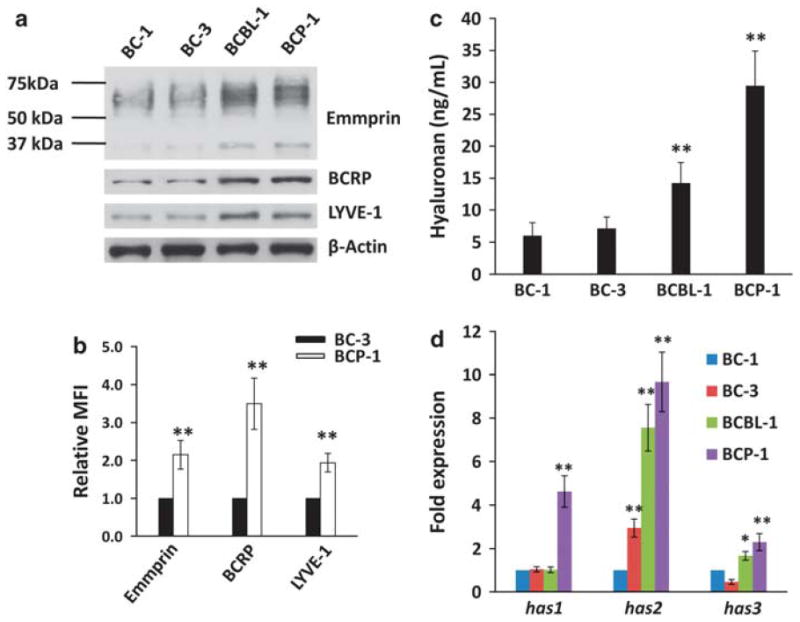
Chemoresistance of PEL cells correlates directly with LYVE-1, emmprin and BCRP expression. (a) Immunoblot analyses were used to detect basal expression of emmprin, LYVE-1 and BCRP for relatively chemosensitive (BC-1 and BC-3) and chemoresistant (BCP-1 and BCBL-1) PEL cells. β-Actin was identified for internal controls. Data shown represent one of three independent experiments. (b) Flow cytometric analyses were used to quantify emmprin, LYVE-1 and BCRP expression on the surface of representative chemosensitive (BC-3) and chemoresistant (BCP-1) PEL cells. Mean fluorescence intensity (MFI), reflecting surface expression of each protein for 10 000 cells in each condition, was calculated for BCP-1 cells relative to BC-3 cells using FlowJo software. (c) Hyaluronan secretion in culture supernatants was quantified as described in the Materials and methods. (d) Transcripts representing three hyaluronan synthase genes (has1–3) were quantified by qRT-PCR, and their expression relative to that for BC-1 cells determined as described in the Materials and methods. Error bars represent the s.e.m. for three independent experiments. *P<0.05; ** P<0.01.
Figure 2.
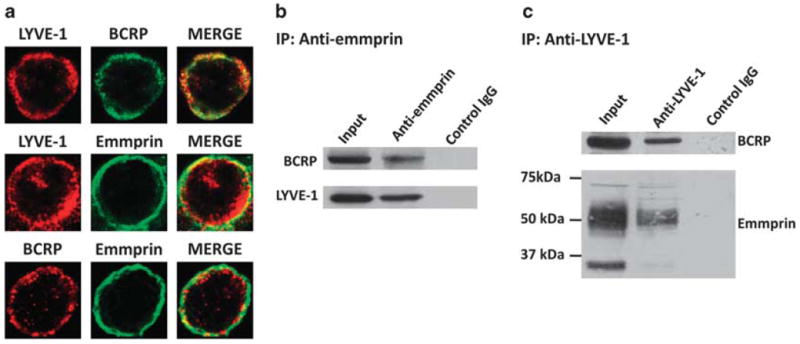
Emmprin, LYVE-1 and BCRP interact on the PEL cell surface. (a) Confocal immunofluorescence assays (IFAs) were performed as described in the Materials and methods to identify expression and localization of emmprin, LYVE-1 and BCRP using BCP-1 cells. Red or green fluorescence represents localization of a single protein, whereas yellow fluorescence represents colocalization of two proteins in merged images. Data shown represent one of three independent experiments and at least 100 cells analyzed for each experiment. (b, c) Co-immunoprecipitation (co-IP) assays were performed as described in the Materials and methods. Proteins were identified within total protein (input) fractions for positive controls, and IgG antibodies of the same subclass were used for negative controls for both anti-emmprin and anti-LYVE-1 co-IP assays.
Emmprin and LYVE-1 regulate BCRP expression and PEL resistance to chemotherapy
Following RNAi resulting in partial inhibition of emmprin expression in PEL cells, immunoblots revealed partial reduction of total BCRP protein expression, but no clearly discernable reduction in LYVE-1 expression (Figure 3a). Inhibition of emmprin expression significantly reduced hyaluronan secretion by chemoresistant PEL cells (Figure 3b). Furthermore, flow cytometry and confocal microscopy demonstrated that inhibition of emmprin significantly reduced BCRP localization on the cell surface, but not LYVE-1 (Figures 3c and d). Doxorubicin is used routinely for the treatment of PEL.7 The microtubule inhibitor paclitaxel also induces apoptosis of human PEL tumors in vitro,40 but paclitaxel is not routinely used for the treatment of PEL due, in part, to the demonstration of PEL resistance to paclitaxel.11 Our viability assays showed that targeting emmprin increased the sensitivity of chemoresistant PEL cells to both doxorubicin and paclitaxel (Figure 3e).
Figure 3.
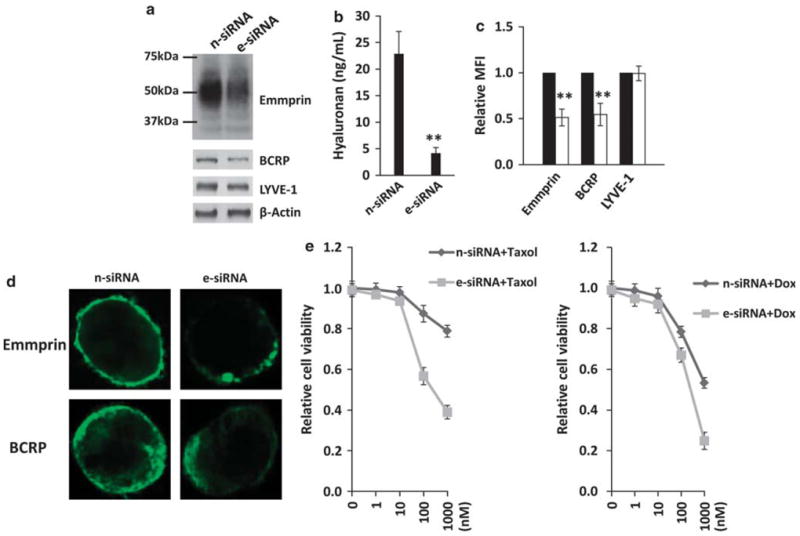
Targeting emmprin reduces BCRP expression, hyaluronan secretion and PEL cell resistance to chemotherapeutic agents. BCP-1 cells were transfected with emmprin-specific small interfering RNA (e-siRNA) or non-target control siRNA (n-siRNA). After 48 h, immunoblot analyses were used to quantify protein expression (a), supernatants used to quantify hyaluronan secretion (b), and flow cytometric analyses used to quantify emmprin, BCRP and LYVE-1 expression on the cell surface (c). For the latter, mean fluorescence intensities representing cell surface expression (MFI), following analysis of 10 000 cells, were determined for e-siRNA-treated BCP-1 cells (white bars) relative to controls (black bars). (d) Confocal IFAs were performed to identify and localize emmprin and BCRP expression as described in the Materials and methods. (e) e-siRNA-transfected or n-siRNA control-transfected cells (24 h) were incubated with the indicated concentrations of paclitaxel (Taxol) or doxorubicin (Dox) for 72 h and relative cell viability quantified using Trypan blue exclusion as described in the Materials and methods. For all experiments, error bars represent the s.e.m. for three independent experiments. **P<0.01.
Using transduction with a recombinant adenovirus encoding emmprin, we found that ectopic overexpression of emmprin increased BCRP expression in chemosensitive PEL cells whereas LYVE-1 remained unaffected (Figure 4a). Furthermore, emmprin overexpression significantly reduced PEL cell sensitivity to both doxorubicin and paclitaxel and, using RNAi, we confirmed that this effect was mediated almost entirely through upregulation of BCRP (Figure 4b). RNAi subsequently confirmed that reducing BCRP expression significantly enhanced PEL cytotoxicity induced by either doxorubicin or paclitaxel (Figures 4c and d). Emmprin overexpression also significantly increased hyaluronan secretion (Figure 5a), and the increase in chemoresistance caused by emmprin overexpression was suppressed by co-administration of oHAs (Figure 5b), suggesting that the chemoprotective effect of emmprin for PEL cells is dependent upon hyaluronan-receptor interactions.
Figure 4.
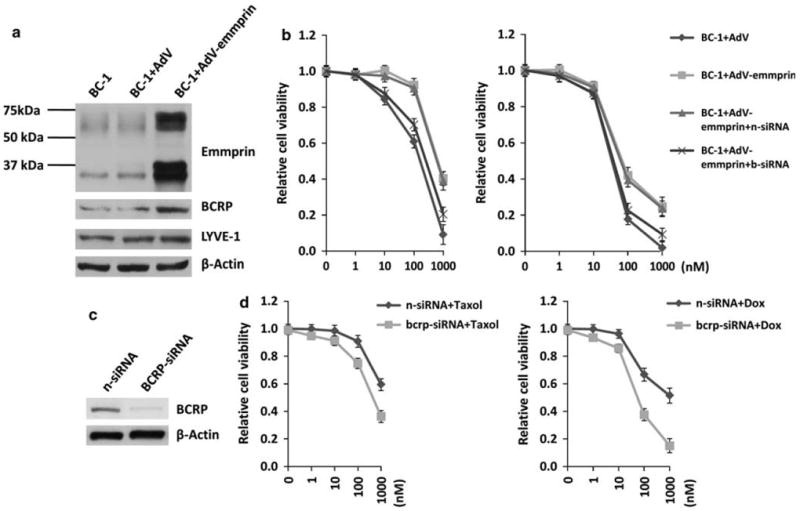
Emmprin induces PEL resistance to chemotherapy through induction of BCRP expression. (a) BC-1 cells were transduced using a recombinant human emmprin-encoding adenovirus (AdV-emmprin) or control adenovirus (AdV), and protein expression quantified 48 h later by immunoblotting. (b) BC-1 cells were transfected with control non-target- (n) or BCRP (b)-specific small interfering RNA (siRNA) for 24 h, and then transduced as in (a) for an additional 48 h before incubation with the indicated concentrations (nm on x axis) of Taxol (left panel) or Dox (right panel) for 72 h each. Relative cell viability was quantified using Trypan blue exclusion. Error bars represent the s.e.m. for three independent experiments. (c) BCBL-1 cells were transfected with BCRP-siRNA or non-target control siRNA (n-siRNA) for 48 h, and then immunoblot analyses were used to detect BCRP expression. (d) Following transfection as in (c), BCBL-1 cells were incubated with Taxol or Dox for 72 h at the indicated concentrations and relative cell viability quantified using Trypan blue exclusion.
Figure 5.
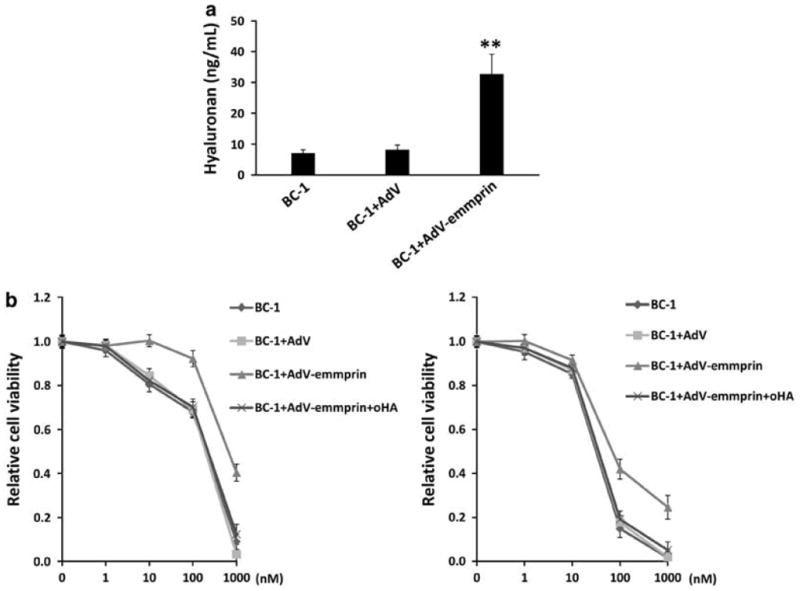
Emmprin induces PEL resistance to chemotherapy through induction of hyaluronan-receptor interactions. (a) BC-1 cells were transduced as in Figure 4 and supernatants used for quantification of hyaluronan secretion after 48 h. (b) BC-1 cells were transduced as above for 48 h and subsequently incubated with either Taxol or Dox at the indicated concentrations in the presence or absence of 100 μg/ml oHA for an additional 72 h. Relative cell viability was quantified using Trypan blue exclusion. Error bars represent the s.e.m. for three independent experiments.
Since we observed LYVE-1 expression on the surface of PEL cells as well as oHA suppression of emmprin-mediated chemoresistance, we hypothesized that inhibition of LYVE-1 expression would also sensitize PEL cells to chemotherapy. We found that RNAi targeting LYVE-1 reduced both total expression and membrane localization of BCRP in PEL cells, but did not affect emmprin expression significantly (Figures 6a–c). Moreover, we found that reduced LYVE-1 expression significantly enhanced PEL cell sensitivity to both doxorubicin and paclitaxel (Figure 6d). Using complimentary flow cytometric assays, we also found that reduction in expression of either emmprin or LYVE-1 led to enhanced apoptosis in the presence of chemotherapeutic agents. However, no effect was observed when either emmprin or LYVE-1 was targeted in the absence of these agents (Figures 7a and b).
Figure 6.
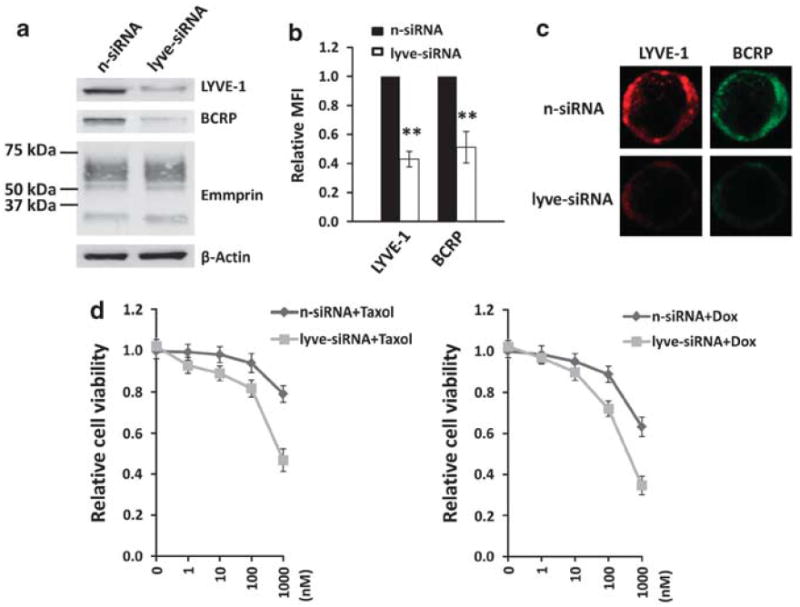
Targeting LYVE-1 reduces BCRP expression and PEL cell resistance to chemotherapeutic agents. BCP-1 cells were transfected with LYVE-1-siRNA or non-target control small interfering RNA (n-siRNA). After 48 h, immunoblot analyses were used to quantify protein expression (a) and flow cytometric assays used to quantify LYVE-1 and BCRP expression on the cell surface (b). For the latter, mean fluorescence intensities representing cell surface expression (MFI), following analysis of 10 000 cells, were determined for LYVE-1-siRNA-treated BCP-1 cells relative to controls. (c) Confocal immunofluorescence assays (IFAs) were used to identify and localize LYVE-1 and BCRP expression as described in the Materials and methods. (d) LYVE-1-siRNA-transfected or n-siRNA control-transfected BCP-1 cells were incubated with Taxol or Dox for 72 h at the indicated concentrations, and cell viability quantified using Trypan blue exclusion. Error bars represent the s.e.m. for three independent experiments. **P<0.01.
Figure 7.
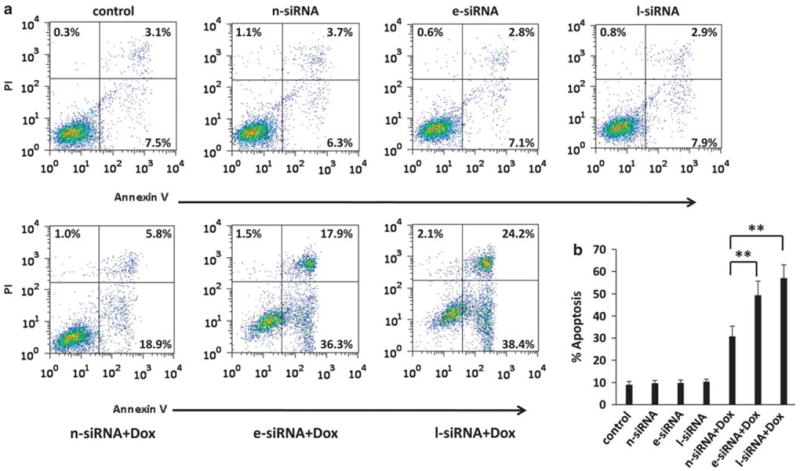
Targeting emmprin or LYVE-1 enhances PEL cell apoptosis induced by chemotherapeutic agents. (a) BCP-1 cells were transfected with emmprin-small interfering RNA (e-siRNA), LYVE-1-siRNA (l-siRNA) or non-target control siRNA (n-siRNA) for 24 h, and then incubated in the presence or absence of 100 nm Dox for an additional 24 h. Apoptosis was quantified by flow cytometry using Annexin V and propidium iodide as described in the Materials and methods. (b) The percentage of total (early + late) apoptotic cells within at least 10 000 cells in each group per experiment was determined as described in the Materials and methods. Error bars represent the s.e.m. for three independent experiments. **P<0.01.
Collectively, these results suggest that cooperative mechanisms involving emmprin and hyaluronan interactions with LYVE-1 regulate PEL chemoresistance, and that upregulation of BCRP is responsible for these effects.
oHAs sensitize PEL cells to chemotherapeutic agents
Published data indicate that oHAs induce apoptosis for lymphoma cell lines,28,41 and as previously stated, we observed that oHAs suppress emmprin-induced chemoresistance for PEL cells (Figure 5b). Therefore, we sought to determine whether oHAs reduce PEL viability through induction of apoptosis, and whether oHAs alone sensitize PEL cells to chemotherapy. In agreement with our results indicating that RNAi targeting emmprin or LYVE-1 alone have no impact on PEL viability, we found that oHAs alone did not induce cytotoxicity for PEL cells (Supplementary Figure S2). However, oHAs significantly enhanced PEL cytotoxicity induced by either doxorubicin or paclitaxel, with this effect being more pronounced for chemoresistant PEL cells (Figures 8a–d and Supplementary Figure S3). Furthermore, oHAs enhanced doxorubicin or paclitaxel induction of PEL apoptosis (Figure 8e). In parallel, we confirmed that oHAs reduced expression of the anti-apoptotic protein Bcl-2 (B-cell lymphoma 2), increased expression of the pro-apoptotic protein Bax and increased expression of the functional, pro-apoptotic cleaved proteins caspase-9 and caspase-3 while reducing the pro-forms of these proteins (Figure 8f). Interestingly, we observed that oHAs suppressed doxorubicin- or paclitaxel-induced expression of emmprin and BCRP but not LYVE-1 (Figure 9a), although oHAs alone had no significant impact on basal expression of emmprin, LYVE-1 or BCRP (Supplementary Figure S4). In addition, oHAs suppressed doxorubicin- or paclitaxel-induced cell surface expression of BCRP (Figures 9b and c). Laser excitation of intrinsic fluorescence for doxorubicin has been recently reported,35 and we confirmed that intracellular accumulation of doxorubicin occurred in a significantly greater number of oHA-treated cells in these assays. Furthermore, intracellular accumulation of doxorubicin correlated with the degree of apoptosis for individual cells as determined by visualization of nuclear fragmentation (Figure 10). Other work has demonstrated that blocking hyaluronan interactions with CD44 disrupts emmprin- and CD44-drug efflux pump complexes on the cell surface,18 and we found that oHAs reduced co-precipitation of LYVE-1 with either emmprin or BCRP (Supplementary Figure S5). However, additional experiments are needed to resolve whether this latter observation is caused, in part, by a reduction of emmprin and BCRP expression with oHAs. Collectively, these data support a role for hyaluronan-receptor interactions in the induction of PEL chemoresistance, and demonstrate that disruption of these interactions enhances chemotherapy-mediated apoptosis for PEL cells.
Figure 8.
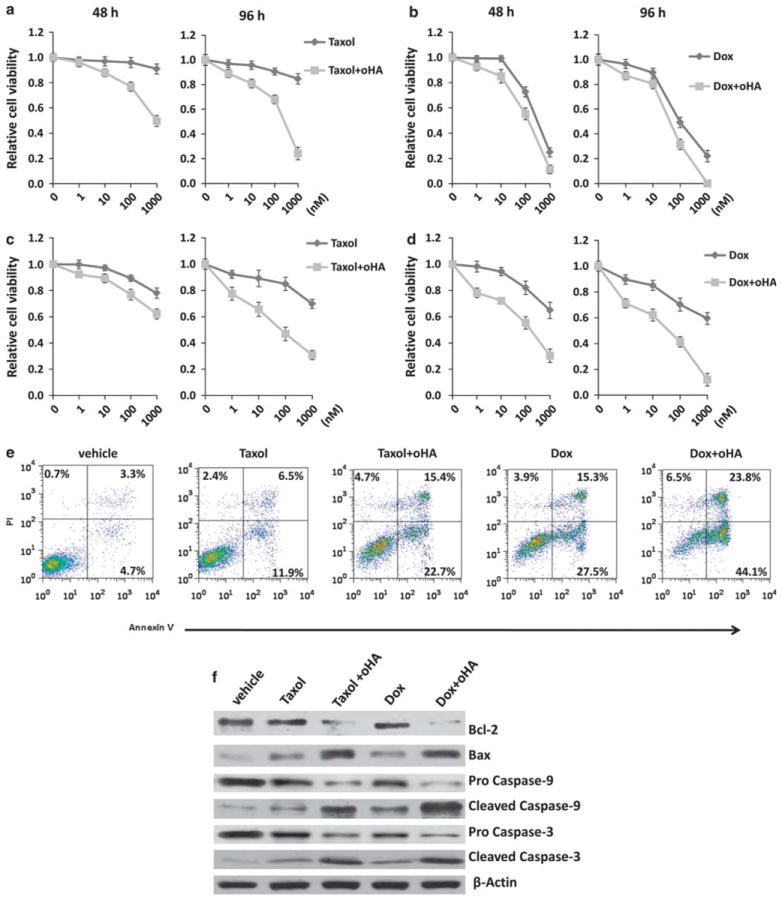
oHAs sensitize chemoresistant PEL cells to chemotherapeutic agents. BCP-1 (a, b) and BCBL-1 cells (c, d) were incubated with either Taxol or Dox at the indicated concentrations and for the indicated times in the presence or absence of 100 μg/ml oHA. Relative cell viability was quantified using Trypan blue exclusion. Error bars represent the s.e.m. for three independent experiments. (e) BCP-1 cells were incubated with 100 nm Taxol or 100 nm Dox in the presence or absence of 100 μg/ml oHA for 48 h, and then apoptosis quantified by flow cytometry as described in the Materials and methods. (f) In parallel with experiments in (e), immunoblots were performed to identify apoptosis-associated protein expression as described in the Materials and methods. Data shown for (e) and (f) represent one of three independent experiments.
Figure 9.
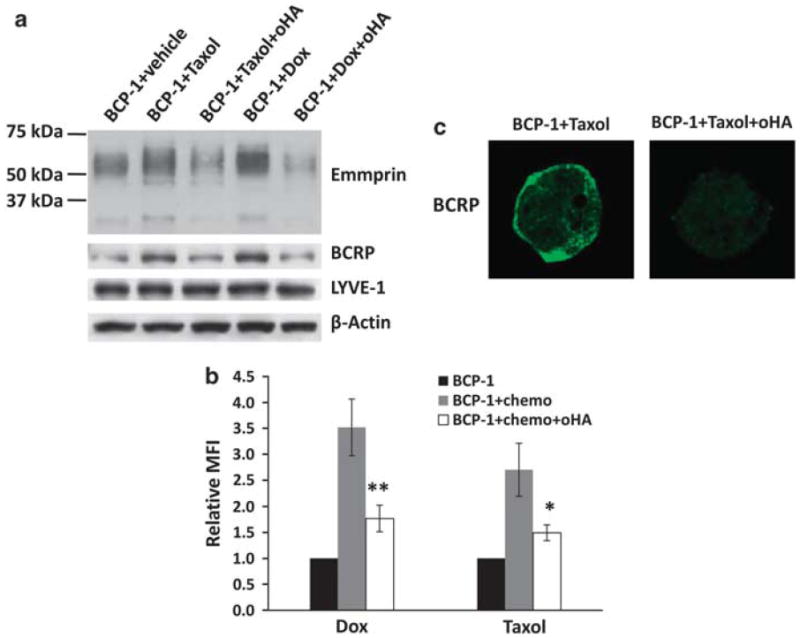
oHAs reduce emmprin and BCRP expression in PEL cells treated with chemotherapeutic agents. (a) BCP-1 cells were incubated with 100 nm Taxol or 100 nm Dox for 96 h in the presence or absence of 100 μg/ml oHA. Immunoblot analyses were used to detect total protein expression, including β-actin for internal controls. Data shown represent one of three independent experiments. (b) Flow cytometry analyses were used to quantify BCRP cell surface expression for similar conditions as in (a). Mean fluorescence intensity (MFI), reflecting surface expression of BCRP for 10 000 cells, was determined for experimental groups relative to untreated BCP-1 control cells. Error bars represent the s.e.m. for three independent experiments *P<0.05; **P<0.01. (c) BCP-1 cells were treated as in (a), and then confocal IFAs performed for identification and localization of BCRP expression as described in the Materials and methods. Data shown represent one of three independent experiments.
Figure 10.
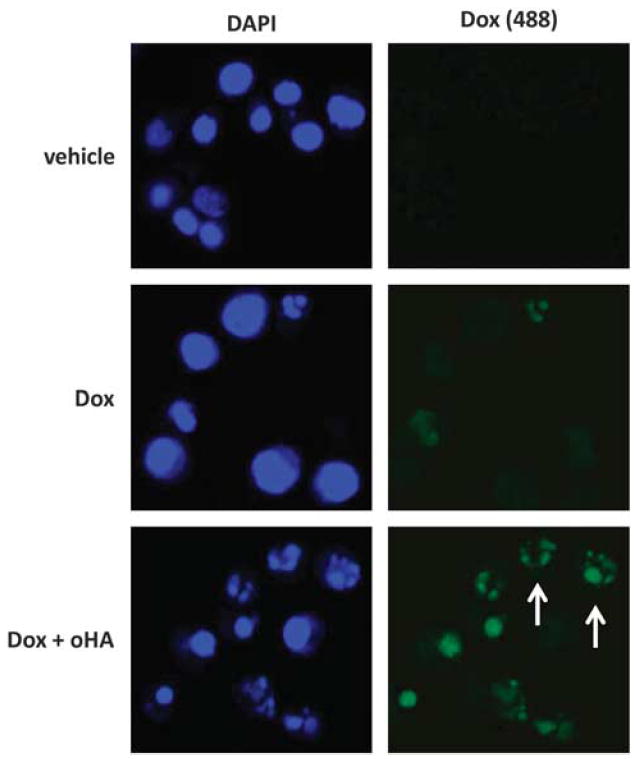
PEL cells incubated with oHAs exhibit increased intracellular accumulation of doxorubicin. BCP-1 cells were incubated with 100 nm Dox for 48 h in the presence or absence of 100 μg/ml oHA, and then confocal IFAs were performed to identify intracellular doxorubicin (green) as described in the Materials and methods. For identification of nuclei (blue), cells were counterstained with 0.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Sigma) in 180 mm Tris-HCl (pH 7.5), and visualization of nuclear fragmentation used to identify cells undergoing apoptosis (arrows). Data shown represent one of three independent experiments.
Discussion
Cytotoxic chemotherapeutic agents represent the current standard of care for PEL, but these agents may aggravate toxicities associated with antiretroviral agents administered to HIV-infected patients and have not improved the poor prognosis for patients with these tumors.4-7 Sensitization of PEL to existing chemotherapies may allow for dose reduction of cytotoxic agents to minimize associated toxicities, as well as augmentation of chemotherapy-mediated PEL apoptosis to improve clinical outcomes. Data from a single report suggest that mutation of p53 leads to doxorubicin resistance for PEL cells.4 A second report found that the KSHV-encoded LANA2 modulates microtubule dynamics through direct binding to polymerized microtubules, thereby interfering with microtubule stabilization by paclitaxel and increasing PEL resistance to this drug.11 However, neither of these mechanisms of resistance can be easily targeted for therapeutic purposes, supporting the need for identification of alternative mechanisms for PEL resistance, specifically those involving potential targets at the cell surface. Emmprin, through interactions with hyaluronan receptors17,23 and membrane-bound transporters,17-19 facilitates tumor cell chemoresistance. In addition, disruption of hyaluronan interactions with its cognate receptors interferes with emmprin-mediated drug resistance,23 in part through disruption of protein complexes containing emmprin.17,18 Our studies were undertaken to determine whether emmprin, the hyaluronan receptor LYVE-1 and the ABC-family multidrug transporter BCRP regulate PEL resistance to chemotherapy. This approach was initially supported by our observation of a direct correlation between PEL resistance to chemotherapeutic agents and expression of emmprin, LYVE-1, and BCRP, as well as hyaluronan secretion (Figure 1), and data supporting interactions for these proteins on the PEL cell surface (Figure 2).
Our data are the first, to our knowledge, establishing roles for either emmprin or LYVE-1 in the regulation of BCRP expression, although data from our group have demonstrated decreased expression of BCRP by glioma cells after oHA treatment.22 Our data are also consistent with previously published data from our group and others indicating that increased emmprin expression stimulates hyaluronan–CD44 interactions,20,23 which in turn increase expression of another ABC family transporter, P-glycoprotein;24,42 however, we find that P-glycoprotein is not expressed to an appreciable extent by PEL cells (not shown). The BCRP promoter contains a CAAT box and Sp1-binding sites.43 Emmprin and LYVE-1 regulate signal transduction pathways23,44-47 that are known to regulate transcriptional activation through cooperative mechanisms involving CAAT box and Sp1 binding.48,49 However, the role of these pathways in transcriptional activation of BCRP requires confirmation. Our studies were not designed to address whether KSHV itself regulates expression of BCRP, emmprin or LYVE-1 in PEL cells. However, we previously reported data revealing that KSHV-encoded LANA induces expression of emmprin.50 Sp1 also induces transcriptional activation of emmprin,51 and LANA interacts directly with Sp1 to promote Sp1-mediated transcriptional activation of telomerase.52 Our future studies will address mechanisms for KSHV-initiated transcriptional activation of these proteins. Of note, our data suggest that neither emmprin nor LYVE-1 regulate expression of one another, although we hypothesize that these two proteins are functionally interdependent by virtue of their interactions.
We found that either oHA treatment or direct LYVE-1 silencing suppresses BCRP expression and enhances PEL cytotoxicity in the presence of chemotherapeutic agents. These data support the possibility that hyaluronan interactions with LYVE-1 on the PEL cell surface facilitate PEL chemoresistance through upregulation of BCRP expression. Although its function as a receptor for hyaluronan is well characterized,29 this is the first report to our knowledge implicating LYVE-1 in downstream regulation of a membrane transport protein important for chemotherapeutic resistance, and the first report detailing a mechanism for LYVE-1 regulation of KSHV-associated cancer pathogenesis despite the fact that LYVE-1 expression has been reported within Kaposi’s sarcoma lesions.32 Published studies implicate interactions between emmprin and the hyaluronan receptor CD44 in the induction of cancer cell chemo-resistance.23,53 In addition, oHAs disrupt emmprin–CD44 interactions17 as well as CD44-mediated intracellular signal transduction and cell pathogenesis relevant to cancer progression.18,20,23,24,28,54,55 However, we found that both total and membrane expression of CD44 are negligible for the PEL cell lines used in our studies (data not shown) in agreement with published results.33 Our data do not exclude the possibility that oHAs enhance PEL cytotoxicity through disruption of hyaluronan interactions with a receptor other than either CD44 or LYVE-1,56,57 or through other mechanisms.
We found that direct targeting of emmprin or LYVE-1 using RNAi, and treatment with oHAs, enhance chemotherapy-induced apoptosis for PEL cells. Given that none of these interventions induced apoptosis in the absence of cytotoxic agents, and that emmprin-enhanced viability for PEL cells was reduced by targeting BCRP, our data suggest that targeting emmprin or LYVE-1 augments chemotherapy-induced PEL apoptosis through inhibition of BCRP expression and drug efflux. This is supported by our observation that chemother-apeutic agents increase emmprin expression by PEL cells in a manner previously observed for other cancer cell types.70 Since emmprin stimulates hyaluronan synthesis,20 and the effect of emmprin on drug resistance is most likely mediated by hyaluronan-receptor interactions,23 it is likely that chemother-apeutic agents also stimulate hyaluronan–LYVE-1 signaling and that oHAs act by interfering with this signaling. In addition, we observed an increase in the number of PEL cells exhibiting intracellular accumulation of doxorubicin in the presence of oHAs, further supporting the conclusion that oHAs inhibit drug efflux through their effects on transporter expression as we and others have observed.18,21,22,24 Emmprin and LYVE-1 also activate signal transduction pathways, including mitogen-activated protein kinase, phosphatidylinositol 3-kinase/Akt and nuclear factor-κB,23,44-47 that regulate apoptosis.58-61 Constitutive activation of these pathways plays a pivotal role in anti-apoptotic signaling and PEL cell survival,62-65 and inhibition of these pathways induces PEL apoptosis.66-69 Therefore, we cannot categorically exclude the possibility that inhibition of emmprin or LYVE-1 also induces PEL apoptosis through interference with signal transduction.
We found that emmprin, LYVE-1 and BCRP colocalize and interact on the PEL cell surface. Recent reports suggest that emmprin interacts with CD44 (ref. 17) and P-glycoprotein,18,19 thereby facilitating drug efflux and resistance to chemotherapy. It is likely that emmprin and CD44 interact with several plasma membrane proteins within the context of lipid rafts rather than through direct binding to one another,55,71,72 and whether emmprin, LYVE-1 and BCRP interact in this manner on the PEL cell surface is currently under investigation. Moreover, oHAs inhibit drug efflux activity and sensitize tumor cells to chemotherapy through disruption of hyaluronan–CD44–drug transporter interactions and internalization of both CD44 and drug transporters18,21,24 in addition to their effects on transporter expression. We found that emmprin or LYVE-1 targeting with RNAi, or treatment with oHAs, reduced total BCRP expression in PEL cells. Using confocal immunofluorescence assays, we also observed a reduction of PEL membrane localization of BCRP with these interventions, but without coincident increases in cytoplasmic BCRP expression; however, these findings do not categorically exclude the possibility that BCRP is internalized and degraded as a result of emmprin or LYVE-1 targeting or oHA treatment. In addition, although we found that oHAs reduced co-immunoprecipitation of emmprin, LYVE-1 and BCRP, we cannot exclude the possibility that the observed reduction in BCRP protein expression with oHA treatment contributes to reduced quantitative interactions between these proteins at the cell surface. Additional experiments should clarify which of these mechanisms for emmprin/LYVE-1 regulation of BCRP play a key role in protecting PEL cells from apoptosis and cytotoxicity induced by chemotherapeutic agents.
In summary, this study provides the first description of cooperative interactions between emmprin, LYVE-1 and BCRP in regulating chemotherapeutic resistance, specifically for PEL. They also support the potential utility of targeting one or more of these intermediates as a therapeutic approach for PEL and other KSHV-associated diseases.
Supplementary Material
Acknowledgments
We thank Dr Dean Kedes (University of Virginia, Charlottesville, VA) and Dr Dirk Dittmer (University of North Carolina, Chapel Hill, NC) for providing PEL cell lines. This work was supported by grants from the National Institutes of Health (R01-CA142362 to CP; R01-CA073839 and R01-CA082867 to BPT), the South Carolina COBRE for Oral Health (P20-RR017696; CP subproject investigator) and the MUSC Hollings Cancer Center (core Grant P30-CA138313).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 2.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86:1276–1280. [PubMed] [Google Scholar]
- 3.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 4.Petre CE, Sin SH, Dittmer DP. Functional p53 signaling in Kaposi’s sarcoma-associated herpesvirus lymphomas: implications for therapy. J Virol. 2007;81:1912–1922. doi: 10.1128/JVI.01757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simonelli C, Spina M, Cinelli R, Talamini R, Tedeschi R, Gloghini A, et al. Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: a single-institution study. J Clin Oncol. 2003;21:3948–3954. doi: 10.1200/JCO.2003.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Boulanger E, Gerard L, Gabarre J, Molina JM, Rapp C, Abino JF, et al. Prognostic factors and outcome of human herpesvirus 8-associated primary effusion lymphoma in patients with AIDS. J Clin Oncol. 2005;23:4372–4380. doi: 10.1200/JCO.2005.07.084. [DOI] [PubMed] [Google Scholar]
- 7.Chen YB, Rahemtullah A, Hochberg E. Primary effusion lymphoma. Oncologist. 2007;12:569–576. doi: 10.1634/theoncologist.12-5-569. [DOI] [PubMed] [Google Scholar]
- 8.Oksenhendler E, Clauvel JP, Jouveshomme S, Davi F, Mansour G. Complete remission of a primary effusion lymphoma with antiretroviral therapy. Am J Hematol. 1998;57:266. doi: 10.1002/(sici)1096-8652(199803)57:3<266::aid-ajh25>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 9.Hocqueloux L, Agbalika F, Oksenhendler E, Molina JM. Long-term remission of an AIDS-related primary effusion lymphoma with antiviral therapy. AIDS. 2001;15:280–282. doi: 10.1097/00002030-200101260-00023. [DOI] [PubMed] [Google Scholar]
- 10.Lim ST, Rubin N, Said J, Levine AM. Primary effusion lymphoma: successful treatment with highly active antiretroviral therapy and rituximab. Ann Hematol. 2005;84:551–552. doi: 10.1007/s00277-005-1040-6. [DOI] [PubMed] [Google Scholar]
- 11.Munoz-Fontela C, Marcos-Villar L, Hernandez F, Gallego P, Rodriguez E, Arroyo J, et al. Induction of paclitaxel resistance by the Kaposi’s sarcoma-associated herpesvirus latent protein LANA2. J Virol. 2008;82:1518–1525. doi: 10.1128/JVI.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biswas C, Zhang Y, DeCastro R, Guo H, Nakamura T, Kataoka H, et al. The human tumor cell-derived collagenase stimulatory factor (renamed EMMPRIN) is a member of the immunoglobulin superfamily. Cancer Res. 1995;55:434–439. [PubMed] [Google Scholar]
- 13.Guo H, Zucker S, Gordon MK, Toole BP, Biswas C. Stimulation of matrix metalloproteinase production by recombinant extracellular matrix metalloproteinase inducer from transfected Chinese hamster ovary cells. J Biol Chem. 1997;272:24–27. [PubMed] [Google Scholar]
- 14.Zucker S, Hymowitz M, Rollo EE, Mann R, Conner CE, Cao J, et al. Tumorigenic potential of extracellular matrix metalloproteinase inducer. Am J Pathol. 2001;158:1921–1928. doi: 10.1016/S0002-9440(10)64660-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirk P, Wilson MC, Heddle C, Brown MH, Barclay AN, Halestrap AP. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000;19:3896–3904. doi: 10.1093/emboj/19.15.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallagher SM, Castorino JJ, Wang D, Philp NJ. Monocarboxylate transporter 4 regulates maturation and trafficking of CD147 to the plasma membrane in the metastatic breast cancer cell line MDA-MB-231. Cancer Res. 2007;67:4182–4189. doi: 10.1158/0008-5472.CAN-06-3184. [DOI] [PubMed] [Google Scholar]
- 17.Slomiany MG, Grass GD, Robertson AD, Yang XY, Maria BL, Beeson C, et al. Hyaluronan, CD44, and emmprin regulate lactate efflux and membrane localization of monocarboxylate transporters in human breast carcinoma cells. Cancer Res. 2009;69:1293–1301. doi: 10.1158/0008-5472.CAN-08-2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slomiany MG, Dai L, Tolliver LB, Grass GD, Zeng Y, Toole BP. Inhibition of functional hyaluronan-CD44 interactions in CD133-positive primary human ovarian carcinoma cells by small hyaluronan oligosaccharides. Clin Cancer Res. 2009;15:7593–7601. doi: 10.1158/1078-0432.CCR-09-2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang WJ, Li QQ, Xu JD, Cao XX, Li HX, Tang F, et al. Interaction between CD147 and P-glycoprotein and their regulation by ubiquitination in breast cancer cells. Chemotherapy. 2008;54:291–301. doi: 10.1159/000151225. [DOI] [PubMed] [Google Scholar]
- 20.Marieb EA, Zoltan-Jones A, Li R, Misra S, Ghatak S, Cao J, et al. Emmprin promotes anchorage-independent growth in human mammary carcinoma cells by stimulating hyaluronan production. Cancer Res. 2004;64:1229–1232. doi: 10.1158/0008-5472.can-03-2832. [DOI] [PubMed] [Google Scholar]
- 21.Slomiany MG, Dai L, Bomar PA, Knackstedt TJ, Kranc DA, Tolliver L, et al. Abrogating drug resistance in malignant peripheral nerve sheath tumors by disrupting hyaluronan-CD44 interactions with small hyaluronan oligosaccharides. Cancer Res. 2009;69:4992–4998. doi: 10.1158/0008-5472.CAN-09-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilg AG, Tye SL, Tolliver LB, Wheeler WG, Visconti RP, Duncan JD, et al. Targeting hyaluronan interactions in malignant gliomas and their drug-resistant multipotent progenitors. Clin Cancer Res. 2008;14:1804–1813. doi: 10.1158/1078-0432.CCR-07-1228. [DOI] [PubMed] [Google Scholar]
- 23.Misra S, Ghatak S, Zoltan-Jones A, Toole BP. Regulation of multidrug resistance in cancer cells by hyaluronan. J Biol Chem. 2003;278:25285–25288. doi: 10.1074/jbc.C300173200. [DOI] [PubMed] [Google Scholar]
- 24.Misra S, Ghatak S, Toole BP. Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J Biol Chem. 2005;280:20310–20315. doi: 10.1074/jbc.M500737200. [DOI] [PubMed] [Google Scholar]
- 25.Torre C, Wang SJ, Xia W, Bourguignon LY. Reduction of hyaluronan-CD44-mediated growth, migration, and cisplatin resistance in head and neck cancer due to inhibition of Rho kinase and PI-3 kinase signaling. Arch Otolaryngol Head Neck Surg. 2010;136:493–501. doi: 10.1001/archoto.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lesley J, Hascall VC, Tammi M, Hyman R. Hyaluronan binding by cell surface CD44. J Biol Chem. 2000;275:26967–26975. doi: 10.1074/jbc.M002527200. [DOI] [PubMed] [Google Scholar]
- 27.Underhill CB, Chi-Rosso G, Toole BP. Effects of detergent solubilization on the hyaluronate-binding protein from membranes of simian virus 40-transformed 3T3 cells. J Biol Chem. 1983;258:8086–8091. [PubMed] [Google Scholar]
- 28.Cordo Russo RI, Garcia MG, Alaniz L, Blanco G, Alvarez E, Hajos SE. Hyaluronan oligosaccharides sensitize lymphoma resistant cell lines to vincristine by modulating P-glycoprotein activity and PI3K/Akt pathway. Int J Cancer. 2008;122:1012–1018. doi: 10.1002/ijc.23122. [DOI] [PubMed] [Google Scholar]
- 29.Jackson DG. Immunological functions of hyaluronan and its receptors in the lymphatics. Immunol Rev. 2009;230:216–231. doi: 10.1111/j.1600-065X.2009.00803.x. [DOI] [PubMed] [Google Scholar]
- 30.Carroll PA, Brazeau E, Lagunoff M. Kaposi’s sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology. 2004;328:7–18. doi: 10.1016/j.virol.2004.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.An FQ, Folarin HM, Compitello N, Roth J, Gerson SL, McCrae KR, et al. Long-term-infected telomerase-immortalized endothelial cells: a model for Kaposi’s sarcoma-associated herpesvirus latency in vitro and in vivo. J Virol. 2006;80:4833–4846. doi: 10.1128/JVI.80.10.4833-4846.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pyakurel P, Pak F, Mwakigonja AR, Kaaya E, Heiden T, Biberfeld P. Lymphatic and vascular origin of Kaposi’s sarcoma spindle cells during tumor development. Int J Cancer. 2006;119:1262–1267. doi: 10.1002/ijc.21969. [DOI] [PubMed] [Google Scholar]
- 33.Boshoff C, Gao SJ, Healy LE, Matthews S, Thomas AJ, Coignet L, et al. Establishing a KSHV+ cell line (BCP-1) from peripheral blood and characterizing its growth in Nod/SCID mice. Blood. 1998;91:1671–1679. [PubMed] [Google Scholar]
- 34.Qin Z, Freitas E, Sullivan R, Mohan S, Bacelieri R, Branch D, et al. Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS Pathog. 2010;6:e1000742. doi: 10.1371/journal.ppat.1000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mellor HR, Callaghan R. Accumulation and distribution of doxorubicin in tumour spheroids: the influence of acidity and expression of P-glycoprotein. Cancer Chemother Pharmacol. 2011 doi: 10.1007/s00280-011-1598-8. e-pub ahead of print 15 March 2011. [DOI] [PubMed] [Google Scholar]
- 36.Li R, Huang L, Guo H, Toole BP. Basigin (murine EMMPRIN) stimulates matrix metalloproteinase production by fibroblasts. J Cell Physiol. 2001;186:371–379. doi: 10.1002/1097-4652(2000)9999:999<000::AID-JCP1042>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 37.Gordon LB, Harten IA, Calabro A, Sugumaran G, Csoka AB, Brown WT, et al. Hyaluronan is not elevated in urine or serum in Hutchinson-Gilford Progeria Syndrome. Hum Genet. 2003;113:178–187. doi: 10.1007/s00439-003-0958-9. [DOI] [PubMed] [Google Scholar]
- 38.Tang W, Chang SB, Hemler ME. Links between CD147 function, glycosylation, and caveolin-1. Mol Biol Cell. 2004;15:4043–4050. doi: 10.1091/mbc.E04-05-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belton RJ, Jr, Chen L, Mesquita FS, Nowak RA. Basigin-2 is a cell surface receptor for soluble basigin ligand. J Biol Chem. 2008;283:17805–17814. doi: 10.1074/jbc.M801876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang YF, Chen CY, Chung SF, Chiou YH, Lo HR. Involvement of oxidative stress and caspase activation in paclitaxel-induced apoptosis of primary effusion lymphoma cells. Cancer Chemother Pharmacol. 2004;54:322–330. doi: 10.1007/s00280-004-0831-0. [DOI] [PubMed] [Google Scholar]
- 41.Alaniz L, Garcia MG, Gallo-Rodriguez C, Agusti R, Sterin-Speziale N, Hajos SE, et al. Hyaluronan oligosaccharides induce cell death through PI3-K/Akt pathway independently of NF-kappaB transcription factor. Glycobiology. 2006;16:359–367. doi: 10.1093/glycob/cwj085. [DOI] [PubMed] [Google Scholar]
- 42.Bourguignon LY, Xia W, Wong G. Hyaluronan-mediated CD44 interaction with p300 and SIRT1 regulates beta-catenin signaling and NFkappaB-specific transcription activity leading to MDR1 and Bcl-xL gene expression and chemoresistance in breast tumor cells. J Biol Chem. 2009;284:2657–2671. doi: 10.1074/jbc.M806708200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–7358. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 44.Venkatesan B, Valente AJ, Prabhu SD, Shanmugam P, Delafontaine P, Chandrasekar B. EMMPRIN activates multiple transcription factors in cardiomyocytes, and induces interleukin-18 expression via Rac1-dependent PI3K/Akt/IKK/NF-kappaB andMKK7/JNK/AP-1 signaling. J Mol Cell Cardiol. 2010;49:655–663. doi: 10.1016/j.yjmcc.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang Y, Nakada MT, Rafferty P, Laraio J, McCabe FL, Millar H, et al. Regulation of vascular endothelial growth factor expression by EMMPRIN via the PI3K-Akt signaling pathway. Mol Cancer Res. 2006;4:371–377. doi: 10.1158/1541-7786.MCR-06-0042. [DOI] [PubMed] [Google Scholar]
- 46.Huang Z, Wang C, Wei L, Wang J, Fan Y, Wang L, et al. Resveratrol inhibits EMMPRIN expression via P38 and ERK1/2 pathways in PMA-induced THP-1 cells. Biochem Biophys Res Commun. 2008;374:517–521. doi: 10.1016/j.bbrc.2008.07.058. [DOI] [PubMed] [Google Scholar]
- 47.Saban MR, Memet S, Jackson DG, Ash J, Roig AA, Israel A, et al. Visualization of lymphatic vessels through NF-kappaB activity. Blood. 2004;104:3228–3230. doi: 10.1182/blood-2004-04-1428. [DOI] [PubMed] [Google Scholar]
- 48.Benjamin JT, Carver BJ, Plosa EJ, Yamamoto Y, Miller JD, Liu JH, et al. NF-kappaB activation limits airway branching through inhibition of Sp1-mediated fibroblast growth factor-10 expression. J Immunol. 2010;185:4896–4903. doi: 10.4049/jimmunol.1001857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stein B, Cogswell PC, Baldwin AS., Jr Functional and physical associations between NF-kappa B and C/EBP family members: a Rel domain-bZIP interaction. Mol Cell Biol. 1993;13:3964–3974. doi: 10.1128/mcb.13.7.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qin Z, Dai L, Slomiany MG, Toole BP, Parsons C. Direct activation of emmprin and associated pathogenesis by an oncogenic herpesvirus. Cancer Res. 2010;70:3884–3889. doi: 10.1158/0008-5472.CAN-09-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kong LM, Liao CG, Fei F, Guo X, Xing JL, Chen ZN. Transcription factor Sp1 regulates expression of cancer-associated molecule CD147 in human lung cancer. Cancer Sci. 2010;101:1463–1470. doi: 10.1111/j.1349-7006.2010.01554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verma SC, Borah S, Robertson ES. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J Virol. 2004;78:10348–10359. doi: 10.1128/JVI.78.19.10348-10359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toole BP, Slomiany MG. Hyaluronan, CD44 and Emmprin: partners in cancer cell chemoresistance. Drug Resist Updat. 2008;11:110–121. doi: 10.1016/j.drup.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghatak S, Misra S, Toole BP. Hyaluronan oligosaccharides inhibit anchorage-independent growth of tumor cells by suppressing the phosphoinositide 3-kinase/Akt cell survival pathway. J Biol Chem. 2002;277:38013–38020. doi: 10.1074/jbc.M202404200. [DOI] [PubMed] [Google Scholar]
- 55.Ghatak S, Misra S, Toole BP. Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J Biol Chem. 2005;280:8875–8883. doi: 10.1074/jbc.M410882200. [DOI] [PubMed] [Google Scholar]
- 56.Zhou B, Weigel JA, Fauss L, Weigel PH. Identification of the hyaluronan receptor for endocytosis (HARE) J Biol Chem. 2000;275:37733–37741. doi: 10.1074/jbc.M003030200. [DOI] [PubMed] [Google Scholar]
- 57.Hamilton SR, Fard SF, Paiwand FF, Tolg C, Veiseh M, Wang C, et al. The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J Biol Chem. 2007;282:16667–16680. doi: 10.1074/jbc.M702078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol. 2010;661:3–38. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- 59.Stiles BL. PI-3-K and AKT: onto the mitochondria. Adv Drug Deliv Rev. 2009;61:1276–1282. doi: 10.1016/j.addr.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 60.Kawauchi K, Ogasawara T, Yasuyama M, Otsuka K, Yamada O. The PI3K/Akt pathway as a target in the treatment of hematologic malignancies. Anticancer Agents Med Chem. 2009;9:550–559. doi: 10.2174/187152009788451851. [DOI] [PubMed] [Google Scholar]
- 61.Shen HM, Tergaonkar V. NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis. 2009;14:348–363. doi: 10.1007/s10495-009-0315-0. [DOI] [PubMed] [Google Scholar]
- 62.Ford PW, Bryan BA, Dyson OF, Weidner DA, Chintalgattu V, Akula SM. Raf/MEK/ERK signalling triggers reactivation of Kaposi’s sarcoma-associated herpesvirus latency. J Gen Virol. 2006;87(Part 5):1139–1144. doi: 10.1099/vir.0.81628-0. [DOI] [PubMed] [Google Scholar]
- 63.Tomlinson CC, Damania B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J Virol. 2004;78:1918–1927. doi: 10.1128/JVI.78.4.1918-1927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cannon ML, Cesarman E. The KSHV G protein-coupled receptor signals via multiple pathways to induce transcription factor activation in primary effusion lymphoma cells. Oncogene. 2004;23:514–523. doi: 10.1038/sj.onc.1207021. [DOI] [PubMed] [Google Scholar]
- 65.Guasparri I, Keller SA, Cesarman E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med. 2004;199:993–1003. doi: 10.1084/jem.20031467. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Sin SH, Roy D, Wang L, Staudt MR, Fakhari FD, Patel DD, et al. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood. 2007;109:2165–2173. doi: 10.1182/blood-2006-06-028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Uddin S, Hussain AR, Al-Hussein KA, Manogaran PS, Wickrema A, Gutierrez MI, et al. Inhibition of phosphatidylinositol 3′-kinase/AKT signaling promotes apoptosis of primary effusion lymphoma cells. Clin Cancer Res. 2005;11:3102–3108. doi: 10.1158/1078-0432.CCR-04-1857. [DOI] [PubMed] [Google Scholar]
- 68.Takahashi-Makise N, Suzu S, Hiyoshi M, Ohsugi T, Katano H, Umezawa K, et al. Biscoclaurine alkaloid cepharanthine inhibits the growth of primary effusion lymphoma in vitro and in vivo and induces apoptosis via suppression of the NF-kappaB pathway. Int J Cancer. 2009;125:1464–1472. doi: 10.1002/ijc.24521. [DOI] [PubMed] [Google Scholar]
- 69.Keller SA, Schattner EJ, Cesarman E. Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood. 2000;96:2537–2542. [PubMed] [Google Scholar]
- 70.Li QQ, Wang WJ, Xu JD, Cao XX, Chen Q, Yang JM, et al. Up-regulation of CD147 and matrix metalloproteinase-2, -9 induced by P-glycoprotein substrates in multidrug resistant breast cancer cells. Cancer Sci. 2007;98:1767–1774. doi: 10.1111/j.1349-7006.2007.00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bourguignon LY, Singleton PA, Diedrich F, Stern R, Gilad E. CD44 interaction with Na+-H+ exchanger (NHE1) creates acidic microenvironments leading to hyaluronidase-2 and cathepsin B activation and breast tumor cell invasion. J Biol Chem. 2004;279:26991–27007. doi: 10.1074/jbc.M311838200. [DOI] [PubMed] [Google Scholar]
- 72.Tang W, Hemler ME. Caveolin-1 regulates matrix metalloproteinases-1 induction and CD147/EMMPRIN cell surface clustering. J Biol Chem. 2004;279:11112–11118. doi: 10.1074/jbc.M312947200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.