

Abstract
Imprinted genes form a special subset of the genome, exhibiting monoallelic expression in a parent-of-origin–dependent fashion. This monoallelic expression is controlled by parental-specific epigenetic marks, which are established in gametogenesis and early embryonic development and are persistent in all somatic cells throughout life. We define this specific set of cis-acting epigenetic regulatory elements as the imprintome, a distinct and specially tasked subset of the epigenome. Imprintome elements contain DNA methylation and histone modifications that regulate monoallelic expression by affecting promoter accessibility, chromatin structure, and chromatin configuration. Understanding their regulation is critical because a significant proportion of human imprinted genes are implicated in complex diseases. Significant species variation in the repertoire of imprinted genes and their epigenetic regulation, however, will not allow model organisms solely to be used for this crucial purpose. Ultimately, only the human will suffice to accurately define the human imprintome.
Keywords: differential methylation, DNA methylation, histone modification, imprinted gene, imprintome, monoallelic expression
Introduction
Imprinted genes are a unique subset of genes that are characterized by monoallelic parent-of-origin–dependent transcription. The imprinted expression of these genes is critical for appropriate fetal growth and development, with regulation based on the establishment, maintenance, and interpretation of parentally derived epigenetic marks. The definition of the epigenome as the totality of epigenetic marks has been in use for more than 10 years and includes the DNA methylation and histone modifications responsible for regulating gene expression (Szyf and Slack 2000). The concept of the imprintome is more recent, having first appeared in print in 2009 (Jirtle 2009) and subsequently used with varying definitions (Cooper and Constancia 2010; Monk 2010). Our discussion of the human imprintome, its composition, and importance to human health is guided by our initial definition. As the term was first coined, the human imprintome is “the environmentally labile cis-acting imprint regulatory elements in the human genome.” Because imprinting is a direct consequence of epigenetic regulation, the imprintome should therefore be considered in terms of the epigenome rather than the genome or transcriptome.
The importance of understanding the imprintome comes from the recognition that the advent of imprinting represents a significant vertical progression in mammalian evolution. The imprintome is critical for development and growth, and understanding of many complex human diseases is improving markedly as the involvement of imprinted genes and imprint regulation is increasingly being established. It is important to distinguish between parent-of-origin–dependent monoallelic gene expression resulting from the imprintome and other genes that are monoallelically expressed but not in a parent-of-origin–dependent manner (Tycko 2010). Random parental monoallelic expression appears to function as a dosage-control mechanism (Gimelbrant et al. 2007) and employs epigenetic regulatory mechanisms that differ from those used by genomically imprinted genes (Luedi et al. 2007). Thus, the imprintome is distinguished from the remainder of the epigenome by its consistency and fidelity: it always exhibits parent-of-origin–specific epigenetic marks with very little spatial, temporal, or interindividual variability.
Although all epigenetic regulation that creates differential allelic expression has implications for development and disease, this review is specifically focused on the phenomenon of genomic imprinting and the imprintome. Current knowledge of the human imprintome will be discussed, including parental-specific imprintome establishment in the germline, the functions of imprint markings in regulating monoallelic expression, current methods of characterizing the imprintome, and the key reason for imprintome study—determinination of alteration in disease states.
Genomic Imprinting
Control of gene expression through shifts in DNA methylation and/or histone modification is common in eukaryotes, but parent-of-origin–specific transcriptional control has only been seen in flowering plants (Kohler and Weinhofer-Molisch 2010) and placental mammals (Killian et al. 2000). Dose control of certain genes is critical in mammals, exemplified by the observations that marsupials and eutherians both undergo X chromosome inactivation in females. There is even some degree of X-linked gene inactivation in the platypus, a nonplacental monotreme (egg-laying mammal) (Deakin et al. 2008). Nevertheless, parent-of-origin control of autosomal gene expression/repression appears to have occurred in concert with the origin of placentation. Imprinting is present not only in eutherian mammals but also in marsupials, which have placentas, albeit smaller ones because of the short intrauterine gestation. Analysis of orthologous imprinted genes in the egg-laying monotremes has thus far failed to identify any that are subject to imprinting, supporting the theory that genomic imprinting arose with the advent of mammalian placentation (Killian et al. 2000, 2001).
Evolution of Parental-Specific Gene Expression
One of the key unanswered questions about genomic imprinting is the reason that it first evolved. The conflict hypothesis of imprint evolution (Wilkins and Haig 2003) proposes that the amount of nutrients available to the fetus can vary because of interactions between the fetus and mother across the placenta, a situation that does not occur in oviparous development. This suggests that males and females can optimize their reproductive success by manipulating the placental resource—in effect, creating a competition between mother and fetus. A strategy that favors the paternal germline is to maximize fetal growth at the expense of the mother as well as offspring of other males. In contrast, the ideal maternal strategy to maximize reproductive success is to restrict growth in order to give an equal chance of survival to all offspring. Thus, a balance of resource utilization must be reached between mother and offspring so that neither benefits to the excessive detriment of the other.
According to the conflict hypothesis, imprinting accomplishes this by taking advantage of silencing methods that had already evolved to regulate gene expression; imprinting has adapted these methods to exploit compartmentalization of DNA in the gametes to establish parent-of-origin epigenetic marks that regulate fetal growth and development. Thus, paternally expressed/maternally silenced genes would tend to be growth promoting, whereas maternally expressed/paternally silenced genes would tend to be growth restrictive. A consequence of the differential epigenetic marking of the parental genomes is the potential for distinct syndromes or diseases whose manifestation depends on the parental origin of the genetic or epigenetic defect. As described below, nearly reciprocal phenotypes often result from such aberrations because of opposite dosage alterations of the same gene or genes.
Species-Specific Imprintomes
Understanding the human imprintome has become a greater necessity because of the demonstration of species-specific differences in imprinting establishment and regulation. Evidence that mouse and human imprinted gene repertoires show only about 30% overlap (Luedi et al. 2005, 2007) should be taken as a caveat for modeling any human system or complex disease that includes imprinted genes. Given the plasticity of the epigenome and, by extension, the imprintome, it can be inferred that epigenetic divergence between species is more rapid and extensive than genetic divergence. If epigenetic changes, specifically imprint regulatory changes, are a driving force in mammalian evolution and species divergence (Proudhon and Bourc’his 2010), then the relevance of any model organism's imprintome in understanding the human imprintome is diminished.
An even closer species comparison emphasizes this point. The divergence of rat and mouse from a common ancestor occurred in the same approximate time frame as the divergence of human and macaque (approximately 20 million years ago) (Springer et al. 2003). Nevertheless, the genetic divergence between mouse and rat is nearly double that between human and macaque (Gibbs et al. 2004), but mouse and rat have similar encephalization quotients (ratio of actual brain size to expected brain size based on body size), whereas human encephalization is at least 6 times that of macaque (Williams 2002). Given the comparative genetic stability between human and monkey, it has been postulated that epigenetics provides the adaptable variability responsible for this sort of difference between species (Keverne 2011).
Imprintome Establishment
The function of the imprintome depends on the physical mechanisms that establish, maintain, and interpret its elements. The initial marks are established during gametogenesis, the only time in which each parent has an independent influence on the imprintome and thus the capability to maximize effects on growth and development of their progeny. Following the merger of parental genomes during fertilization, subsequent epigenetic imprint marks can be established, which are also maintained in somatic cells. These somatic imprints retain parent-of-origin specificity because they are created based on the original imprint marks established in the gametes.
Imprintome creation involves both DNA methylation and histone modifications, with methylation being more conducive for study for a variety of reasons. DNA methylation analysis is more straightforward and definitive than identification of histone modifications, which requires indirect detection. Also, histones cannot be as precisely localized relative to DNA sequence and have complex combinations of modifications that may be difficult to distinguish. Possibly the best reason for studying DNA methylation is that it may be the most consistent marking of the imprint control regions (ICRs[1]) and the genes they regulate. Imprintome methylation established in the gametes and postfertilization is highly stable, regardless of gene expression level, and shows only occasional tissue-specific or intraindividual variability (Woodfine et al. 2011).
Creation of Imprint Marks
DNA methylation occurs at cytosines in CpG dinucleotides, producing 5-methylcytosine (5mC1). There are three known enzymes with DNA methyltransferase activity: DNMT1, DNMT3A, and DNMT3B (Miranda and Jones 2007). DNMT3A and DNMT3B catalyze de novo methylation—newly established methylation marks during gametogenesis and early development and aberrantly produced marks in carcinogenesis (Ooi et al. 2009). DNMT1, with its high preference for hemimethylated substrates, primarily maintains these methylation patterns in somatic cells during DNA replication (Bestor 2000). In mammals, 60–90% of CpG sites are methylated (Bird 1986), but in contrast, CpG islands, which have GC content greater than 55%, are typically hypomethylated compared with their CpG poor counterparts within intergenic and intronic regions (Takai and Jones 2002).
The mechanisms that specifically direct and establish the fundamental regulatory elements of the imprintome during gametogenesis are becoming much better understood. For DNA methylation, a heterodimer is formed between DNMT3A and the regulatory subunit DNMT3L, with two catalytic sites with spacing equivalent to dsDNA bases positioned one helical turn apart, about 8 to 12 base pairs (Jia et al. 2007). Based on this spacing, it was hypothesized that a bound DNTM3A/DNMT3L complex could simultaneously methylate two CpG sites. Support for this theory comes from observations that the methylation complex has preferential activity for CpG sites with 8– to 12–base pair spacing, a pattern that is present in a subset of imprinted regions but not in a sample set of CpG islands (Jia et al. 2007). It turns out that this specific subset of imprinted loci are methylated during oogenesis (i.e., are maternally imprinted), and this maternal specific methylation is disrupted by Dnmt3L mutations (Bourc’his et al. 2001).
Although specifically spaced CpGs are a preferred target of the de novo methylation machinery, their presence is not sufficient for methylation. Interestingly, investigations designed to determine sequence features defining imprinted genes and the differentially methylated regions (DMRs1) that regulate these genes found no strong sequence conservation. However, functionally orthologous DMRs in different species all have tandem repeats, but because the sequence motifs are highly divergent across species, it appears that it is not the specific sequences that are important, only repeat presence (Paulsen et al. 2001, 2005). Such repeats were also among the features found significant for predicting novel imprinted human genes, with the greatest importance attached to the presence of endogenous retroviruses (Luedi et al. 2007).
Observations such as these, along with evolutionary comparisons that show lack of repeat elements at the nonimprinted platypus orthologues of imprinted eutherian genes (Renfree et al. 2009), led to the host-defense or “parasite” theory for the origins of imprint marks (Labialle and Cavaille 2011). This hypothesis posits that methylation silencing originated as a mechanism to block the spread of transposable elements. This is not inconsistent with the parental conflict theory, which proposes adaptation of existing silencing mechanisms in the genesis of imprinted genes. The basis of imprinting could result from subsequent spreading of methylation from “parasite” sequences to nearby genes if a selective advantage was conferred (Barlow 1993; Ferguson-Smith and Surani 2001).
Another analysis comparing characteristics of known imprinted genes to autosomal genes found significant increases in intron length and intergenic distance for imprinted genes in the human but not in the mouse (Hutter et al. 2010). These results are consistent with longer intergenic regions having a proportionately greater probability of containing regulatory motifs. Another explanation is that the increased length is due to transposed elements and that these motifs are carried along with the transposons. This is another example of how epigenetic and imprintome differences may play significant roles in speciation and underscores that the results of imprintome studies may not translate well across species.
Gametic Imprint Methylation
Maternally and paternally established methylation marks in ICRs are distinct from each other. Along with the previously mentioned 8– to 12–base pair spacing between methylated CpG dinucleotides, paternally methylated ICRs often overlap CpG-rich promoter regions (Bourc’his and Bestor 2006). In contrast, maternally methylated ICRs tend to be located in intergenic regions with low CpG content (Bourc’his and Bestor 2006) and have increased conservation of noncoding sequences near exon–intron boundaries (Hutter et al. 2010). The ability of algorithms to use known imprinted genes as a training set and correctly predict maternal or paternal expression based on positions and frequencies of repeat elements (Luedi et al. 2007) also indicates sexual dimorphism in imprint regulation. Results such as these provide a starting point to determine the criteria by which parent-specific imprintome methylation is established.
Somatic Imprint Methylation
As previously discussed, methylation imprint marks are established independently by each parent during gametogenesis. After fertilization, both genomes are demethylated, but there are still parent-of-origin–specific effects. The paternal genome is actively demethylated (Mayer et al. 2000; Santos et al. 2002) by a process that begins with oxidation of 5mC to 5-hydroxymethylcytosine (5hmC1) (Wossidlo et al. 2011). In contrast, active demethylation of the maternal genome is blocked by the protective factor PGC7/Dppa3/Stella (Wossidlo et al. 2011), such that demethylation is passive because of nonrenewal of methylation after DNA replication. Methylation of gametic imprintome marks is preserved through this process and also by the binding of PGC7/Dppa3/Stella (Nakamura et al. 2007). Genomic methylation is reestablished after implantation, and it is also at this time that somatic imprints are created, dependent on the gametic marks. Like the gametic marks, the somatic imprint marks are maintained across cell types and throughout life, but consistency of somatic marks is not always as rigid as those established during gametogenesis (Woodfine et al. 2011). A possible reason for the creation of somatic imprintome elements is to impose coordinate regulation because many imprinted genes exist in clusters. In this way, one gametic mark regulating one transcript can lead to the establishment of multiple somatic marks that then regulate multiple genes over a wider genomic region.
Somatic DMRs are another area indicative of disparity between the mouse and human imprintomes, with several well-characterized mouse somatic marks not present in humans. In mouse, the maternally expressed genes Igf2r and Cdkn1c have somatic DMRs and corresponding paternally expressed noncoding RNAs (ncRNAs1)—Air and Kcnq1ot1, respectively. Allele-specific expression of the ncRNAs is controlled by gametic DMRs within Igf2r and Cdkn1c, and expression of Air and Kcnq1ot1 leads to the establishment of somatic DMRs in the promoters of the corresponding coding genes (Du et al. 2003; Sleutels et al. 2002). Once somatic DMRs are established, their maintenance does not require either the gametic DMR or ncRNA transcription (Green et al. 2007). In contrast with the mouse system, human IGF2R does have the gametic DMR but does not normally express an Air equivalent, correspondingly lacks the somatic DMR in the promoter region, and is biallelically expressed, except for sporadic monoallelic expression observed in placenta (Monk et al. 2006). Alternatively, the human ncRNA KCNQ1OT1 has expression and function comparable with mouse Kcnq1ot1, leading to monoalletic expression of CDKN1C from the maternal allele. However, there is still a species distinction because paternal KCNQ1OT1 expression in the human does not produce somatic methlyation at the CDKN1C promoter (Figure 1A) (Onyango et al. 2000). The mechanisms for monoalleleic expression of these genes, including the implications of DMR and ncRNA differences in humans, will be discussed below along with other mechanisms of controlling monoallelic expression.
Figure 1.

Mechanisms of monoallelic gene expression controlled by parent-of-origin imprintome elements. (A) Differential germline methylation controls expression of an antisense noncoding RNA that represses neighboring genes in an imprinted cluster. Example shown: CDKN1C/KCNQ1. Light oval, unmethylated imprint control region (ICR); dark oval, methylated ICR. (B) Differential methylation controls expression, and transcriptional interference affects promoter usage of oppositely transcribed genes. Example shown: NNAT/BLCAP. Light oval, unmethylated ICR; dark oval, methylated ICR; p1, efficient promoter, used in maternal monoallelic expression; p2, inefficient promoter, used in paternal monoallelic expression. (C) Parent-of-origin–specific methylation prevents CTCF binding and cohesion-mediated chromatin remodeling. Differently arranged chromatin loops allow or prevent promoter–enhancer interaction. Example shown: IGF2/H19/HOTS. Light oval, unmethylated ICR; dark oval, methylated ICR; filled circles, H19 promoter methylation.
There are other human imprinted genes known to have associated somatic DMRs. Maternally methylated somatic DMRs are present at MEST, GRB10, and DIRAS3, and paternally established somatic DMRs are associated with IGF2, MEG3, and DLK1 (Woodfine et al. 2011). Comparisons in different tissue types show that although methylation of the paternal DMRs is mostly consistent, the maternal DMRs have more variable methylation across tissue types (Woodfine et al. 2011). Although the somatic DMRs are parental allele specific, established in a gametic methylation-dependent manner, and are in clusters including ncRNAs or antisense transcripts, little is known about the mechanisms by which gametic imprints are translated into somatic DNA marks (John and Lefebvre 2011).
Interactions of DNA Methylation with Histone Modifications
Another important component of the imprintome is histone modification, a key regulator of “open” and “closed” chromatin structure and thus gene expression. Although histone modifications include methylation, acetylation, phosphorylation, sumoylation, and ubiquitilation (Talbert and Henikoff 2010), a particular group of histone modifications with high relevance to gene regulation has been characterized (Izzo and Schneider 2010; Sakabe and Nobrega 2010; Shu et al. 2011). Relevant modifications include methylation and acetylation, which have been most intensively examined for their role in controlling epigenetic programming. Importantly, histone modifications do not normally function independently from DNA methylation.
Methylated DNA is typically associated with chromatin that has deacetylated histones H3 and H4 (Kacem and Feil 2009) and methylated lysine or arginine, principally H3K9, H4K20, and H2A/H4R3 (Henckel et al. 2009; Pannetier et al. 2008). This association is primarily because of the methyl-CpG-binding protein MeCP2, which recruits both histone deacetylases (Jones et al. 1998; Nan et al. 1998) and histone methyltransferase (Fuks et al. 2003) (Figure 2). This histone modification pattern promotes closed chromatin and suppresses gene transcription. Conversely, unmethylated DMRs have characteristic histone acetylation and H3K4 methylation, which results in open chromatin and enhanced gene transcription. Interestingly, whereas DNA methylation has an active role in creating closed chromatin, unmethylated DNA may regulate open chromatin by inaction, by not recruiting histone deacetylase and histone methyltransferase complexes.
Figure 2.

Interactions of DNA methylation and histone modifications. (A) Closed chromatin has CpG methylation (star) generated by DNA methyltransferase (DNMT) and histone methylation at H3K9, H4K20, and H2A/H4R3, with recruitment of histone deacetylase (HDAC) and methyltransferase (HMT) by methylated CpG by means of MeCP2. Transcription factors (TFs) are inhibited from binding, and gene expression is silenced. (B) Open, unmethylated chromatin does not recruit HDAC and HMT, and H4K3 methylation blocks DNMT binding. TFs bind unmethylated chromatin, recruiting histone acetyltransferase (HAT), which promotes transcriptional activation.
There is also a growing body of work characterizing 5hmC and its role in active DNA demethylation. It is proposed that 5hmC is an intermediate product of demethylation, the result of oxidation by Tet3 (Iqbal et al. 2011). 5hmC is then converted back to cytosine by deamination by AID, which is followed by thymine DNA glycosylase excision repair (Cortellino et al. 2011). This mechanism could then keep chromatin open, again by removing the substrate for histone deacetylase and histone methyltransferase.
The interactions also operate in the opposite direction, as histone modifications and chromatin structure can control de novo DNA methylation. H3K4 trimethylation (H3K4me3) is found in active promoter regions and can block DNA methylation in a locus-specific manner (Ikegami et al. 2009). The DNMT3L/DNMT3A complex that is required for de novo CpG methylation specifically binds the amino terminus of histone H3, but the trimethylation of H3K4 blocks binding of the methylation complex. The requirement of appropriately modified chromatin for de novo methylation (Ooi et al. 2007; Otani et al. 2009) means that DNA methylation and chromatin modification could be a cyclical process. This circular system could control the targeting for imprintome element establishment, with feedback maintaining both fidelity and errors across generations.
One direct demonstration of these correlations in imprinted genes comes from a study of chromatin in sperm and the mapping of paternal marks. Multiple imprinted transcripts with paternal expression, such as MEST and PEG3, and the antisense ncRNAs MESTIT1, KCNQOT1, and GNASAS show both unmethylated DNA and high levels of H3K4me3. In contrast, paternally silenced genes are methylated in sperm and have high levels of H3K9me3, a mark not present at paternally expressed genes (Hammoud et al. 2009). Finally, although maternally expressed MEG3 is unmethylated in sperm, it has both active and silencing marks—H3K4me3 and H3K27me3, respectively. Subsequently, in the embryo, MEG3 loses the activating H3K4 methylation concurrent with gaining CpG methylation (da Rocha et al. 2008; Glazov et al. 2008; Hammoud et al. 2009).
In a system specific to sperm, CTCFL (CTCF binding protein-like, also named BORIS), a protein normally expressed in testes, binds DNA at CTCF motifs (Loukinov et al. 2002) and targets both paternal-specific DNA methylation and histone modification by recruiting the protein arginine methyltransferase PRMT7, as well as DNMT3L, DNMT3A, and DNMT3B (Jelinic et al. 2006). This results in the sequence-specific establishment of DNA methylation in conjunction with H4R3 methylation.
Imprintome Regulation of Imprinted Gene Expression
Regulation by ncRNA
Several distinct methods of using imprintome elements to regulate monoallelic expression exist, as reviewed by Lewis and Reik (2006). One is the antisense RNA mechanism exemplified by mouse Igf2r (Sleutels et al. 2002) and mouse and human CDKN1C (Mancini-Dinardo et al. 2006) (Figure 1A). Multiple imprinted gene clusters use this mechanism of gene control in which imprinted expression of one ncRNA controls expression of other imprinted genes through somatic establishment of DNA methylation and repressive chromatin structure (Koerner et al. 2009).
As previously mentioned, mouse Igf2r and human IGF2R do not have identical ncRNA regulatory systems. No human equivalent to Air has been observed in normal somatic cells, and there is no AIR-dependent somatic DMR in the IGF2R promoter, consistent with the lack of IGF2R imprinting (Vu et al. 2006). A human AIR transcript has been detected in some Wilms’ tumors, using the IGF2R intronic DMR as a promoter, and this human AIR shows conservation of regulatory sequences with mouse Air (Yotova et al. 2008). Therefore, human AIR may function in sporadic IGF2R imprinting, with only very specific spatial and temporal expression.
Another ncRNA example in human is KCNQ1OT1, which is maternally silenced by gametic DNA methylation (Figure 1A). Expression of the paternal allele functions in cis to establish imprintome elements regulating other genes in the KCNQ1 imprinted cluster (Mancini-Dinardo et al. 2006), which is the largest known imprinted gene cluster. This cluster shows significant species differences in imprinted gene regulation; of the 14 genes in this cluster, six are ubiquitously imprinted in mouse and humans, whereas the other eight are imprinted only in mouse placenta (Frost et al. 2010). Paternal KCNQ1OT1 expression inhibits the paternal copy of the growth-regulating gene CDKN1C, which results in maternal-specific expression beyond the immediate area of KCNQ1 (Figure 1A) (Koerner et al. 2009). Mouse studies have shown that Dnmt1 deletion results in expression of the normally silenced maternal Kcnq1ot1 allele and subsequent silencing of all 14 imprinted genes on the maternal allele (Green et al. 2007). Conversely, deletions of paternal Kcnq1ot1 abolish imprint regulation of the six ubiquitously imprinted genes but not the genes with placental-specific imprinting (Mohammad et al. 2010). The similar effects of deletion of either Dnmt1 or Kcnq1ot1 on methylation and monoalleleic expression of the same genes was explained by determination that the key regulatory region of Kcnq1ot1 is a silencing domain that recruits Dnmt1 to somatic DMRs (Mohammad et al. 2010). Because human CDKN1C does not have a somatic DMR comparable with mouse Cdkn1c, how KCNQ1OT1 functions in the silencing mechanism for CDKN1C is unknown. Therefore, if the human KCNQ1OT1 transcript also functions by recruitment of DNMT1, as in the mouse system, any somatic methylation has yet to be determined.
Regulation by Other Imprinted Genes
The imprinted gene ZAC is also involved in regulating the CDKN1C cluster by acting directly on KCNQ1OT1. Paternally expressed ZAC encodes a transcription factor that binds the unmethylated (paternal) KCNQ1OT1 promoter, activating expression and subsequently downregulating CDKN1C (Arima et al. 2005). Therefore, there is a pathway regulating genes in this imprinted cluster that uses these two paternally expressed transcripts, with one enhancing the other.
Neuronatin (NNAT) is similarly imprinted in both mouse and human with maternally established methylation and paternal expression (Evans et al. 2001). NNAT is contained within the sole intron of the tumor supressor gene BLCAP, in an antisense orientiation (Figure 1B), and its expression seems to regulate monoallelic expression of BLCAP. BLCAP is usually biallelically expressed and is only known to be monoallelically expressed in the brain, possibly because of the very high level of NNAT transcription in the brain (Schulz et al. 2009). BLCAP has two transcription start sites, one each upstream and downstream of NNAT. Paternally expressed NNAT transcribes through the downstream start site and promoter of BLCAP, presumably silencing it by transcriptional interference (Shearwin et al. 2005). Consequently, when BLCAP is imprinted, there are actually two imprinted transcripts, one expressed maternally from the downstream promoter, and the other expressed paternally from the upstream promoter (Schulz et al. 2009).
Regulation by Chromatin Conformation
A second method for regulating monoallelic gene expression uses methylation to alter a “handle” used to create long-distance DNA interactions that bring promoters and enhancers together. The reciprocally imprinted IGF2–H19 transcript pair (Figure 1C) is a well-defined example of this system and is controlled by paternal gametic methylation, unlike the previous examples. IGF2 is expressed from the paternal allele, whereas the ncRNA H19 is expressed from the maternal allele; however, the silencing mechanism is different from the opposite parental allelic transcription of Igf2r and Air. A primary difference was seen in mouse studies, which showed that transcription of the noncoding H19 does not have a role in repression of Igf2 (Schmidt et al. 1999).
A paternally methylated ICR is 2 kb upstream of the noncoding transcript H19, amid a series of six binding sites for the transcription factor CTCF. This ICR acts as a regulator of both H19 and IGF2 but through independent mechanisms. One mechanism has gametic ICR methylation that leads to paternal somatic methylation in the H19 promoter, which silences H19, and silencing is maintained even after deletion of the ICR in somatic cells (Srivastava et al. 2000). Only recently, a novel coding gene, HOTS, which overlaps H19 in an antisense orientation, has been identified (Onyango and Feinberg 2011). HOTS is monoallelically expressed from the maternal allele, like H19 (Figure 1C), and observed loss of imprinting of IGF2 is associated with loss of expression for both H19 and HOTS, indicating that HOTS is regulated by the same mechanisms as H19 (Onyango and Feinberg 2011). The fact that HOTS is a coding gene with apparent tumor suppressor activity (Onyango and Feinberg 2011), whereas H19 is a noncoding transcript, suggests that HOTS is the true tumor suppressor of the H19 locus (Yoshimizu et al. 2008). It can then be hypothesized that it is the HOTS transcript that is the true target of the regulatory imprintome marks and thus the feature behind evolution of imprintome features and monoallelic expression of the locus.
IGF2 expression is controlled by a second mechanism involving ICR methylation, which blocks CTCF binding so that only the unmethylated maternal ICR is accessible. CTCF binding of the maternal allele acts as an insulator, separating IGF2 from a downstream enhancer (Figure 1C) (Bell and Felsenfeld 2000; Hark et al. 2000; Szabo et al. 2000). Insulation by CTCF binding also requires colocalization of cohesin, which mediates sister chromatid cohesion but also has roles in gene regulation in organisms from yeast to human (Dorsett 2011). The requirement for cohesin indicates that the insulation is not a linear DNA roadblock, but rather is due to altered higher-order chromatin structure mediated by CTCF binding (Nativio et al. 2009; Wallace and Felsenfeld 2007).
Examination of the role of CTCF and cohesin in chromatin structure and looping at the human IGF2–H19 locus has identified allelic differences in CTCF–cohesin interactions with chromatin and, subsequently, allele-specific chromatin looping (Nativio et al. 2009). There are three biallelic CTCF–cohesin complexes across this region that interact on both alleles, and allele-specific interaction of CTCF–cohesin bound to the maternal allele is proposed to control interaction of IGF2 and the enhancer. The enhancer and IGF2 promoter are in contact on only one allele, presumed to be the methylated paternal allele that expresses IGF2. Conversely, interaction between the ICR and a CTCF–cohesin binding site downstream of H19 also occurs in only one allele, presumably maternal (Figure 1C). A proposed model is that multiple CTCF–cohesin complexes come together in a chromatin “hub” and that maternal allele ICR–H19 binding creates a specific loop that separates IGF2 from the enhancer (Nativio et al. 2009).
Taken together, these findings describe a complex system in which DNA methylation is responsible for repression of paternal H19 (and presumably HOTS), most likely through closed chromatin structure. At the same time, paternal IGF2 expression requires interaction between the promoter and a remote enhancer. This interaction is preserved by gametic DNA methylation that blocks CTCF–cohesin binding and formation of an alternate chromatin loop. Therefore, one parentally established DMR is the initiator of a system that ultimately controls reciprocal imprinted expression of multiple transcripts by two mechanisms. One mechanism uses promoter repression; the other uses methylation-mediated chromatin conformation. Once established, neither system appears to affect regulation of the other's target.
Complex hybrid mechanistic imprinting systems may also function in regulation of the CDKN1C cluster and explain imprint regulation in the absence of a somatic DMR at the CDKN1C promoter in human. Silencer sequences have been found in both the kvDMR at the KCNQ1OT1 promoter and in CDKN1C, and CTCF binding has been detected for these sites (Du et al. 2003). This raises the possibility that CDKN1C cluster regulation also includes both ncRNA-mediated somatic imprintome marking and allele-specific chromatin conformation.
Human chromosome 14q32.2 contains an imprinting cluster with some similarities to IGF2–H19 because both imprinted domains contain paternally expressed genes (DLK1 and RTL1, and IGF2, respectively) and maternally expressed genes (MEG3 and MEG8, and H19, respectively). There are multiple maternally expressed small nucleolar RNAs and microRNAs in the 14q32.2 cluster. Two known DMRs reside in this cluster: IG-DMR, an intergenic DMR of gametic origin, and the somatic MEG3-DMR. IG-DMR methylation determines the MEG3-DMR methylation state, which appears responsible for parent-of-origin–specific expression of multiple genes in this cluster (Kagami et al. 2010). How MEG3-DMR methylation controls imprinting is not as well understood as for other regions, but CTCF and cohesin are involved (Kernohan et al. 2010). Methylation-dependent interactions of CTCF with the mouse equivalent of the MEG3-DMR have been identified, with CTCF binding preferentially to the unmethylated allele, similar to the IGF2–H19 locus (Lin et al. 2011). Thus, chromatin conformation may be involved in imprinting regulation of this gene cluster as it is for IGF2-H19.
Regulation by Intragenic Methylation
The tumor suppressor gene RB1 was only recently found to be imprinted. Imprinting at this locus is regulated by a differentially methylated CpG island in intron 2 that does not exist in the nonimprinted mouse homologue (Kanber et al. 2009). Also, unlike clustered imprinted gene regulation, RB1 imprinted expression appears to be entirely dependent on this DMR, with no ncRNA involvement. The CpG island is derived from a retrotransposed pseudogene and exists in both primates and New World monkeys but not in mice or rats (Buiting et al. 2010), which means RB1 imprinting is a much more recent event than imprinting of the previously described genes. This CpG island has created an alternate RB1 promoter that is silent when methylated on the maternal allele. It is hypothesized that transcriptional interference (Shearwin et al. 2005), as mentioned with NNAT and BLCAP regulation, is involved, with transcription from the intronic promoter blocking usage of the full-length promoter (Figure 1B).
Transposed Imprinted Genes
Finally, a study of imprinted retrotransposed genes that exist in both human and mouse shows yet another pattern of regulation, that of the isolated imprinted gene. A number of imprinted retrogenes that have transposed into biallelically expressed genes have DMRs, and imprint regulation does not extend beyond the retrogene (Monk et al. 2011). Histone analysis of two such human genes, NAPIL5 and MCTS2, found allele specificity of the activating mark H3K4me2 in both the human and mouse orthologues, but the mouse chromatin marks are bivalent, with repressive histone marks that the human genes lack (Monk et al. 2011). The human DMRs are significantly shorter than the mouse DMRs and have lost approximately half of the CpG sites. This comparison is in contrast with other human imprinted genes, which usually have longer DMRs than the mouse counterparts. It is suggested that these shorter human DMRs are not recognized for the recruitment of histone-modifying enzymes, thus the lack of repressive marks. Nevertheless, this has no effect on maintenance of DNA methylation, so although imprinting has been preserved, the imprintome marks have diverged across species.
Identification of the Human Imprintome
A literature search identified 22 DMRs associated with known human imprinted genes (Table 1); this represents fewer than half of the genes identified experimentally as being imprinted. Identification of most of these DMRs was by screening known imprinted genes for differential methylation by dye-terminator sequencing after bisulfite conversion (Tremblay et al. 1997). Sequencing by this method is capable of qualitatively identifying stretches of CpGs with intermediate methylation in genomic DNA. Confirmation of these regions is usually by sequencing |of clones to definitively identify parental allele-specific stretches of hyper- and hypomethylation (Tremblay et al. 1997).
Table 1.
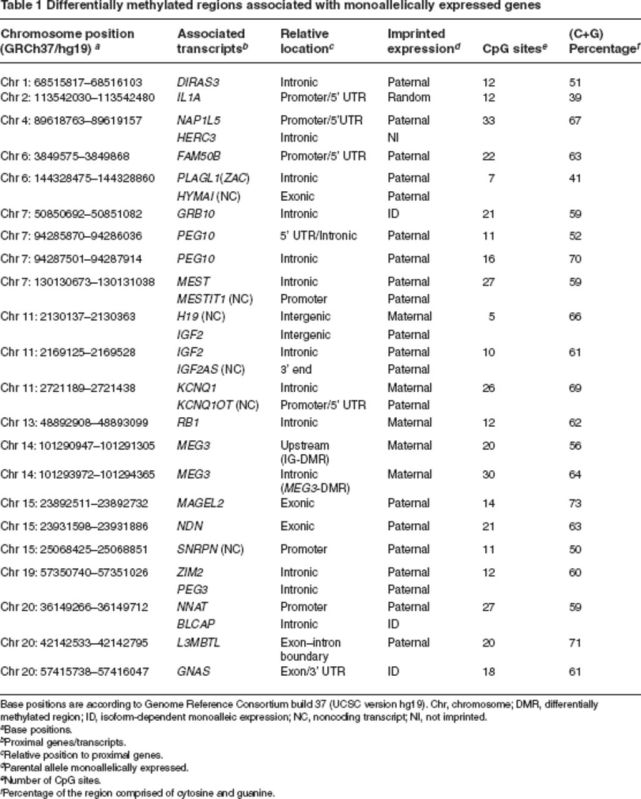
|
Newer methods, including pyrosequencing and mass spectrometry–based methylation analysis using a Sequenom MassArray system (Sequenom, San Diego, CA) are capable of quantitative methylation analysis. We have used this method to confirm the differential methylation status of the reported DMRs and to define the range of differential methylation (Table 1). For the chromosomal regions listed, all or nearly all of the contained CpG sites are 35–65% methylated in multiple cell types. Unfortunately, this method is unable to determine contiguous allele-specifc methylation, so cloning is still necessary to definitively identify an imprint regulatory DMR.
Next-Generation Sequencing of Methylation
The latest methods for high-throughput, parallel, next-generation sequencing (NGS1) are allele specific, generate discrete sequences from single DNA strands, and are also quantitative. Applications for targeted sequencing of methylated regions include sequencing of methylated DNA captured by either immunoprecipitation with 5mC specific antibodies (MeDIP-seq) (Down et al. 2008) or affinity purification by the MDB domain of MeCP2 (MethylCap-seq) (Brinkman et al. 2010). Similar methods could also be used to map 5hmC with the appropriate antibodies. These methods provide powerful whole-genome coverage, but the length of each sequence read is much shorter than other methods, reducing the number of consecutive CpG sites analyzed. Another limitation is that capture efficiency depends on CpG density, so regions with higher CpG content are overrepresented. Thus, imprintome elements with low CpG content will not be as effectively captured and sequenced and may be missed without great depth of sequencing.
Direct whole-genome sequencing on bisulfite-converted DNA by NGS is also possible and would avoid any pulldown biases but trade one drawback for another. With the bisulfite conversion of all cytosines not in CpG pairs, and many that are, there are only three bases in most sequence outputs. The resulting sequences are ambiguous for genome alignment, particularly with the short reads of NGS, making analysis of de novo sequencing results challenging. Finally, bisulfite conversion and sequencing can not distinguish between 5hmC and 5mC, a difference that may be highly significant for regulation.
The very latest methods, such as Pacific Biosciences SMRT sequencing, sequence single molecules in real time; nanopore sequencing, which is still in the experimental stages, also has the same potential. Of greatest significance, these methods have the potential to directly identify methylated, and possibly also hydroxymethylated, bases in single-strand sequencing without bisulfite conversion by variation in DNA polymerase kinetics (Flusberg et al. 2010) or altered electrical current across a nanopore (Wallace et al. 2010). These high-throughput methods hold the promise of unambiguous and unbiased sequencing capable of identifying differential methylation.
Array Detection of Methylation
Until NGS whole-genome methylation sequencing is validated and standardized, the most cost-effective method for comparative methylation analysis with complete genomic coverage is MeDIP chromatin immunoprecipitation, using affinity pulldowns as described previously, but with analysis on whole genome tiling arrays. Methylated DNA collected by MeDIP is quantitated by hybridization to genome arrays, providing relative methylation levels for screening purposes. Imprintome DMRs are consistent across tissues, time, and individuals, so false positives can be reduced by using diverse samples to eliminate variability. Although the completeness of genome-wide detection by NGS depends on the depth and redundancy of sequencing, the entire genome is interrogated equally when arrays are scanned. The resolution of this method is limited to array probe length, but more significant is the already-described challenge of distinguishing DMRs, with their typically low CpG density, from nonimprintome methylation. This requires multiple samples to provide sufficient power to distinguish between hypermethylation and differential methylation. Also, although the same issues of low capture efficiency that affect NGS detection sensitivity apply, with extreme read depth required to give a desirable probability of coverage, array hybridization will interrogate the entire genome.
To eliminate variability from nonimprintome methylation, methylation mapping of gametic cells may be the most informative because this approach detects only the original parentally established marks. A major drawback of this approach is that, although sperm DNA is plentiful, human oocyte DNA is difficult to obtain in sufficient quantity for this type of assay. Analysis of sperm DNA alone has two limitations. First, it contains DNA methylation other than the DMR-establishing marks. Second, the majority of known DMRs are established by maternal methylation, so sperm analysis would likely miss most novel DMRs. The ideal screen would use oocyte DNA to subtract common methylation and thereby identify paternal- and maternal-specific DNA methylation marks.
A successful approach has been used that sidesteps the oocyte DNA limitations by using tissues of uniparental origin—paternally derived hydatidiform moles and maternally derived mature cystic ovarian teratomas (Choufani et al. 2011). This approach found novel DMRs associated with known imprinted genes and led to the validation of AXL, for which paternal-specific methylation was found, as a novel maternally expressed imprinted gene.
Detection of Histone Modifications
Genome-wide interrogation of DNA methylation is a more straightforward process than the determination of histone modification status. Nevertheless, whole genome arrays and NGS have the potential to screen for chromatin structure or histone modifications indicative of imprinting. Chromatin immunoprecipitation (ChIP) with antibodies specific to histone modifications, followed by either microarray analysis (ChIP-chip) or sequencing (ChIP-seq), can identify differentially marked regions comparably with the methylation screens described. Using such a screen, DMR imprint regulatory elements that contain overlapping active and repressive histone marks were found (Dindot et al. 2009). Another investigation showed that many zinc finger genes also contain contradictory activating and repressing histone modifications in 3’ exons, although these genes are biallelically expressed (Blahnik et al. 2011). With the validity of the technique established for detecting such overlapping marks, further work will determine what ICRs and DMRs may be ascertained and how these marks in biallelically expressed genes might be related to imprint regulation.
Informatic Approaches
Informatic approaches have also proven useful in the ascertainment of imprinted genes and imprintome elements. The list of potential imprinted human genes from the previously mentioned prediction study (Luedi et al. 2007) has resulted in the confirmation of several novel imprinted genes (KCNK9, DLGAP, and FAM50B) and a DMR for FAM50B linked to monoallelic expression (Zhang et al. 2011). Continuation of this approach using the convergence of informatic predictions, methylation analysis, and histone modification studies could have a great deal of power to specifically identify imprintome elements.
Imprintome Dysregulation in Diseases and Neurologic Disorders
The most important reason for mapping and understanding the human imprintome is to improve human health. Because a monoallelically expressed imprinted gene is functionally haploid, loss or alteration of the expressed allele is essentially a dominant effect. In particular, early studies identified parent-of-origin–specific deletions associated with Prader-Willi syndrome (PWS1) and Angelman syndrome (AS1) (Knoll et al. 1989; Niikawa and Ishikiriyama 1985), and these deletions were determined to affect imprinted genes (Buiting 2010). Also, uniparental disomy (UPD1) was subsequently found affecting the same regions in some patients (Yamazawa et al. 2010), effectively duplicating the effect of a parental-specific deletion.
The fraction of known and predicted imprinted genes involved in growth and development results in many of these genes also being involved in mental and physical developmental disorders. Additionally, one implication of the conflict hypothesis is that different brain regions are maternally or paternally influenced in development, with particular implications for autism and schizophrenia (Crespi and Badcock 2008). The significance of epigenetic dysregulation of imprinted genes in cancer is well established, with new data rapidly accumulating. Cancers may result from epigenetic variance in the gametes, as for developmental disorders, but the environmentally labile nature of the epigenome and imprintome also provides a mechanism for exposure-mediated carcinogenesis in somatic cells. Because of the stability of cytosine methylation and the relative ease of its determination, even a subset of the complete human imprintome, as provided in Table 1, could be a valuable useful research tool for defining those imprinted genes that are epigenetically dysregulated in diseases and by environmental exposures. The final section of this review will discuss known imprinted gene–related disorders and imprintome aberrations involved in the etiology of diseases and neurologic disorders.
The Imprintome in Growth and Developmental Disorders
The necessity of tight expression control over the growth-promoting gene Igf2 and the growth-restricting gene Igf2r is demonstrated by deletions of these genes in the mouse. When the expressed paternal allele of Igf2 is deleted, the resulting mice are viable and fertile but are significantly smaller, with body weight approximately 60% that of wild-type mice (DeChiara et al. 1991). Deletion of the expressed maternal allele of Igf2r causes fetal overgrowth and perinatal lethality. The affected offspring are approximately 30% larger than wild-type siblings, with kinked tails, polydactyly, and overgrowth of internal organs (Lau et al. 1994; Wylie et al. 2003).
Such studies cannot be done in humans, but a number of human diseases have been connected to similar dysregulation of imprinted genes. Beckwith-Wiedemann syndrome (BWS1) is characterized by fetal overgrowth and childhood tumors, showing similarities to the overgrowth of mice expressing both copies of Igf2 due to H19 deletion (Leighton et al. 1995). A number of chromosomal rearrangements and abnormalities in imprinted domains on 11p15.5, including UPD (Yamazawa et al. 2010), can cause BWS (Choufani et al. 2010). However, early studies of IGF2 imprinting in human disease also found hypermethylation of the H19 ICR in association with bialleleic expression of IGF2. This effect was observed in some BWS cases (Reik et al. 1995) and other overgrowth situations (Morison et al. 1996), as well as Wilms’ tumor of the kidney (Moulton et al. 1994; Steenman et al. 1994), but without any related genetic mutations. Further studies have identified mutations that affect ICR methylation in cis (Weksberg et al. 2010), but in most cases, there are no apparent genetic correlations to hypomethylation, indicating an epigenetic cause.
Epigenetic alterations to the imprintome in BWS are even more common in the neighboring imprinted domain containing CDKN1C and KCNQ1. Approximately 50% of BWS patients have loss of methylation at KvDMR and associated expression of the normally silenced KCNQ1OT1 antisense transcript. This abnormal methylation and expression has been associated with silencing of CDKN1C (Cerrato et al. 2005), which also correlates with loss of imprinting of IGF2 (Engel et al. 2000) and potentially alters expression of other imprinted genes in this region.
Silver-Russell syndrome (SRS) has phenotypes and apparent causes reciprocal to BWS. The primary characteristic of SRS is pre- and postnatal growth restriction, and approximately 44% of cases have hypomethylation of the H19 ICR, resulting in repression of both IGF2 alleles (Eggermann et al. 2010). Additionally, approximately 7% of SRS patients with H19 hypomethylation also have hypomethylated DMRs of other imprinted loci, but the nearby KvDMR shows no significant methylation changes (Penaherrera et al. 2010). These observations of aberrant methylation in SRS correlate to chromosome 7 maternal uniparental disomy, which is seen in 5% of SRS cases (Abu-Amero et al. 2010), has no associated altered methylation at H19 on the maternal chromosome, and results in a similar developmental phenotype as altered methylation (Wakeling et al. 2010).
Examination of the histones and chromatin at the IGF2–H19 locus in BWS and SRS show correspondence between DMR status and elements of the imprintome and regulatory system. Gain of methylation at the ICR in BWS subjects corresponds to histone modification changes that essentially convert the ICR signature of the silenced maternal allele to that of the expressed paternal allele. Likewise, the loss of methylation in SRS converts the paternal imprintome marks to that of the silenced maternal allele. Finally, examination of CTCF–cohesin binding in BWS and SRS found that methylation and histone modification changes led to altered chromatin conformation as well, apparently altering chromatin structure and consequent expression to that of the other parental allele (Nativio et al. 2011).
PWS and AS merit mention because they are also developmental disorders resulting from inappropriate expression of imprinted genes (Buiting 2010). Both syndromes are linked to the same region of chromosome 15 (15q11–q13), which contains both paternally and maternally expressed genes. Loss of expression by deletion, UPD, or other chromosomal rearrangements results in PWS when it is the paternally expressed genes (including MKRN3, MAGEL2, NDN, SNRPN, and multiple small nucleolar RNAs) that are lost, with deficiency of SNORD116 small nucleolar RNAs giving PWD or PWS-like phenotypes (Sahoo et al. 2008). AS results when the maternally expressed genes are lost, with loss of UBE3A having been observed as sufficient for the AS phenotype (Kishino et al. 1997). However, imprintome defects are rarely involved in these conditions because nearly all cases (approximately 99%) of PWS are due to deletion or UPD, and approximately 80% of AS cases are due to deletion, UPD, or UBE3A mutations, with only 3–5% of AS cases known to involve imprinting defects (Horsthemke and Wagstaff 2008).
Deletion analyses have identified an imprinting center near SNRPN (PWS/AS-IC) with separate PWS and AS critical regions that control parent-of-origin–specific expression of PWS- and AS-related genes (Buiting et al. 1995; Horsthemke and Wagstaff 2008). There are apparent epigenetic defects that involve this imprinting center because abnormal methylation, but no detectable deletions, has been seen in a number of individuals with overlapping AS and PWS phenotypes (Gillessen-Kaesbach et al. 1999). Perhaps the most intriguing epigenetic effect is seen in patients with hypomethylation of multiple imprinted loci (HIL), which has been observed in subsets of patients with transient neonatal diabetes mellitus (Mackay et al. 2008), BWS (Bliek et al. 2009), and SRS-like growth restriction (Turner et al. 2010). It may be that HIL cases are more common than suspected and may be present in a significant proportion of patients, particularly those with disorders that are difficult to classify or that have overlapping phenotypes. In one individual with phenotypes of both PWS and BWS, hypomethylation was observed at both the AS/PWS and BWS loci, as well as at H19, PEG3, NESPAS, and GNAS (Baple et al. 2011). The new methods of methylation analysis can determine the true frequency of HIL in patients with developmental disorders and determine the full scope of imprintome defects in these syndromes.
The Imprintome and Cancer
Abnormal gene expression is known to be central to cancer origin and progression, and the central roles of imprinted genes in growth and regulation, combined with the importance of tight transcriptional control over imprinting, make this class of genes highly relevant to cancer. Imprintome dysregulation of IGF2 associated with Wilms’ tumors has already been mentioned, and at least 24 other known imprinted human genes show altered expression or methylation in tumors (Table 2) (Hubertus et al. 2011; Monk 2010). Significantly, multiple imprinted genes have tumor-suppressive functions, such as ZAC/PLAGL1, BLCAP, RB1, and TP73. Probably most relevant to studying the imprintome in cancer is altered DMR methylation and loss of imprinting in multiple cancer types for 12 imprinted domains that involve nearly half of the 60 known human imprinted genes (Monk 2010).
Table 2.

|
IGF2 is the imprinted gene with the most extensively documented cancer involvement, with loss of imprinting observed in most cancer types examined (Monk 2010) as well as loss of long-distance CTCF–cohesin-mediated interactions (Vu et al. 2010). H19 was believed to have tumor supressor activity based on observations when H19 expression was lost (Yoshimizu et al. 2008), but it now appears that the true tumor suppressor is the H19 antisense gene HOTS (Onyango and Feinberg 2011). Because H19 and HOTS show matching monoalleleic expression patterns, the observed loss of H19 expression presumably included loss of HOTS as well. This loss of expression of a tumor suppressor in combination with loss of IGF2 imprinting, and therefore overexpression, that typically occurs when H19 is repressed may be why this imprinted pair is at the center of so many cancer types.
By the same mechanisms as in developmental disorders, CDKN1C also has a role in cancer and has regulatory function in multiple hallmarks of cancer, including apoptosis and metastasis (Kavanagh and Joseph 2011), and hypomethylation of KvDMR and subsequent silencing of CDKN1C frequently occur in sporadic cancers (Monk 2010).
As discussed in the review by Monk (2010), there may be multiple mechanisms responsible for DMR alterations connected to loss of imprinting in the genesis of cancer. Driver mutations destabilizing methylation establishment and maintenance, microsatellite instability, and CpG island methylator phenotypes could be responsible (Hinoue et al. 2009; Kane et al. 1997; Kim et al. 2003; Ogino et al. 2006). Current unanswered questions include whether tumor suppressor genes are prone to epimutations, thus disrupting entire pathways, and whether epimutations are mutally exclusive—that is, do multiple hits in a pathway provide any advantage to tumor growth in comparison to single hits? Also, referring back to the two-way interaction between DNA methylation and histone modifications, knowing which imprintome element is the target of primary epimutations in carcinogenesis may help guide diagnostic, therapeutic, and perhaps even preventative strategies.
The Imprintome and Psychiatric Disorders
The theory of parental conflict in the evolution of imprinted genes has implications for brain development and psychiatric disease; this subject is comprehensively covered in other reviews (Badcock and Crespi 2008; Crespi 2008; Goos and Ragsdale 2008). Connections have been found linking increased relative effects of maternally expressed genes in psychotic spectrum conditions, such as schizophrenia, bipolar disorder, depression, PWS, and Klinefelter syndrome. Conversely, autism spectrum conditions, including autism, Asperger syndrome, Rett syndrome, Turner syndrome, AS, and BWS, often show increased effects from paternally expressed genes. These disease effects parallel the parental-specific effects of imprinted genes on development in that psychotic spectrum conditions are associated with undergrowth in brain development, whereas autism spectrum conditions can involve brain and body overgrowth (Crespi and Badcock 2008).
Most telling are multiple recent studies on parent-of-origin effects, and the connection between imprinted genes and rare copy number variants observed in autism and schizophrenia (Depienne et al. 2009; Fradin et al. 2010; Guffanti et al. 2011; Hogart et al. 2009; Ingason et al. 2011; Kato et al. 2008). These studies often focus on copy number variants in 15q11–15q13, the PWS critical region, known to contain an imprinted gene cluster. Screens for de novo copy number mutations in autism and schizophrenia probands have also led to 15q11–15q13, identifying associated copy number variants in this region (Sanders et al. 2011; Stefansson et al. 2008). Copy number alteration of imprinted genes is considered a strong contributing factor for these conditions because of the importance of expression control for imprinted genes. Because these studies detect only genomic alterations, no imprintome alterations are yet known, but effective changes in copy number due to epigenetic alterations could have a similar effect. Definition of the imprintome and development of cost-effective technologies for screening in large numbers of affected and unaffected individuals will determine the scope of involvement for these genes and their epigenetic regulation in psychiatric disorders.
Studies into the role of dysregulation of imprinted genes in psychiatric diseases have built in limitations in that brain-specific expression profiles can not be measured in living individuals. This means the relevant correlations can not be made between disease state and abnormal expression in brain tissue. However, given the consistency of parentally established epigenetic marks, inherited epigenetic abnormalities should be detectable in peripheral tissues that are amenable to analysis. Likewise, inherited or de novo genetic mutations affecting imprinted genes, particularly copy number variants, as previously mentioned, will also be detectable. Therefore, even in the absence of expression data, screening affected individuals for these types of disruptions should prove highly informative for determining the role of imprinted genes and the imprintome in pyschiatric diseases.
Master Imprintome Regulators and Disease
Finally, special notice must be given to recent determinations regarding imprinted “master regulator” genes. Given the number of genes and processes affected by these regulators, changes in their expression could have drastic and far-reaching effects. The imprinted transcription factor ZAC deserves special attention because it functions as a central regulator of other imprinted genes, giving it a wide influence over development and health. Mouse studies have identified a network of imprinted genes that shows interactions between and coregulation of Zac1, Igf2–H19, Cdkn1c, Rtl1, Gnas, and Dlk1 (Gabory et al. 2009; Varrault et al. 2006), and Zac1 directly regulates Igf2–H19 by binding a shared enhancer (Varrault et al. 2006). Through its already described interactions with KCNQ1OT1, ZAC may contribute to BWS, and it is a strong candidate for transient neonatal diabetes mellitus, probably through loss of imprinting and bialleleic expression (Mackay et al. 2002; Varrault et al. 2001). ZAC is also a tumor supressor gene, with loss of expression associated with carcinogenesis in basal and squamous cell carcinomas, pituitary adenomas, and breast cancer (Basyuk et al. 2005; Bilanges et al. 1999; Koy et al. 2004; Pagotto et al. 2000). More specific to imprintome studies, epigenetic silencing of ZAC by hypermethylation has been observed in several cancer types, specifically as an early event in ovarian cancer (Kamikihara et al. 2005).
Another imprinted central regulator of note is KLF14, which has robust monoalleleic maternal expression in all mouse and human embryonic tissues tested. KLF14 appears to have originated as a retrotransposon of KLF16 shortly after the divergence of marsupials and eutherians and appears to have human-specific accelerated evolution (Parker-Katiraee et al. 2007). KLF14 is another imprinted transcription factor, which regulates gene expression in adipose tissue related to metabolic phenotypes, including type 2 diabetes risk; body mass index; high-density lipoprotein, triglyceride, and fasting insulin levels; and insulin sensitivity (Small et al. 2011). Thus far, 46 genes are known to have interactions in trans with KLF14 (Small et al. 2011), and a marker proximal to KLF14 shows parent-of-origin–specific association to basal cell carcinoma risk (Stacey et al. 2009). Dnmt3a knockout studies in mice indicate that maternal methylation is required for Klf14 expression, but the methylation site has yet to be determined (Parker-Katiraee et al. 2007). Identification of the human imprintome elements responsible for KLF14 monoallelic expression will be valuable in screening for epigenetic roles in metabolic syndrome.
Summary
Theodosius Dobzhansky's (1973) elegant paper entitled “Nothing in Biology Makes Sense except in the Light of Evolution” introduced a concept that is now generally accepted. It might not be too audacious to likewise now state that nothing in human development and disease makes sense except in the light of the imprintome because it plays such a fundamental role in normal fetal growth and development. Moreover, there is now compelling information indicating that cancer and psychiatric disorders, previously considered to be complex genetic diseases, arise in part because of the epigenetic deregulation of imprinted gene expression. Consequently, it is critically important to identify the complete repertoire of regulatory elements that comprise the human imprintome. Presently, the most promising experimental approach for accomplishing this task is genome-wide comparative mapping of parent-of-origin–dependent differential methylation and corresponding histone modifications in human gametes and primary somatic cells. Given the significant variation in imprintomes among species, it is also essential that this comprehensive epigenetic analysis be performed in humans. Without a proper human imprintome map, the ability to optimally diagnose, prevent, and treat human diseases will remain elusive.
Acknowledgments
This work was supported by grants from the National Institute of Health (1R01ES016772, 1R01DK085173, 1R01CA15566401, 1R01CA142983, DOE DE-0FG02-05ER64101); the Ester B. O’Keeffe Foundation Award; and Fred and Alice Stanback.
Biography
David A. Skaar, PhD, is senior research associate in the Department of Oncology; Yue Li, PhD, is a research associate, and Cathrine Hoyo, PhD, is an associate professor, both in the Department of Community and Family Medicine; and Susan K. Murphy, PhD, is an associate professor in the Department of Obstetrics and Gynecology, all at the Duke University Medical Center, Durham, North Carolina. Autumn J. Bernal, PhD, is a health scientist at ChemRisk, Aliso Viejo, California. Randy L. Jirtle, PhD, is visiting professor in the Department of Oncology, University of Wisconsin-Madison, Madison, Wisconsin.
Footnotes
Abbreviations that appear ≥3x throughout this article: 5hmC, 5-hydroxymethylcytosine; 5mC, 5-methylcytosine; AS, Angelman syndrome; BWS, Beckwith-Wiedemann syndrome; DMR, differentially methylated region; ICR, imprint control regions; ncRNA, noncoding RNA; NGS, next-generation sequencing; PWS, Prader-Willi syndrome; UPD, uniparental disomy.
References
- Abu-Amero S, Wakeling EL, Preece M, Whittaker J, Stanier P, Moore GE. 2010. Epigenetic signatures of Silver-Russell syndrome. J Med Genet 47:150–154 [DOI] [PubMed] [Google Scholar]
- Arima T, Kamikihara T, Hayashida T, Kato K, Inoue T, Shirayoshi Y, Oshimura M, Soejima H, Mukai T, Wake N. 2005. ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res 33:2650–2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badcock C, Crespi B. 2008. Battle of the sexes may set the brain. Nature 454:1054–1055 [DOI] [PubMed] [Google Scholar]
- Baple EL, Poole RL, Mansour S, Willoughby C, Temple IK, Docherty LE, Taylor R, Mackay DJ. 2011. An atypical case of hypomethylation at multiple imprinted loci. Eur J Hum Genet 19:360–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow DP. 1993. Methylation and imprinting: From host defense to gene regulation? Science 260:309–310 [DOI] [PubMed] [Google Scholar]
- Basyuk E, Coulon V, Le Digarcher A, Coisy-Quivy M, Moles JP, Gandarillas A, Journot L. 2005. The candidate tumor suppressor gene ZAC is involved in keratinocyte differentiation and its expression is lost in basal cell carcinomas. Mol Cancer Res 3:483–492 [DOI] [PubMed] [Google Scholar]
- Bell AC, Felsenfeld G. 2000. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405:482–485 [DOI] [PubMed] [Google Scholar]
- Bestor TH. 2000. The DNA methyltransferases of mammals. Hum Mol Genet 9:2395–2402 [DOI] [PubMed] [Google Scholar]
- Bilanges B, Varrault A, Basyuk E, Rodriguez C, Mazumdar A, Pantaloni C, Bockaert J, Theillet C, Spengler D, Journot L. 1999. Loss of expression of the candidate tumor suppressor gene ZAC in breast cancer cell lines and primary tumors. Oncogene 18:3979–3988 [DOI] [PubMed] [Google Scholar]
- Bird AP. 1986. CpG-rich islands and the function of DNA methylation. Nature 321:209–213 [DOI] [PubMed] [Google Scholar]
- Blahnik KR, Dou L, Echipare L, Iyengar S, O’Geen H, Sanchez E, Zhao Y, Marra MA, Hirst M, Costello JF, Korf I, Farnham PJ. 2011. Characterization of the contradictory chromatin signatures at the 3’ exons of zinc finger genes. PLoS One 6:e17121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliek J, Verde G, Callaway J, Maas SM, De Crescenzo A, Sparago A, Cerrato F, Russo S, Ferraiuolo S, Rinaldi MM, Fischetto R, Lalatta F, Giordano L, Ferrari P, Cubellis MV, Larizza L, Temple IK, Mannens MM, Mackay DJ, Riccio A. 2009. Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur J Hum Genet 17:611–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourc’his D, Bestor TH. 2006. Origins of extreme sexual dimorphism in genomic imprinting. Cytogenet Genome Res 113:36–40 [DOI] [PubMed] [Google Scholar]
- Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. 2001. Dnmt3L and the establishment of maternal genomic imprints. Science 294:2536–2539 [DOI] [PubMed] [Google Scholar]
- Brinkman AB, Simmer F, Ma K, Kaan A, Zhu J, Stunnenberg HG. 2010. Whole-genome DNA methylation profiling using MethylCap-seq. Methods 52:232–236 [DOI] [PubMed] [Google Scholar]
- Buiting K. 2010. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet 154C:365–376 [DOI] [PubMed] [Google Scholar]
- Buiting K, Kanber D, Horsthemke B, Lohmann D. 2010. Imprinting of RB1 (the new kid on the block). Brief Funct Genomics 9:347–353 [DOI] [PubMed] [Google Scholar]
- Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S, Nicholls RD, Horsthemke B. 1995. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet 9:395–400 [DOI] [PubMed] [Google Scholar]
- Cerrato F, Sparago A, Di Matteo I, Zou X, Dean W, Sasaki H, Smith P, Genesio R, Bruggemann M, Reik W, Riccio A. 2005. The two-domain hypothesis in Beckwith-Wiedemann syndrome: Autonomous imprinting of the telomeric domain of the distal chromosome 7 cluster. Hum Mol Genet 14:503–511 [DOI] [PubMed] [Google Scholar]
- Choufani S, Shapiro JS, Susiarjo M, Butcher DT, Grafodatskaya D, Lou Y, Ferreira JC, Pinto D, Scherer SW, Shaffer LG, Coullin P, Caniggia I, Beyene J, Slim R, Bartolomei MS, Weksberg R. 2011. A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res 21:465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choufani S, Shuman C, Weksberg R. 2010. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 154C:343–354 [DOI] [PubMed] [Google Scholar]
- Cooper WN, Constancia M. 2010. How genome-wide approaches can be used to unravel the remaining secrets of the imprintome. Brief Funct Genomics 9:315–328 [DOI] [PubMed] [Google Scholar]
- Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A. 2011. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146:67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi B. 2008. Genomic imprinting in the development and evolution of psychotic spectrum conditions. Biol Rev Camb Philos Soc 83:441–493 [DOI] [PubMed] [Google Scholar]
- Crespi B, Badcock C. 2008. Psychosis and autism as diametrical disorders of the social brain. Behav Brain Sci 31:241–261discussion 261–320 [DOI] [PubMed] [Google Scholar]
- da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. 2008. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet 24:306–316 [DOI] [PubMed] [Google Scholar]
- Deakin JE, Hore TA, Koina E, Marshall Graves JA. 2008. The status of dosage compensation in the multiple X chromosomes of the platypus. PLoS Genet 4:e1000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChiara TM, Robertson EJ, Efstratiadis A. 1991. Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64:849–859 [DOI] [PubMed] [Google Scholar]
- Depienne C, Moreno-De-Luca D, Heron D, Bouteiller D, Gennetier A, Delorme R, Chaste P, Siffroi JP, Chantot-Bastaraud S, Benyahia B, Trouillard O, Nygren G, Kopp S, Johansson M, Rastam M, Burglen L, Leguern E, Verloes A, Leboyer M, Brice A, Gillberg C, Betancur C. 2009. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry 66:349–359 [DOI] [PubMed] [Google Scholar]
- Dindot SV, Person R, Strivens M, Garcia R, Beaudet al. 2009. Epigenetic profiling at mouse imprinted gene clusters reveals novel epigenetic and genetic features at differentially methylated regions. Genome Res 19:1374–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky Z. 1973. Nothing in biology makes sense except in light of evolution. Am Biol Teacher 35:125–129 [Google Scholar]
- Dorsett D. 2011. Cohesin: Genomic insights into controlling gene transcription and development. Curr Opin Genet Dev 21:199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Graf S, Johnson N, Herrero J, Tomazou EM, Thorne NP, Backdahl L, Herberth M, Howe KL, Jackson DK, Miretti MM, Marioni JC, Birney E, Hubbard TJ, Durbin R, Tavare S, Beck S. 2008. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 26:779–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Beatty LG, Zhou W, Lew J, Schoenherr C, Weksberg R, Sadowski PD. 2003. Insulator and silencer sequences in the imprinted region of human chromosome 11p15.5. Hum Mol Genet 12:1927–1939 [DOI] [PubMed] [Google Scholar]
- Eggermann T, Begemann M, Spengler S, Schroder C, Kordass U, Binder G. 2010. Genetic and epigenetic findings in Silver-Russell syndrome. Pediatr Endocrinol Rev 8:86–93 [PubMed] [Google Scholar]
- Engel JR, Smallwood A, Harper A, Higgins MJ, Oshimura M, Reik W, Schofield PN, Maher ER. 2000. Epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. J Med Genet 37:921–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans HK, Wylie AA, Murphy SK, Jirtle RL. 2001. The neuronatin gene resides in a “micro-imprinted” domain on human chromosome 20q11.2. Genomics 77:99–104 [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith AC, Surani MA. 2001. Imprinting and the epigenetic asymmetry between parental genomes. Science 293:1086–1089 [DOI] [PubMed] [Google Scholar]
- Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7:461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradin D, Cheslack-Postava K, Ladd-Acosta C, Newschaffer C, Chakravarti A, Arking DE, Feinberg A, Fallin MD. 2010. Parent-of-origin effects in autism identified through genome-wide linkage analysis of 16,000 SNPs. PLoS One 5:e12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JM, Udayashankar R, Moore HD, Moore GE. 2010. Telomeric NAP1L4 and OSBPL5 of the KCNQ1 cluster, and the DECORIN gene are not imprinted in human trophoblast stem cells. PLoS One 5:e11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. 2003. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 278:4035–4040 [DOI] [PubMed] [Google Scholar]
- Gabory A, Ripoche MA, Le Digarcher A, Watrin F, Ziyyat A, Forne T, Jammes H, Ainscough JF, Surani MA, Journot L, Dandolo L. 2009. H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development 136:3413–3421 [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, Okwuonu G, Hines S, Lewis L, DeRamo C, Delgado O, Dugan-Rocha S, Miner G, Morgan M, Hawes A, Gill R, Celera, Holt RA, Adams MD, Amanatides PG, Baden-Tillson H, Barnstead M, Chin S, Evans CA, Ferriera S, Fosler C, Glodek A, Gu Z, Jennings D, Kraft CL, Nguyen T, Pfannkoch CM, Sitter C, Sutton GG, Venter JC, Woodage T, Smith D, Lee HM, Gustafson E, Cahill P, Kana A, Doucette-Stamm L, Weinstock K, Fechtel K, Weiss RB, Dunn DM, Green ED, Blakesley RW, Bouffard GG, De Jong PJ, Osoegawa K, Zhu B, Marra M, Schein J, Bosdet I, Fjell C, Jones S, Krzywinski M, Mathewson C, Siddiqui A, Wye N, McPherson J, Zhao S, Fraser CM, Shetty J, Shatsman S, Geer K, Chen Y, Abramzon S, Nierman WC, Havlak PH, Chen R, Durbin KJ, Egan A, Ren Y, Song XZ, Li B, Liu Y, Qin X, Cawley S, Cooney AJ, D’Souza LM, Martin K, Wu JQ, Gonzalez-Garay ML, Jackson AR, Kalafus KJ, McLeod MP, Milosavljevic A, Virk D, Volkov A, Wheeler DA, Zhang Z, Bailey JA, Eichler EE, Tuzun E, Birney E, Mongin E, Ureta-Vidal A, Woodwark C, Zdobnov E, Bork P, Suyama M, Torrents D, Alexandersson M, Trask BJ, Young JM, Huang H, Wang H, Xing H, Daniels S, Gietzen D, Schmidt J, Stevens K, Vitt U, Wingrove J, Camara F, Mar Alba M, Abril JF, Guigo R, Smit A, Dubchak I, Rubin EM, Couronne O, Poliakov A, Hubner N, Ganten D, Goesele C, Hummel O, Kreitler T, Lee YA, Monti J, Schulz H, Zimdahl H, Himmelbauer H, Lehrach H, Jacob HJ, Bromberg S, Gullings-Handley J, Jensen-Seaman MI, Kwitek AE, Lazar J, Pasko D, Tonellato PJ, Twigger S, Ponting CP, Duarte JM, Rice S, Goodstadt L, Beatson SA, Emes RD, Winter EE, Webber C, Brandt P, Nyakatura G, Adetobi M, Chiaromonte F, Elnitski L, Eswara P, Hardison RC, Hou M, Kolbe D, Makova K, Miller W, Nekrutenko A, Riemer C, Schwartz S, Taylor J, Yang S, Zhang Y, Lindpaintner K, Andrews TD, Caccamo M, Clamp M, Clarke L, Curwen V, Durbin R, Eyras E, Searle SM, Cooper GM, Batzoglou S, Brudno M, Sidow A, Stone EA, Payseur BA, Bourque G, Lopez-Otin C, Puente XS, Chakrabarti K, Chatterji S, Dewey C, Pachter L, Bray N, Yap VB, Caspi A, Tesler G, Pevzner PA, Haussler D, Roskin KM, Baertsch R, Clawson H, Furey TS, Hinrichs AS, Karolchik D, Kent WJ, Rosenbloom KR, Trumbower H, Weirauch M, Cooper DN, Stenson PD, Ma B, Brent M, Arumugam M, Shteynberg D, Copley RR, Taylor MS, Riethman H, Mudunuri U, Peterson J, Guyer M, Felsenfeld A, Old S, Mockrin S, Collins F. 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428:493–521 [DOI] [PubMed] [Google Scholar]
- Gillessen-Kaesbach G, Demuth S, Thiele H, Theile U, Lich C, Horsthemke B. 1999. A previously unrecognised phenotype characterised by obesity muscular hypotonia and ability to speak in patients with Angelman syndrome caused by an imprinting defect. Eur J Hum Genet 7:638–644 [DOI] [PubMed] [Google Scholar]
- Gimelbrant A, Hutchinson JN, Thompson BR, Chess A. 2007. Widespread monoallelic expression on human autosomes. Science 318:1136–1140 [DOI] [PubMed] [Google Scholar]
- Glazov EA, McWilliam S, Barris WC, Dalrymple BP. 2008. Origin, evolution, and biological role of miRNA cluster in DLK-DIO3 genomic region in placental mammals. Mol Biol Evol 25:939–948 [DOI] [PubMed] [Google Scholar]
- Goos LM, Ragsdale G. 2008. Genomic imprinting and human psychology: Cognition, behavior and pathology. Adv Exp Med Biol 626:71–88 [DOI] [PubMed] [Google Scholar]
- Green K, Lewis A, Dawson C, Dean W, Reinhart B, Chaillet JR, Reik W. 2007. A developmental window of opportunity for imprinted gene silencing mediated by DNA methylation and the Kcnq1ot1 noncoding RNA. Mamm Genome 18:32–42 [DOI] [PubMed] [Google Scholar]
- Guffanti G, Strik Lievers L, Bonati MT, Marchi M, Geronazzo L, Nardocci N, Estienne M, Larizza L, Macciardi F, Russo S. 2011. Role of UBE3A and ATP10A genes in autism susceptibility region 15q11-q13 in an Italian population: A positive replication for UBE3A. Psychiatry Res 185:33–38 [DOI] [PubMed] [Google Scholar]
- Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR. 2009. Distinctive chromatin in human sperm packages genes for embryo development. Nature 460:473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405:486–489 [DOI] [PubMed] [Google Scholar]
- Henckel A, Nakabayashi K, Sanz LA, Feil R, Hata K, Arnaud P. 2009. Histone methylation is mechanistically linked to DNA methylation at imprinting control regions in mammals. Hum Mol Genet 18:3375–3383 [DOI] [PubMed] [Google Scholar]
- Hinoue T, Weisenberger DJ, Pan F, Campan M, Kim M, Young J, Whitehall VL, Leggett BA, Laird PW. 2009. Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PLoS One 4:e8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogart A, Leung KN, Wang NJ, Wu DJ, Driscoll J, Vallero RO, Schanen NC, LaSalle JM. 2009. Chromosome 15q11-13 duplication syndrome brain reveals epigenetic alterations in gene expression not predicted from copy number. J Med Genet 46:86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsthemke B, Wagstaff J. 2008. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet A 146A:2041– 2052 [DOI] [PubMed] [Google Scholar]
- Hubertus J, Lacher M, Rottenkolber M, Muller-Hocker J, Berger M, Stehr M, von Schweinitz D, Kappler R. 2011. Altered expression of imprinted genes in Wilms tumors. Oncol Rep 25:817–823 [DOI] [PubMed] [Google Scholar]
- Hutter B, Bieg M, Helms V, Paulsen M. 2010. Imprinted genes show unique patterns of sequence conservation. BMC Genomics 11:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami K, Ohgane J, Tanaka S, Yagi S, Shiota K. 2009. Interplay between DNA methylation histone modification and chromatin remodeling in stem cells and during development. Int J Dev Biol 53:203–214 [DOI] [PubMed] [Google Scholar]
- Ingason A, Kirov G, Giegling I, Hansen T, Isles AR, Jakobsen KD, Kristinsson KT, le Roux L, Gustafsson O, Craddock N, Moller HJ, McQuillin A, Muglia P, Cichon S, Rietschel M, Ophoff RA, Djurovic S, Andreassen OA, Pietilainen OP, Peltonen L, Dempster E, Collier DA, St Clair D, Rasmussen HB, Glenthoj BY, Kiemeney LA, Franke B, Tosato S, Bonetto C, Saemundsen E, Hreidarsson SJ, Nothen MM, Gurling H, O’Donovan MC, Owen MJ, Sigurdsson E, Petursson H, Stefansson H, Rujescu D, Stefansson K, Werge T. 2011. Maternally derived microduplications at 15q11-q13: Implication of imprinted genes in psychotic illness. Am J Psychiatry 168:408–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Jin SG, Pfeifer GP, Szabo PE. 2011. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci U S A 108:3642–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo A, Schneider R. 2010. Chatting histone modifications in mammals. Brief Funct Genomics 9:429–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinic P, Stehle JC, Shaw P. 2006. The testis-specific factor CTCFL cooperates with the protein methyltransferase PRMT7 in H19 imprinting control region methylation. PLoS Biol 4:e355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. 2007. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449:248–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirtle RL. 2009. Epigenome: The program for human health and disease. Epigenomics 1:13–16 [DOI] [PubMed] [Google Scholar]
- John RM, Lefebvre L. 2011. Developmental regulation of somatic imprints. Differentiation 81:270–280 [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. 1998. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19:187–191 [DOI] [PubMed] [Google Scholar]
- Kacem S, Feil R. 2009. Chromatin mechanisms in genomic imprinting. Mamm Genome 20:544–556 [DOI] [PubMed] [Google Scholar]
- Kagami M, O’Sullivan MJ, Green AJ, Watabe Y, Arisaka O, Masawa N, Matsuoka K, Fukami M, Matsubara K, Kato F, Ferguson-Smith AC, Ogata T. 2010. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: Hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet 6:e1000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamikihara T, Arima T, Kato K, Matsuda T, Kato H, Douchi T, Nagata Y, Nakao M, Wake N. 2005. Epigenetic silencing of the imprinted gene ZAC by DNA methylation is an early event in the progression of human ovarian cancer. Int J Cancer 115:690–700 [DOI] [PubMed] [Google Scholar]
- Kanber D, Berulava T, Ammerpohl O, Mitter D, Richter J, Siebert R, Horsthemke B, Lohmann D, Buiting K. 2009. The human retinoblastoma gene is imprinted. PLoS Genet 5:e1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. 1997. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 57:808–811 [PubMed] [Google Scholar]
- Kato C, Tochigi M, Ohashi J, Koishi S, Kawakubo Y, Yamamoto K, Matsumoto H, Hashimoto O, Kim SY, Watanabe K, Kano Y, Nanba E, Kato N, Sasaki T. 2008. Association study of the 15q11-q13 maternal expression domain in Japanese autistic patients. Am J Med Genet B Neuropsychiatr Genet 147B:1008–1012 [DOI] [PubMed] [Google Scholar]
- Kavanagh E, Joseph B. 2011. The hallmarks of CDKN1C (p57, KIP2) in cancer. Biochim Biophys Acta 1816:50–56 [DOI] [PubMed] [Google Scholar]
- Kernohan KD, Jiang Y, Tremblay DC, Bonvissuto AC, Eubanks JH, Mann MR, Berube NG. 2010. ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev Cell 18:191–202 [DOI] [PubMed] [Google Scholar]
- Keverne EB. 2011. Epigenetics and brain evolution. Epigenomics 3:183–191 [DOI] [PubMed] [Google Scholar]
- Killian JK, Byrd JC, Jirtle JV, Munday BL, Stoskopf MK, MacDonald RG, Jirtle RL. 2000. M6P/IGF2R imprinting evolution in mammals. Mol Cell 5:707–716 [DOI] [PubMed] [Google Scholar]
- Killian JK, Nolan CM, Stewart N, Munday BL, Andersen NA, Nicol S, Jirtle RL. 2001. Monotreme IGF2 expression and ancestral origin of genomic imprinting. J Exp Zool 291:205–212 [DOI] [PubMed] [Google Scholar]
- Kim H, Kim YH, Kim SE, Kim NG, Noh SH. 2003. Concerted promoter hypermethylation of hMLH1, p16INK4A, and E-cadherin in gastric carcinomas with microsatellite instability. J Pathol 200:23–31 [DOI] [PubMed] [Google Scholar]
- Kishino T, Lalande M, Wagstaff J. 1997. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15:70–73 [DOI] [PubMed] [Google Scholar]
- Knoll JH, Nicholls RD, Magenis RE, Graham JM, Jr, Lalande M, Latt SA. 1989. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. Am J Med Genet 32:285–290 [DOI] [PubMed] [Google Scholar]
- Koerner MV, Pauler FM, Huang R, Barlow DP. 2009. The function of non-coding RNAs in genomic imprinting. Development 136:1771–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler C, Weinhofer-Molisch I. 2010. Mechanisms and evolution of genomic imprinting in plants. Heredity 105:57–63 [DOI] [PubMed] [Google Scholar]
- Koy S, Hauses M, Appelt H, Friedrich K, Schackert HK, Eckelt U. 2004. Loss of expression of ZAC/LOT1 in squamous cell carcinomas of head and neck. Head Neck 26:338–344 [DOI] [PubMed] [Google Scholar]
- Labialle S, Cavaille J. 2011. Do repeated arrays of regulatory small-RNA genes elicit genomic imprinting?: Concurrent emergence of large clusters of small non-coding RNAs and genomic imprinting at four evolutionarily distinct eutherian chromosomal loci. Bioessays 33:565–573 [DOI] [PubMed] [Google Scholar]
- Lau MM, Stewart CE, Liu Z, Bhatt H, Rotwein P, Stewart CL. 1994. Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev 8:2953–2963 [DOI] [PubMed] [Google Scholar]
- Leighton PA, Saam JR, Ingram RS, Stewart CL, Tilghman SM. 1995. An enhancer deletion affects both H19 and Igf2 expression. Genes Dev 9:2079–2089 [DOI] [PubMed] [Google Scholar]
- Lewis A, Reik W. 2006. How imprinting centres work. Cytogenet Genome Res 113:81–89 [DOI] [PubMed] [Google Scholar]
- Lin S, Ferguson-Smith AC, Schultz RM, Bartolomei MS. 2011. Nonallelic transcriptional roles of CTCF and cohesins at imprinted loci. Mol Cell Biol 31:3094–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukinov DI, Pugacheva E, Vatolin S, Pack SD, Moon H, Chernukhin I, Mannan P, Larsson E, Kanduri C, Vostrov AA, Cui H, Niemitz EL, Rasko JE, Docquier FM, Kistler M, Breen JJ, Zhuang Z, Quitschke WW, Renkawitz R, Klenova EM, Feinberg AP, Ohlsson R, Morse HC, 3rd, Lobanenkov VV. 2002. BORIS, a novel male germ-line-specific protein associated with epigenetic reprogramming events shares the same 11-zinc-finger domain with CTCF the insulator protein involved in reading imprinting marks in the soma. Proc Natl Acad Sci U S A 99:6806–6811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ. 2007. Computational and experimental identification of novel human imprinted genes. Genome Res 17:1723–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedi PP, Hartemink AJ, Jirtle RL. 2005. Genome-wide prediction of imprinted murine genes. Genome Res 15:875–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP, Hahnemann JM, Kordonouri O, Masoud AF, Oestergaard E, Storr J, Ellard S, Hattersley AT, Robinson DO, Temple IK. 2008. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet 40:949–951 [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Coupe AM, Shield JP, Storr JN, Temple IK, Robinson DO. 2002. Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum Genet 110:139–144 [DOI] [PubMed] [Google Scholar]
- Mancini-Dinardo D, Steele SJ, Levorse JM, Ingram RS, Tilghman SM. 2006. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev 20:1268–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. 2000. Demethylation of the zygotic paternal genome. Nature 403:501–502 [DOI] [PubMed] [Google Scholar]
- Miranda TB, Jones PA. 2007. DNA methylation: The nuts and bolts of repression. J Cell Physiol 213:384–390 [DOI] [PubMed] [Google Scholar]
- Mohammad F, Mondal T, Guseva N, Pandey GK, Kanduri C. 2010. Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 137:2493–2499 [DOI] [PubMed] [Google Scholar]
- Monk D. 2010. Deciphering the cancer imprintome. Brief Funct Genomics 9:329–339 [DOI] [PubMed] [Google Scholar]
- Monk D, Arnaud P, Apostolidou S, Hills FA, Kelsey G, Stanier P, Feil R, Moore GE. 2006. Limited evolutionary conservation of imprinting in the human placenta. Proc Natl Acad Sci U S A 103:6623–6628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk D, Arnaud P, Frost JM, Wood AJ, Cowley M, Martin-Trujillo A, Guillaumet-Adkins A, Iglesias Platas I, Camprubi C, Bourc’his D, Feil R, Moore GE, Oakey RJ. 2011. Human imprinted retrogenes exhibit non-canonical imprint chromatin signatures and reside in non-imprinted host genes. Nucleic Acids Res 39:4577–4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morison IM, Becroft DM, Taniguchi T, Woods CG, Reeve AE. 1996. Somatic overgrowth associated with overexpression of insulin-like growth factor II. Nat Med 2:311–316 [DOI] [PubMed] [Google Scholar]
- Moulton T, Crenshaw T, Hao Y, Moosikasuwan J, Lin N, Dembitzer F, Hensle T, Weiss L, McMorrow L, Loew T, Kraus W, Gerard W, Tycko B. 1994. Epigenetic lesions at the H19 locus in Wilms’ tumour patients. Nat Genet 7:440–447 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Arai Y, Umehara H, Masuhara M, Kimura T, Taniguchi H, Sekimoto T, Ikawa M, Yoneda Y, Okabe M, Tanaka S, Shiota K, Nakano T. 2007. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol 9:64–71 [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. 1998. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386–389 [DOI] [PubMed] [Google Scholar]
- Nativio R, Sparago A, Ito Y, Weksberg R, Riccio A, Murrell A. 2011. Disruption of genomic neighbourhood at the imprinted IGF2-H19 locus in Beckwith-Wiedemann syndrome and Silver-Russell syndrome. Hum Mol Genet 20:1363–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nativio R, Wendt KS, Ito Y, Huddleston JE, Uribe-Lewis S, Woodfine K, Krueger C, Reik W, Peters JM, Murrell A. 2009. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet 5:e1000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikawa N, Ishikiriyama S. 1985. Clinical and cytogenetic studies of the Prader-Willi syndrome: Evidence of phenotype-karyotype correlation. Hum Genet 69:22–27 [DOI] [PubMed] [Google Scholar]
- Ogino S, Kawasaki T, Kirkner GJ, Ogawa A, Dorfman I, Loda M, Fuchs CS. 2006. Down-regulation of p21 (CDKN1A/CIP1) is inversely associated with microsatellite instability and CpG island methylator phenotype (CIMP) in colorectal cancer. J Pathol 210:147–154 [DOI] [PubMed] [Google Scholar]
- Onyango P, Feinberg AP. 2011. A nucleolar protein H19 opposite tumor suppressor (HOTS), is a tumor growth inhibitor encoded by a human imprinted H19 antisense transcript. Proc Natl Acad Sci U S A 108:16759–16764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyango P, Miller W, Lehoczky J, Leung CT, Birren B, Wheelan S, Dewar K, Feinberg AP. 2000. Sequence and comparative analysis of the mouse 1-megabase region orthologous to the human 11p15 imprinted domain. Genome Res 10:1697–1710 [DOI] [PubMed] [Google Scholar]
- Ooi SK, O’Donnell AH, Bestor TH. 2009. Mammalian cytosine methylation at a glance. J Cell Sci 122:2787–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH. 2007. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448:714–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani J, Nankumo T, Arita K, Inamoto S, Ariyoshi M, Shirakawa M. 2009. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep 10:1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagotto U, Arzberger T, Theodoropoulou M, Grubler Y, Pantaloni C, Saeger W, Losa M, Journot L, Stalla GK, Spengler D. 2000. The expression of the antiproliferative gene ZAC is lost or highly reduced in nonfunctioning pituitary adenomas. Cancer Res 60:6794–6799 [PubMed] [Google Scholar]
- Pannetier M, Julien E, Schotta G, Tardat M, Sardet C, Jenuwein T, Feil R. 2008. PR-SET7 and SUV4-20H regulate H4 lysine-20 methylation at imprinting control regions in the mouse. EMBO Rep 9:998–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker-Katiraee L, Carson AR, Yamada T, Arnaud P, Feil R, Abu-Amero SN, Moore GE, Kaneda M, Perry GH, Stone AC, Lee C, Meguro-Horike M, Sasaki H, Kobayashi K, Nakabayashi K, Scherer SW. 2007. Identification of the imprinted KLF14 transcription factor undergoing human-specific accelerated evolution. PLoS Genet 3:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen M, Khare T, Burgard C, Tierling S, Walter J. 2005. Evolution of the Beckwith-Wiedemann syndrome region in vertebrates. Genome Res 15:146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen M, Takada S, Youngson NA, Benchaib M, Charlier C, Segers K, Georges M, Ferguson-Smith AC. 2001. Comparative sequence analysis of the imprinted Dlk1-Gtl2 locus in three mammalian species reveals highly conserved genomic elements and refines comparison with the Igf2-H19 region. Genome Res 11:2085–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penaherrera MS, Weindler S, Van Allen MI, Yong SL, Metzger DL, McGillivray B, Boerkoel C, Langlois S, Robinson WP. 2010. Methylation profiling in individuals with Russell-Silver syndrome. Am J Med Genet A 152A:347–355 [DOI] [PubMed] [Google Scholar]
- Proudhon C, Bourc’his D. 2010. [Evolution of genomic imprinting in mammals: What a zoo!]. Med Sci (Paris) 26:497–503 [DOI] [PubMed] [Google Scholar]
- Reik W, Brown KW, Schneid H, Le Bouc Y, Bickmore W, Maher ER. 1995. Imprinting mutations in the Beckwith-Wiedemann syndrome suggested by altered imprinting pattern in the IGF2-H19 domain. Hum Mol Genet 4:2379–2385 [DOI] [PubMed] [Google Scholar]
- Renfree MB, Papenfuss AT, Shaw G, Pask AJ. 2009. Eggs, embryos and the evolution of imprinting: Insights from the platypus genome. Reprod Fertil Dev 21:935–942 [DOI] [PubMed] [Google Scholar]
- Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaudet al. 2008. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet 40:719–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe NJ, Nobrega MA. 2010. Genome-wide maps of transcription regulatory elements. Wiley Interdiscip Rev Syst Biol Med 2:422–437 [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, Mason CE, Bilguvar K, Celestino-Soper PB, Choi M, Crawford EL, Davis L, Davis Wright NR, Dhodapkar RM, Dicola M, Dilullo NM, Fernandez TV, Fielding-Singh V, Fishman DO, Frahm S, Garagaloyan R, Goh GS, Kammela S, Klei L, Lowe JK, Lund SC, McGrew AD, Meyer KA, Moffat WJ, Murdoch JD, O’Roak BJ, Ober GT, Pottenger RS, Raubeson MJ, Song Y, Wang Q, Yaspan BL, Yu TW, Yurkiewicz IR, Beaudet al, Cantor RM, Curland M, Grice DE, Gunel M, Lifton RP, Mane SM, Martin DM, Shaw CA, Sheldon M, Tischfield JA, Walsh CA, Morrow EM, Ledbetter DH, Fombonne E, Lord C, Martin CL, Brooks AI, Sutcliffe JS, Cook EH, Jr, Geschwind D, Roeder K, Devlin B, State MW. 2011. Multiple recurrent de novo CNVs including duplications of the 7q11.23 Williams syndrome region are strongly associated with autism. Neuron 70:863–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos F, Hendrich B, Reik W, Dean W. 2002. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol 241:172–182 [DOI] [PubMed] [Google Scholar]
- Schmidt JV, Levorse JM, Tilghman SM. 1999. Enhancer competition between H19 and Igf2 does not mediate their imprinting. Proc Natl Acad Sci U S A 96:9733–9738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz R, McCole RB, Woodfine K, Wood AJ, Chahal M, Monk D, Moore GE, Oakey RJ. 2009. Transcript- and tissue-specific imprinting of a tumour suppressor gene. Hum Mol Genet 18:118–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearwin KE, Callen BP, Egan JB. 2005. Transcriptional interference—A crash course. Trends Genet 21:339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu W, Chen H, Bo X, Wang S. 2011. Genome-wide analysis of the relationships between DNaseI HS histone modifications and gene expression reveals distinct modes of chromatin domains. Nucleic Acids Res 39:7428–7443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleutels F, Zwart R, Barlow DP. 2002. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415:810–813 [DOI] [PubMed] [Google Scholar]
- Small KS, Hedman AK, Grundberg E, Nica AC, Thorleifsson G, Kong A, Thorsteindottir U, Shin SY, Richards HB, Soranzo N, Ahmadi KR, Lindgren CM, Stefansson K, Dermitzakis ET, Deloukas P, Spector TD, McCarthy MI. 2011. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet 43:561–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer MS, Murphy WJ, Eizirik E and O’Brien SJ. 2003. Placental mammal diversification and the Cretaceous-Tertiary boundary. Proc Natl Acad Sci U S A 100:1056–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava M, Hsieh S, Grinberg A, Williams-Simons L, Huang SP, Pfeifer K. 2000. H19 and Igf2 monoallelic expression is regulated in two distinct ways by a shared cis acting regulatory region upstream of H19. Genes Dev 14:1186–1195 [PMC free article] [PubMed] [Google Scholar]
- Stacey SN, Sulem P, Masson G, Gudjonsson SA, Thorleifsson G, Jakobsdottir M, Sigurdsson A, Gudbjartsson DF, Sigurgeirsson B, Benediktsdottir KR, Thorisdottir K, Ragnarsson R, Scherer D, Hemminki K, Rudnai P, Gurzau E, Koppova K, Botella-Estrada R, Soriano V, Juberias P, Saez B, Gilaberte Y, Fuentelsaz V, Corredera C, Grasa M, Hoiom V, Lindblom A, Bonenkamp JJ, van Rossum MM, Aben KK, de Vries E, Santinami M, Di Mauro MG, Maurichi A, Wendt J, Hochleitner P, Pehamberger H, Gudmundsson J, Magnusdottir DN, Gretarsdottir S, Holm H, Steinthorsdottir V, Frigge ML, Blondal T, Saemundsdottir J, Bjarnason H, Kristjansson K, Bjornsdottir G, Okamoto I, Rivoltini L, Rodolfo M, Kiemeney LA, Hansson J, Nagore E, Mayordomo JI, Kumar R, Karagas MR, Nelson HH, Gulcher JR, Rafnar T, Thorsteinsdottir U, Olafsson JH, Kong A, Stefansson K. 2009. New common variants affecting susceptibility to basal cell carcinoma. Nat Genet 41:909–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenman MJ, Rainier S, Dobry CJ, Grundy P, Horon IL, Feinberg AP. 1994. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms’ tumour. Nat Genet 7:433–439 [DOI] [PubMed] [Google Scholar]
- Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Moller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Muhleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nothen MM, Peltonen L, Collier DA, St Clair D, Stefansson K. 2008. Large recurrent microdeletions associated with schizophrenia. Nature 455:232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo P, Tang SH, Rentsendorj A, Pfeifer GP, Mann JR. 2000. Maternal-specific footprints at putative CTCF sites in the H19 imprinting control region give evidence for insulator function. Curr Biol 10:607–610 [DOI] [PubMed] [Google Scholar]
- Szyf M, Slack AD. 2000. Mechanisms of epigenetic silencing of the c21 gene in Y1 adrenocortical tumor cells. Endocr Res 26:921–930 [DOI] [PubMed] [Google Scholar]
- Takai D, Jones PA. 2002. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A 99:3740–3745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S. 2010. Histone variants—Ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 11:264–275 [DOI] [PubMed] [Google Scholar]
- Tremblay KD, Duran KL, Bartolomei MS. 1997. A 5#57401; 2-kilobase-pair region of the imprinted mouse H19 gene exhibits exclusive paternal methylation throughout development. Mol Cell Biol 17:4322–4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CL, Mackay DM, Callaway JL, Docherty LE, Poole RL, Bullman H, Lever M, Castle BM, Kivuva EC, Turnpenny PD, Mehta SG, Mansour S, Wakeling EL, Mathew V, Madden J, Davies JH, Temple IK. 2010. Methylation analysis of 79 patients with growth restriction reveals novel patterns of methylation change at imprinted loci. Eur J Hum Genet 18:648–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko B. 2010. Allele-specific DNA methylation: Beyond imprinting. Hum Mol Genet 19:R210–R220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrault A, Bilanges B, Mackay DJ, Basyuk E, Ahr B, Fernandez C, Robinson DO, Bockaert J, Journot L. 2001. Characterization of the methylation-sensitive promoter of the imprinted ZAC gene supports its role in transient neonatal diabetes mellitus. J Biol Chem 276:18653–18656 [DOI] [PubMed] [Google Scholar]
- Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, Pavlidis P, Journot L. 2006. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell 11:711–722 [DOI] [PubMed] [Google Scholar]
- Vu TH, Jirtle RL, Hoffman AR. 2006. Cross-species clues of an epigenetic imprinting regulatory code for the IGF2R gene. Cytogenet Genome Res 113:202–208 [DOI] [PubMed] [Google Scholar]
- Vu TH, Nguyen AH, Hoffman AR. 2010. Loss of IGF2 imprinting is associated with abrogation of long-range intrachromosomal interactions in human cancer cells. Hum Mol Genet 19:901–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeling EL, Amero SA, Alders M, Bliek J, Forsythe E, Kumar S, Lim DH, MacDonald F, Mackay DJ, Maher ER, Moore GE, Poole RL, Price SM, Tangeraas T, Turner CL, Van Haelst MM, Willoughby C, Temple IK, Cobben JM. 2010. Epigenotype-phenotype correlations in Silver-Russell syndrome. J Med Genet 47:760–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace EV, Stoddart D, Heron AJ, Mikhailova E, Maglia G, Donohoe TJ, Bayley H. 2010. Identification of epigenetic DNA modifications with a protein nanopore. Chem Commun (Camb) 46:8195–8197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JA, Felsenfeld G. 2007. We gather together: Insulators and genome organization. Curr Opin Genet Dev 17:400–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksberg R, Shuman C, Beckwith JB. 2010. Beckwith-Wiedemann syndrome. Eur J Hum Genet 18:8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins JF, Haig D. 2003. What good is genomic imprinting: The function of parent-specific gene expression. Nat Rev Genet 4:359–368 [DOI] [PubMed] [Google Scholar]
- Williams MF. 2002. Primate encephalization and intelligence. Med Hypotheses 58:284–290 [DOI] [PubMed] [Google Scholar]
- Woodfine K, Huddleston JE, Murrell A. 2011. Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue. Epigenetics Chromatin 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, Walter J. 2011. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun 2:241. [DOI] [PubMed] [Google Scholar]
- Wylie AA, Pulford DJ, McVie-Wylie AJ, Waterland RA, Evans HK, Chen YT, Nolan CM, Orton TC, Jirtle RL. 2003. Tissue-specific inactivation of murine M6P/IGF2R. Am J Pathol 162:321–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazawa K, Ogata T, Ferguson-Smith AC. 2010. Uniparental disomy and human disease: An overview. Am J Med Genet C Semin Med Genet 154C:329–334 [DOI] [PubMed] [Google Scholar]
- Yoshimizu T, Miroglio A, Ripoche MA, Gabory A, Vernucci M, Riccio A, Colnot S, Godard C, Terris B, Jammes H, Dandolo L. 2008. The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci U S A 105:12417–12422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yotova IY, Vlatkovic IM, Pauler FM, Warczok KE, Ambros PF, Oshimura M, Theussl HC, Gessler M, Wagner EF, Barlow DP. 2008. Identification of the human homolog of the imprinted mouse Air non-coding RNA. Genomics 92:464–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Skaar DA, Li Y, Huang D, Price TM, Murphy SK, Jirtle RL. 2011. Novel retrotransposed imprinted locus identified at human 6p25. Nucleic Acids Res 39:5388–5400 [DOI] [PMC free article] [PubMed] [Google Scholar]