

Abstract
Focal adhesions mediate force transfer between ECM-integrin complexes and the cytoskeleton. Although vinculin has been implicated in force transmission, few direct measurements have been made, and there is little mechanistic insight. Using vinculin-null cells expressing vinculin mutants, we demonstrate that vinculin is not required for transmission of adhesive and traction forces but is necessary for myosin contractility-dependent adhesion strength and traction force and for the coupling of cell area and traction force. Adhesion strength and traction forces depend differentially on vinculin head (VH) and tail domains. VH enhances adhesion strength by increasing ECM-bound integrin–talin complexes, independently from interactions with vinculin tail ligands and contractility. A full-length, autoinhibition-deficient mutant (T12) increases adhesion strength compared with VH, implying roles for both vinculin activation and the actin-binding tail. In contrast to adhesion strength, vinculin-dependent traction forces absolutely require a full-length and activated molecule; VH has no effect. Physical linkage of the head and tail domains is required for maximal force responses. Residence times of vinculin in focal adhesions, but not T12 or VH, correlate with applied force, supporting a mechanosensitive model for vinculin activation in which forces stabilize vinculin’s active conformation to promote force transfer.
Keywords: cell adhesion, fibronectin
Integrin-mediated adhesion to ECM provides mechanical anchorage and signals that direct cell migration, proliferation, and differentiation (1, 2), processes central to tissue organization, maintenance, and repair. After ligand binding, integrins cluster into focal adhesion (FA) complexes that transmit adhesive and traction forces (3–6). FAs consist of integrins and actins separated by a ∼40 nm core that includes cytoskeleton (CSK) elements, such as vinculin and talin, and signaling molecules, including focal adhesion kinase and paxillin (7). FAs mediate responses to internal and external stresses by modulating force transfer between integrins and the CSK (8–10). This function has been likened to a “mechanical clutch” between an engine and transmission (11).
On the basis of its structure and binding partners, vinculin represents an attractive candidate for orchestrator of clutch function. Vinculin consists of a globular head (VH) linked to a tail domain (VT) by a proline-rich strap (12). VH contains talin, α-actinin, and α- and β-catenin binding sites; actin, paxillin, and phosphatidylinositol 4,5-bisphosphate (PIP2) binding sites are in VT; and vasodilator-stimulated phosphoprotein (VASP), actin-related protein 2/3 (Arp2/3), and vinexin binding sites reside in the proline-rich region. Interactions with these partners are regulated by an autoinhibited conformation arising from high-affinity intramolecular head–tail binding (13, 14). Activation of vinculin can occur by simultaneous binding to talin and actin or α-catenin and actin (15, 16). Vinculin is activated when localized to FAs (17). Vinculin forms a complex with β1 integrin and talin (18) and interacts with talin to enhance integrin activation (19). Therefore, vinculin has the required molecular properties to mechanically link integrin–ECM complexes to the actomyosin CSK in a regulated manner.
In addition to studies on vinculin’s effects on muscle function (20–22), which may or may not be related to its mechanical functions, vinculin’s role in force transmission has largely been inferred from studies with vinculin-deficient cells showing altered FA assembly and aberrant migration (23, 24). For instance, VH drives FA growth via interactions with talin, whereas VT colocalizes to actin filaments (25), but whether these interactions mediate force transfer is unknown. Vinculin-deficient cells do exhibit reduced cortical CSK stiffness and adhesive force (26, 27), and vinculin is a force-carrying component between FAs and the CSK (28). Although these studies implicate vinculin in force transmission, few such measurements have been made, and some have provided evidence against a role of vinculin in force coupling (29). Moreover, possible roles played by vinculin domains and autoinhibition in mechanotransduction are largely unexplored.
In this study we used stable lines of vinculin-null cells expressing vinculin mutants and two force-measuring platforms to directly analyze whether and how vinculin transmits force. We found that although vinculin is not essential for transmission of traction and adhesive forces, it regulates the coupling of cell area and traction force and is required for myosin contractility-dependent traction forces and adhesion strength. In addition, we found that adhesion strength and traction forces depend to different extents on VH and VT, but maximal force transmission requires the talin/α-actinin–binding site on VH, physical connection of VH and VT, and release of the autoinhibitory head–tail interaction. Finally, we discovered a linear relationship between the traction force at an FA and the residence time for vinculin at that FA, providing evidence for a mechanosensitive model for vinculin activation in which forces applied across vinculin maintain the molecule in its active conformation to increase residence times at FAs to promote force transfer.
Results
Stable Expression of Vinculins in Vinculin-Null Cells.
We expressed WT and mutant vinculins fused to enhanced green fluorescent protein (eGFP) in vinculin-null cells using a tetracycline-regulated retroviral system (Fig. 1A and Fig. S1A). This strategy has major advantages over routine approaches using transient expression in vinculin-expressing cells: (i) experiments are based on the same cell population, eliminating batch-to-batch variability in expression levels; (ii) reexpression of target vinculins in cells lacking endogenous expression avoids confounding effects of endogenous vinculin; and (iii) the retroviral system has high transduction efficiencies, resulting in a polyclonal population of engineered cells and avoiding issues associated with clonal lines. We applied this system to two vinculin-null mouse embryonic fibroblast lines [MEF1 (15, 30) and MEF2 (13, 23, 31)] to rule out artifacts of a particular line. After transduction, WT vinculin-eGFP positive cell populations were enriched by FACS. Western blotting confirmed expression of vinculin constructs in both lines of transduced vinculin-null MEFs (Fig. 1B). Expression levels for WT were comparable to levels in vinculin+/+ cells derived from littermate controls. Culturing MEF2 cells in the presence of anhydrotetracycline significantly repressed levels of vinculin-eGFP expression. eGFP-vinculin localized to FAs, demonstrating proper function for the expressed proteins (Fig. 1C).
Fig. 1.
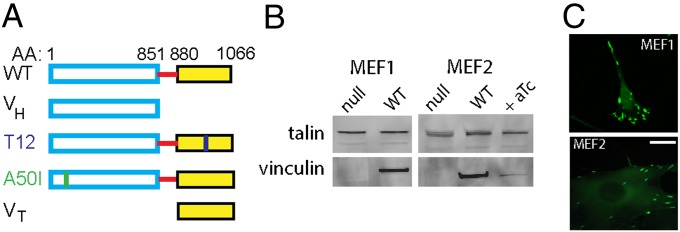
Vinculin-null cells engineered to express vinculin variants. (A) Vinculin variants fused to eGFP: WT, head domain (VH), auto-inhibition mutant (T12), talin-binding mutant (A50I), and tail domain (VT). (B) Western blot analysis of engineered cell lines confirmed expression of vinculin constructs. Vinculin expression was repressed in presence of anhydrotetracycline (aTc, 100 ng/mL). (C) eGFP-vinculin localized to FAs for both MEF1- and MEF2-derived lines. (Scale bar, 10 µm.)
To investigate the contributions of vinculin domains to force transmission, cell lines expressing eGFP-vinculin mutants were derived from MEF1 vinculin-null cells. We first examined two mutants: (i) a molecule comprising only VH (1–851) and lacking most of the proline-rich strap and actin-binding tail, and (ii) a full-length variant (T12) with mutations along the head–tail interface that reduce head–tail binding affinity 100-fold and render the molecule in an active conformation that can readily bind talin and actin (Fig. 1A). These mutants have been characterized for their binding to talin and actin and recruitment to FAs (13, 25, 31). Equivalent expression levels were observed among cell lines (Fig. S1B). VH-expressing cells were more round than WT-expressing cells, and the VH construct localized to large radial FAs (Fig. S1C). T12-expressing cells displayed more and larger FAs than WT-expressing cells (Fig. S1C). These phenotypes are consistent with observations for transiently transfected cells (13, 25).
Vinculin Activation Increases Traction Forces and Regulates Coupling Between Cell Area and Total Traction Force.
We used microfabricated postarray deflection devices (mPADs) to measure traction forces. When seeded overnight onto fibronectin (FN)-coated mPADs, cells spread and developed FAs (Fig. 2A and Fig. S2A). T12-expressing cells exhibited higher spread areas compared with other lines, and VH-expressing cells spread more than null but not WT-expressing cells (Fig. 2B). Treatment with blebbistatin (20 µM, 30 min), an inhibitor of myosin contractility, reduced cell area for null, WT-, and T12-expressing cells but not VH-expressing cells. We measured post deflections for null and vinculin-expressing lines. Fig. 2A and Fig. S2A present images (Upper) for FN-coated posts (red) and eGFP-vinculin (green) recruited to FAs, with the cell outlined in yellow and force vectors (cyan, Lower) calculated from post deflections. The magnitude of traction forces varied significantly across a single cell, with the highest forces at the cell periphery (Fig. 2A and Fig. S2A). Fig. 2C presents box-whisker plots for the total traction force per cell, which represents the sum of the magnitudes of the force vectors for each cell and is commonly used for reporting traction forces (32). Traction forces are dynamic, and the data in Fig. 2C represent a “snapshot” of the traction forces in a cell population at equilibrium (overnight culture). Vinculin-null cells generate considerable traction forces (∼100 nN), indicating that vinculin is not required for force transmission at FAs. WT expression increased the total traction force by 40% compared with vinculin-null controls. This result demonstrates that vinculin enhances the transmission of traction forces. In contrast, VH expression had no effect on the total traction force compared with null cells, showing that, despite localization to FAs, VH by itself does not influence traction forces. T12-expressing cells exhibited twofold higher total traction forces than null cells, and the total traction force was 40% higher than that generated by WT-expressing cells. This result shows that disruption of vinculin head–tail inhibition enhances the transmission of traction forces. Blebbistatin reduced traction forces by 30% in WT- and T12-expressing cells, but the total traction force in null and VH-expressing cells was insensitive to blebbistatin. This result shows that transmission of myosin contractility-dependent traction forces at FAs requires a full-length vinculin molecule containing both VH and VT.
Fig. 2.

Vinculin regulates traction forces. (A) Cells spread on mPADs (posts labeled red) showing localization of vinculin (eGFP) to FAs (Upper) and spreading (yellow outline) and force vectors (cyan arrows) (Lower). (Scale bar, 4 µm.) (B) Box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile) for cell area (>24 cells per condition). Kruskal-Wallis P < 0.0001, *P < 0.05 vs. null, #P < 0.05 vs. WT, †P < 0.05 vs. VH; §P < 0.05 blebb vs. control. (C) Box-whisker plot (mean, 10th, 25th, 75th, and 90th percentile) for total traction force per cell (>24 cells). Kruskal-Wallis P < 0.0001, *P < 0.022 vs. null, #P < 0.01 vs. WT, †P < 0.01 vs. VH; §P < 0.05 blebb vs. control. (D) Relationship between traction force and cell area (>24 cells per condition) showing linear regression fits.
We examined the relationship between cell area and traction force because Fu et al. (32) showed tight coupling between cell area and CSK tension, suggesting that cell area–traction force coupling represents a robust metric to analyze force responses to vinculin expression. Fig. 2D and Fig. S2B plot cell area and corresponding traction force for individual cells as well as regression lines. There is a strong correlation between cell area and traction force for null, WT-, and T12-expressing cells. Vinculin-null cells displayed a linear relationship between cell area and traction force, indicating that vinculin is dispensable for cell area–traction force coupling. This result supports a role for other FA components in the transmission of traction forces, such as direct talin–actin force transfer (33). However, WT expression significantly enhances coupling between cell area and traction force, as demonstrated by the twofold increase in the regression slope compared with null cells. T12 expression results in stronger coupling between cell area and traction force compared with WT, showing that vinculin head–tail inhibition plays a critical role in regulating traction forces. VH-expressing cells showed no coupling between cell area and traction force. This result indicates that VH disrupts basal cell area–traction force coupling, demonstrating that both VH and VT are required for vinculin-enhanced coupling between cell area and total traction force. Although blebbistatin reduces cell area and traction force, it does not disrupt the relationship between cell area and traction force (Fig. S2C).
How Vinculin Head and Tail Domains and Autoinhibition Contribute to Adhesion Strength.
We measured the steady-state (16 h after seeding) adhesion strength of cells expressing WT to FN using a spinning disk device. Whereas traction force measurements report on forces applied to the substrate arising from actomyosin contractility or actin polymerization, the adhesion strength assay measures the amount of force required to detach the cell from the ECM. The spinning disk exposes cells to a hydrodynamic shear force that increases linearly with radial position from the disk center and provides sensitive measurements of adhesion strength (Fig. S3A).
WT-expressing cells were cultured overnight on FN-coated micropatterned islands to eliminate differences in adhesive area and cell shape. This is an important consideration because expression of these constructs produces changes in cell area, a parameter that also regulates adhesion strength (6). Cells remained constrained to the micropatterned area as single cells. Expression of WT in vinculin-null MEF1 and MEF2 cells increased adhesion strength by 25% and 27%, respectively, over null controls (Fig. 3A). To test whether the increases in adhesion strength were caused by vinculin expression, we cultured WT-MEF2 cells in anhydrotetracycline to suppress expression. Under these conditions, the adhesion strength returned to the levels of null cells (Fig. 3A). Studies with blocking antibody demonstrated that adhesion to FN was mediated by β1 integrin (Fig. S3B). These results demonstrate that vinculin directly modulates adhesion strength and that this system provides direct measurements of β1 integrin–FN-mediated adhesion strength.
Fig. 3.

Vinculin head and tail domains have distinct contributions to adhesion strength. (A) Expression of WT in vinculin-null cells increased adhesion strength over controls (*P < 0.03 vs. null, #P < 0.05 vs. null, +aTc). aTc-induced suppression of WT expression returned adhesion strength to null levels. (B) VH expression increased adhesion strength by 25%, whereas T12 increased adhesion strength by 50% compared with null controls. ANOVA P < 0.0001, *P < 0.05 vs. null, #P < 0.05 vs. WT, and †P < 0.05 vs. VH.
We next examined the adhesion strength of vinculin-null cells expressing vinculin mutants. VH expression increased adhesion strength by 25% compared with null controls (Fig. 3B), indicating that recruitment of VH to adhesive complexes increases adhesion strength independently from VT. VH increased adhesion strength to equivalent levels as WT. T12 expression increased adhesion strength by 50% over null cells (Fig. 3B), doubling the increase in adhesion strength by either WT or VH. This result indicates that regulation of vinculin autoinhibition plays an important role in the generation of adhesion strength and that the active vinculin conformation presenting head and tail domains results in maximal adhesion strength.
We hypothesized that binding of VH to talin or α-actinin was essential for vinculin-dependent increases in adhesion strength. We examined the effect of expressing a full-length, talin-/α-actinin-binding deficient mutant (A50I). No differences in adhesion strength were observed between A50I-expressing and null cells (Fig. S3C), indicating that vinculin binding to one or both of these ligands is essential for vinculin-mediated adhesion strength.
Physical Linkage Between Vinculin Head and Tail Domains Is Required for Maximal Adhesion Strength.
We postulated that the increased adhesion strength for T12-expressing cells relative to VH-expressing cells arises from differences in load transfer from the integrin–ECM complexes to the actin CSK via vinculin. We tested this model by independently expressing VH and VT in the same cell. We transiently transfected MEF1 cells with plasmids encoding for VH, T12, or VT or cotransfected plasmids for VH and VT. Transfected cells were enriched by flow cytometry sorting and seeded on FN islands. Image analysis demonstrated that vinculin mutants localized to FN patterns in a similar way as those in the stable lines (Fig. S4A). Cotransfected VH and VT localized to the FN island, but there was no strong colocalization because these two domains are not physically linked (Fig. S4B). Coexpression of separate VH and VT did not alter adhesion strength compared with expression of either domain, and adhesion strength was 25% lower than that for T12 expression (Fig. S4C). Expression of VT resulted in similar levels of adhesion strength as VH. This result was unexpected because VT does not bind to the integrin–talin complex or α-actinin-rich lamellopodia protrusions (14, 34). A likely explanation for the effects of VT is that this domain enhances adhesion strength by cross-linking actin to increase cortical CSK stiffness and load sharing among integrin bonds. Indeed, there is evidence that VT enhances actin cross-linking and cortical CSK stiffness (29). These data show that the physical linkage between vinculin head and tail domains is required for maximal adhesion strength, indicating that force transfer from the adhesive clusters to the actin CSK via vinculin contributes to adhesion strength.
Vinculin Head–Tail Autoinhibition Regulates the Number of Integrin–FN Complexes and Recruitment of Vinculin and Talin.
Because adhesion strength is regulated by the number/distribution of integrin–ECM complexes, FA assembly, and CSK interactions (6), we analyzed integrin binding and FA assembly to gain insights into possible reasons for the differences in adhesion strength. We first examined the effects of vinculin mutants on the levels and distribution of integrin–FN complexes using a cross-linking and detergent extraction method to selectively retain integrin–FN complexes. Fig. 4A presents images of single cells adhering to FN islands and immunostained for β1 integrin, and Fig. 4 B and C plot the fraction of the adhesive area occupied by integrin–FN complexes and the intensity of integrin staining over the micropatterned area. Vinculin-null cells assembled integrin β1–FN complexes along the periphery of the adhesive area, with minimal staining in the interior. WT expression did not change the spatial distribution or area occupied by integrin–FN complexes but resulted in a 15% increase in intensity. In contrast, VH expression resulted in a fourfold increase in the area occupied by integrin–FN complexes, mostly localized to the periphery of the adhesive area, and a 40% increase in intensity compared with the null control. T12 expression yielded a fourfold increase in the area of integrin–FN complexes and a 50% increase in intensity compared with the null control. These results demonstrate that WT has a modest effect in regulating the number and spatial distribution of integrin–FN complexes and that presentation of VH, either alone or in a mutant with disrupted head–tail binding, significantly increases the number and spatial distribution of integrin–FN complexes. Furthermore, the lack of differences in integrin–FN complexes between VH and T12 indicates that VT does not contribute significantly to the assembly or stability of integrin–FN complexes. These results indicate that vinculin head–tail inhibition to control exposure of VH plays a major regulatory role in controlling the number and spatial distribution of ECM–integrin complexes.
Fig. 4.

Vinculin head–tail interaction regulates integrin–FN complexes and FA assembly. (A) Immunostaining for β1 integrin for cells adhering to FN micropatterned islands. Staining is shown as grayscale on white background to facilitate visualization. (Scale bar, 5 µm.) (B) Fraction of adhesive area occupied by integrin–FN complexes. ANOVA P < 0.0001, *P < 0.05 vs. null, #P < 0.05 vs. WT. (C) Intensity of integrin staining over micropatterned area. ANOVA P < 0.0001, *P < 0.05 vs. null, #P < 0.05 vs. WT (>60 cells per condition). (D) (Upper) Immunostaining for talin for cells adhering to FN islands. (Scale bar, 5 µm.) (Lower) Area of talin staining normalized to total adhesive area. ANOVA P < 0.0001, *P < 0.05 vs. null, #P < 0.05 vs. WT. (E) (Upper) Immunostaining for vinculin for cells on FN islands (Scale bar, 5 µm.) (Lower) Area of vinculin staining normalized to total adhesive area. ANOVA P < 0.0001, *P < 0.05 vs. null, #P < 0.05 vs. WT (>20 cells per condition).
We examined the role of vinculin autoinhibition on FA assembly by measuring recruitment of talin and the vinculin constructs to FAs. For all cells, talin and vinculin staining was restricted to the circumference of the micropatterned area (Fig. 4 D and E), consistent with the staining patterns for integrin–FN complexes. In addition to circumferential staining, T12- and VH-expressing cells exhibited small vinculin and talin clusters in the interior of the adhesive area. Analysis of the adhesive area occupied by talin showed that WT expression had modest effects compared with null cells (Fig. 4D). In contrast, expression of VH or T12 significantly enhanced the area occupied by talin and vinculin compared with WT and null cells (Fig. 4 D and E). These results demonstrate that WT has a modest effect in regulating FA assembly and that presentation of VH, either alone or in a mutant with disrupted head–tail binding, significantly increases the area occupied by FAs. Furthermore, the lack of differences between VH and T12 indicates that VT does not contribute significantly to FA assembly. These results suggest that vinculin head–tail autoinhibition plays an important function in regulating FA assembly by controlling the exposure of VH, resulting in an increase of both talin and vinculin recruitment to FAs.
Because there were no major differences in integrin–FN complexes and FA assembly between null and WT-expressing cells, we attribute the increased adhesion strength for WT to enhanced force distribution at the adhesive interface due to WT-mediated local cortical CSK stiffening and load transfer to the CSK. In contrast, the increased adhesion strength for VH compared with the null control likely results from the higher number of integrin–FN complexes and enhanced FA assembly. Compared with VH, T12 further increases adhesion strength without altering the levels of integrin–FN complexes or talin/vinculin recruitment to FAs. We attribute this additional increase in adhesion strength for T12 to VT binding to the actin CSK to enhance load transfer.
Effect of Contractility on Adhesion Strength Is Mediated by Vinculin Autoinhibition.
The increase in adhesion strength for T12 compared with VH suggests that force transfer from FAs to the CSK via VT is required for maximal adhesion strength. Because myosin contractility is critical to force generation (4, 5, 26, 35), we analyzed the contributions of myosin contractility to adhesion strength in the context of vinculin expression. Cells were seeded overnight on FN islands and exposed to blebbistatin (20 µM) for 30 min before measuring adhesion strength. Blebbistatin had no effect on adhesion strength for vinculin-null cells (Fig. 5). In contrast, blebbistatin reduced adhesion strength by 30% in WT-expressing cells. This result is consistent with our work demonstrating that vinculin is required for contractility-dependent adhesion strength (26). In this earlier study, we showed that the reductions in adhesion strength in response to contractility inhibition were associated with loss of vinculin and talin from FAs, in agreement with contractility-dependent FA assembly (36).
Fig. 5.
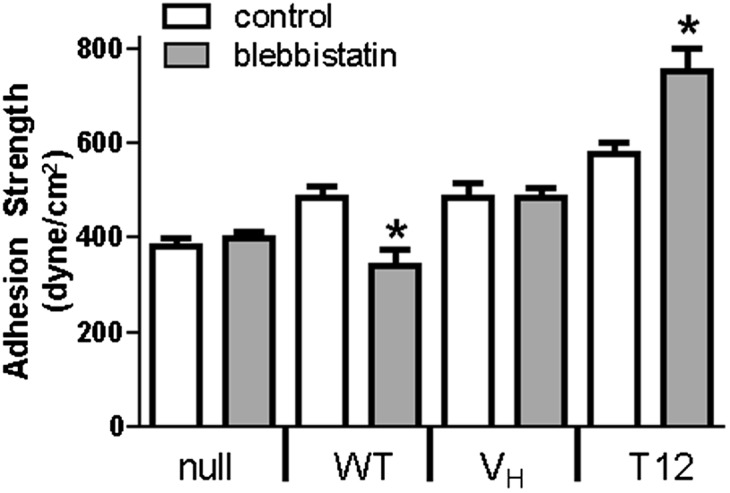
Effect of contractility on adhesion strength is mediated by vinculin autoinhibition. *P < 0.05 vs. control.
We hypothesized that the adhesion strength of VH-expressing cells would be insensitive to blebbistatin treatment because this mutant cannot interact with the actin CSK because it lacks binding sites for Arp2/3, paxillin, and actin. Indeed, blebbistatin treatment had no effect on the adhesion strength of VH-expressing cells (Fig. 5), indicating that the potential VASP-binding site in VH is not capable of mediating interaction with actomyosin. Moreover, integrin–FN complex assembly and recruitment of talin and VH to FAs were insensitive to blebbistatin (Fig. S5). This response differs from the effects of blebbistatin on WT vinculin-expressing cells and indicates that contractility-mediated changes in adhesion strength and FA assembly require a full-length, actin-binding vinculin molecule. We next analyzed the adhesion strength of T12-expressing cells with blebbistatin treatment. Surprisingly, blebbistatin enhanced adhesion strength for T12-expressing cells by 25% compared with untreated controls, in stark contrast to the effects of blebbistatin on WT-expressing cells (Fig. 5). Blebbistatin did not alter integrin–FN complex formation or vinculin and talin recruitment to FAs for T12-expressing cells (Fig. S5), so this enhancement in adhesion strength cannot be attributed to changes in integrin–FN bonds or FA assembly. These results indicate that vinculin autoinhibitory regulation is critical to contractility-mediated changes in adhesion strength, demonstrating a role for activated vinculin in force transfer. Furthermore, these data suggest that T12 promotes adhesion strength better than VH through the ability of VT to interact with the actin CSK and by the inability of T12 to substantially regain the autoinhibited conformation.
Vinculin Residence Times in FAs Correlate with Applied Force and Require Autoinhibitory Head–Tail Interactions.
The striking difference in the effects of blebbistatin on the adhesion strength of WT- vs. T12-expressing cells implicates head–tail interactions in the regulation of contractility-mediated enhancements in adhesive force. We hypothesized that forces applied across vinculin maintain the molecule in its active conformation and counterbalance the high-affinity head–tail inhibition. For WT, inhibition of contractility unloads the vinculin molecule and promotes rebinding between VH and VT, resulting in vinculin inactivation and FA disassembly. For the T12 mutant, the head–tail interaction is reduced 100-fold, resulting in a molecule that, although not constitutively open (13), is much easier to open and harder to reclose. This mutant retains the WT affinity of VH for talin and VT for actin. Because of the ∼100-fold reduced head–tail autoinhibition, the T12 mutant would not reclose significantly when unloaded, and it would therefore transmit adhesive forces even during blebbistatin-mediated inhibition of contractility.
To test this model, we examined the relationship between vinculin residence times at FAs and applied force by performing fluorescence recovery after photobleaching (FRAP) experiments on cells on mPADs. We examined recovery times after photobleaching for eGFP-vinculin–containing FAs associated with posts with known deflections. In this fashion we could monitor vinculin dynamics at FAs under force. FRAP movies for WT-vinculin FAs under different traction forces are provided (Movies S1–S4), and Fig. S6A presents images of WT-containing FAs on mPADs before and after photobleaching. Fig. 6A displays FRAP recovery curves for WT in FAs transmitting different forces. The applied force remained constant over the 3-min FRAP experiment (Fig. S6 B and C). The half-life recovery time (t1/2) vs. traction force for individual FAs in WT-expressing cells is plotted in Fig. 6B. Strikingly, we observed a linear relationship between applied force and recovery time for WT. A simple explanation for the strong correlation between vinculin residence time in FAs and applied force is that tension applied across the vinculin molecule maintains vinculin in the active conformation to increase its residence time in FAs. Blebbistatin (20 µM) eliminates the linear relationship between recovery time and force (Fig. 6C), consistent with our model. We also examined the recovery time vs. force relationship for cells expressing T12 and VH (Fig. 6D). Both VH and T12 exhibited twofold slower recovery times compared with WT, consistent with previous data (25, 31). However, in stark contrast to WT, recovery times for T12 and VH did not correlate with applied forces at FAs. These results demonstrate that the vinculin head–tail interaction is critical to the coupling of vinculin residence time at FAs and applied force. Importantly, the lack of correlation of recovery times with force for VH demonstrates that VT is required for FA residence time–force coupling. Additionally, this result rules out the explanation that this phenomenon arises from force-mediated exposure of vinculin binding sites on talin (37). These findings support a mechanosensitive model for vinculin activation in which forces applied across vinculin maintain the molecule in its active conformation to increase residence times at FAs to transfer force.
Fig. 6.

Vinculin residence times in FAs correlate with applied force and require head–tail interactions. FRAP was performed on FAs on mPAD posts with known applied forces. (A) FRAP recovery curves for WT localized to FAs transmitting different forces. (B) Correlation between recovery time (t1/2) and applied force (>15 cells analyzed per condition) for WT. Linear regression: t1/2 = 1.56 × force + 20.0, P < 0.0001. (C) Blebbistatin treatment (20 µM) eliminates linear relationship between recovery time and force (no linear dependence, P = 0.75). (D) No correlation between t1/2 and applied force was observed for T12 (P = 0.45) or VH (P = 0.35). Different y axis scales were used between WT and VH, T12 for ease of visualization.
Discussion
How vinculin and its interactions with binding partners transmit force remains poorly understood. Here we clearly demonstrate that vinculin regulates both traction forces and adhesion strength to ECM and dissect the contributions of different vinculin domains to these force outputs. The vinculin-dependent enhancements in traction force and adhesion strength quantify vinculin’s contributions in force transmission and provide a mechanical basis to explain the effects of vinculin deletion on impaired cell spreading, migration, and muscle contraction. We show that vinculin regulates the coupling between cell area and traction force. The coupling between cell area and traction force reflects an integrated feedback response regulating cell shape and has been implicated in rigidity sensing (32). Our finding that full-length vinculin enhances cell area–traction force coupling but VH completely disrupts this coupling indicates that vinculin is a key regulator of these mechanical responses and identifies a unique function for vinculin in mechanosensing.
Although VH drives FA growth and VT localizes to actin filaments (25), we demonstrate distinct contributions to force transmission for each domain. Vinculin transmits force by increasing ECM-bound integrin–talin complexes via VH, whereas VT transfers force to the actin CSK (Fig. S7). These mechanical functions require the talin/α-actinin-binding site on VH. We note that vinculin-dependent changes in force transmission do not scale proportionally with changes in integrin-FN binding and FA assembly owing to biomechanical considerations, including spatial location of integrin–FN complexes and cortical CSK stiffness that result in nonlinear bond loading (6, 38). We also discovered an important role for vinculin’s head–tail autoinhibitory interaction in regulating traction forces, adhesion strength, and the coupling between cell area and traction force. Finally, maximal adhesion strength requires VH and VT to be physically coupled, indicating that force transfer occurs through the vinculin molecule rather than independent contributions from each domain.
Although myosin contractility is critical to traction forces and adhesion strength, the contribution of vinculin to myosin contractility-dependent adhesive forces is unknown. We demonstrate that a full-length vinculin molecule containing both VH and VT is required for myosin contractility-dependent effects on traction force and adhesion strength, suggesting that force transfer occurs through the vinculin molecule. For WT-expressing cells, blebbistatin treatment reduced adhesion strength to vinculin-null levels, whereas blebbistatin did not alter adhesion strength in vinculin-null cells. Surprisingly, blebbistatin enhanced adhesion strength for T12-expressing cells, indicating that head–tail autoinhibition regulates the vinculin-dependent effects of myosin contractility on adhesion strength. One explanation for the blebbistatin-dependent increases in adhesion strength for T12 is that inhibition of contractility reduces the internal force applied to FAs, thereby increasing the force that can be supported by the external ECM–integrin linkage at FAs (39). The requirement for vinculin in myosin contractility-dependent adhesive forces establishes a unique function for vinculin in mechanotransduction beyond regulation of FA assembly (25, 28).
By applying FRAP to an FA under force, we discovered that vinculin residence time at an FA correlates linearly with the force applied to that FA. Vinculin residence time–force coupling requires a full-length molecule, head–tail autoinhibition, and myosin contractility. These results directly relate vinculin dynamics to force and complement studies showing that contractility influences FA dynamics (40). Our data support a model for vinculin stabilization in which forces applied across vinculin maintain the molecule in its active conformation to increase residence times at FAs to promote force transfer (Fig. S7). Direct measurements of the forces experienced by vinculin in the context of adhesion strength and traction forces are still needed to fully validate this model. Vinculin’s binding partners and phosphorylation sites could provide indirect mechanisms for its force-regulated recruitment and activity.
Our findings support a mechanosensitive model for vinculin activation. Structural and biochemical data support a role for talin and actin binding in vinculin activation (13, 15, 41, 42). Given the requirements for the talin-binding site on VH and the actin-binding VT for force transmission, it is likely that coordinated activation by talin and actin provides a major mechanism driving vinculin activation and that force transmission across vinculin stabilizes its active conformation. This mechanism provides an explanation for the observation that vinculin recruitment to FAs is separable from mechanical loading (28). Force-dependent vinculin activation identifies another mechanism that complements mechanosensitive pathways at FAs, such as integrin–FN binding (43, 44) and talin stretching to expose binding sites (37).
The improved understanding of vinculin’s contributions to force transmission provided by this work has several implications. First, vinculin regulates the transmission of adhesive forces by modulating ECM–integrin complexes via VH and transmitting forces from these complexes to the actin CSK via VT. Second, vinculin regulates cell area–traction force coupling and myosin contractility-dependent adhesion strength and traction forces. As such, vinculin likely provides “fine tuning” control required for coordinated processes like migration and contraction. Finally, force-based regulation of vinculin activation provides a mechanism to generate local mechanosensitive responses at FAs such as force-dependent FA growth (8, 36). Mechano-regulation of vinculin residence times at FAs represents a pathway for coordinated assembly of FAs at the leading edge and disassembly of FAs at the rear of the cell. In fact, experiments with a force sensor revealed high forces across vinculin at the leading edge and low forces at the trailing edge (28), consistent with our model. Collectively, this work provides these important insights into how vinculin’s structure and binding partners interact with contractility to regulate force transmission.
Methods
Vinculin lines were generated by retroviral transduction of vinculin-null cells and FACS selection. Cell adhesion strength and traction forces were measured with a spinning disk device and mPADs, respectively. FRAP was performed on cells on mPADs. Detailed methods are presented in SI Methods.
Supplementary Material
Acknowledgments
Funding was provided by National Institutes of Health Grants R01-GM065918 (to A.J.G.), R01-EB00262, R01-GM74048 (both to C.S.C.), and R01-GM41605 (to S.W.C.), RESBIO Technology Resource for Polymeric Biomaterials, Human Frontier Science Program Grant RGP0013 (to J.F.C.), and National Science Foundation Career Award DMR-0955811 (to J.E.C.) and CMMI-1129611 (to J.F.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1216209110/-/DCSupplemental.
References
- 1.Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Wickström SA, Radovanac K, Fässler R. Genetic analyses of integrin signaling. Cold Spring Harb Perspect Biol. 2011;3(2):a005116. doi: 10.1101/cshperspect.a005116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beningo KA, Dembo M, Kaverina I, Small JV, Wang YL. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J Cell Biol. 2001;153(4):881–888. doi: 10.1083/jcb.153.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balaban NQ, et al. Force and focal adhesion assembly: A close relationship studied using elastic micropatterned substrates. Nat Cell Biol. 2001;3(5):466–472. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- 5.Tan JL, et al. Cells lying on a bed of microneedles: An approach to isolate mechanical force. Proc Natl Acad Sci USA. 2003;100(4):1484–1489. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallant ND, Michael KE, García AJ. Cell adhesion strengthening: Contributions of adhesive area, integrin binding, and focal adhesion assembly. Mol Biol Cell. 2005;16(9):4329–4340. doi: 10.1091/mbc.E05-02-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanchanawong P, et al. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468(7323):580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riveline D, et al. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153(6):1175–1186. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galbraith CG, Yamada KM, Sheetz MP. The relationship between force and focal complex development. J Cell Biol. 2002;159(4):695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 1997;88(1):39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- 11.Smilenov LB, Mikhailov A, Pelham RJ, Marcantonio EE, Gundersen GG. Focal adhesion motility revealed in stationary fibroblasts. Science. 1999;286(5442):1172–1174. doi: 10.1126/science.286.5442.1172. [DOI] [PubMed] [Google Scholar]
- 12.Ziegler WH, Liddington RC, Critchley DR. The structure and regulation of vinculin. Trends Cell Biol. 2006;16(9):453–460. doi: 10.1016/j.tcb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Cohen DM, Chen H, Johnson RP, Choudhury B, Craig SW. Two distinct head-tail interfaces cooperate to suppress activation of vinculin by talin. J Biol Chem. 2005;280(17):17109–17117. doi: 10.1074/jbc.M414704200. [DOI] [PubMed] [Google Scholar]
- 14.Johnson RP, Craig SW. F-actin binding site masked by the intramolecular association of vinculin head and tail domains. Nature. 1995;373(6511):261–264. doi: 10.1038/373261a0. [DOI] [PubMed] [Google Scholar]
- 15.Chen H, Choudhury DM, Craig SW. Coincidence of actin filaments and talin is required to activate vinculin. J Biol Chem. 2006;281(52):40389–40398. doi: 10.1074/jbc.M607324200. [DOI] [PubMed] [Google Scholar]
- 16.Peng X, Maiers JL, Choudhury D, Craig SW, DeMali KA. α-Catenin uses a novel mechanism to activate vinculin. J Biol Chem. 2012;287(10):7728–7737. doi: 10.1074/jbc.M111.297481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Cohen DM, Choudhury DM, Kioka N, Craig SW. Spatial distribution and functional significance of activated vinculin in living cells. J Cell Biol. 2005;169(3):459–470. doi: 10.1083/jcb.200410100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horwitz AF, Duggan K, Buck CA, Beckerle MC, Burridge K. Interaction of plasma membrane fibronectin receptor with talin—a transmembrane linkage. Nature. 1986;320(6062):531–533. doi: 10.1038/320531a0. [DOI] [PubMed] [Google Scholar]
- 19.Ohmori T, et al. Vinculin activates inside-out signaling of integrin αIIbβ3 in Chinese hamster ovary cells. Biochem Biophys Res Commun. 2010;400(3):323–328. doi: 10.1016/j.bbrc.2010.08.056. [DOI] [PubMed] [Google Scholar]
- 20.Zemljic-Harpf AE, et al. Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol Cell Biol. 2007;27(21):7522–7537. doi: 10.1128/MCB.00728-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y, Zhang W, Gunst SJ. Activation of vinculin induced by cholinergic stimulation regulates contraction of tracheal smooth muscle tissue. J Biol Chem. 2011;286(5):3630–3644. doi: 10.1074/jbc.M110.139923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barstead RJ, Waterston RH. Vinculin is essential for muscle function in the nematode. J Cell Biol. 1991;114(4):715–724. doi: 10.1083/jcb.114.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu W, Baribault H, Adamson ED. Vinculin knockout results in heart and brain defects during embryonic development. Development. 1998;125(2):327–337. doi: 10.1242/dev.125.2.327. [DOI] [PubMed] [Google Scholar]
- 24.Xu W, Coll JL, Adamson ED. Rescue of the mutant phenotype by reexpression of full-length vinculin in null F9 cells; effects on cell locomotion by domain deleted vinculin. J Cell Sci. 1998;111(Pt 11):1535–1544. doi: 10.1242/jcs.111.11.1535. [DOI] [PubMed] [Google Scholar]
- 25.Humphries JD, et al. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J Cell Biol. 2007;179(5):1043–1057. doi: 10.1083/jcb.200703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dumbauld DW, et al. Contractility modulates cell adhesion strengthening through focal adhesion kinase and assembly of vinculin-containing focal adhesions. J Cell Physiol. 2010;223(3):746–756. doi: 10.1002/jcp.22084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diez G, Auernheimer V, Fabry B, Goldmann WH. Head/tail interaction of vinculin influences cell mechanical behavior. Biochem Biophys Res Commun. 2011;406(1):85–88. doi: 10.1016/j.bbrc.2011.01.115. [DOI] [PubMed] [Google Scholar]
- 28.Grashoff C, et al. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466(7303):263–266. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mierke CT, et al. Mechano-coupling and regulation of contractility by the vinculin tail domain. Biophys J. 2008;94(2):661–670. doi: 10.1529/biophysj.107.108472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeMali KA, Barlow CA, Burridge K. Recruitment of the Arp2/3 complex to vinculin: Coupling membrane protrusion to matrix adhesion. J Cell Biol. 2002;159(5):881–891. doi: 10.1083/jcb.200206043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen DM, Kutscher B, Chen H, Murphy DB, Craig SW. A conformational switch in vinculin drives formation and dynamics of a talin-vinculin complex at focal adhesions. J Biol Chem. 2006;281(23):16006–16015. doi: 10.1074/jbc.M600738200. [DOI] [PubMed] [Google Scholar]
- 32.Fu J, et al. Mechanical regulation of cell function with geometrically modulated elastomeric substrates. Nat Methods. 2010;7(9):733–736. doi: 10.1038/nmeth.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang G, Giannone G, Critchley DR, Fukumoto E, Sheetz MP. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature. 2003;424(6946):334–337. doi: 10.1038/nature01805. [DOI] [PubMed] [Google Scholar]
- 34.Menkel AR, et al. Characterization of an F-actin-binding domain in the cytoskeletal protein vinculin. J Cell Biol. 1994;126(5):1231–1240. doi: 10.1083/jcb.126.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gardel ML, et al. Traction stress in focal adhesions correlates biphasically with actin retrograde flow speed. J Cell Biol. 2008;183(6):999–1005. doi: 10.1083/jcb.200810060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133(6):1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.del Rio A, et al. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323(5914):638–641. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans EA. Detailed mechanics of membrane-membrane adhesion and separation. II. Discrete kinetically trapped molecular cross-bridges. Biophys J. 1985;48(1):185–192. doi: 10.1016/S0006-3495(85)83771-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coyer SR, et al. Nanopatterning reveals an ECM area threshold for focal adhesion assembly and force transmission that is regulated by integrin activation and cytoskeleton tension. J Cell Sci. 2012;125(Pt 21):5110–5123. doi: 10.1242/jcs.108035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolfenson H, Bershadsky A, Henis YI, Geiger B. Actomyosin-generated tension controls the molecular kinetics of focal adhesions. J Cell Sci. 2011;124(9):1425–1432. doi: 10.1242/jcs.077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Izard T, et al. Vinculin activation by talin through helical bundle conversion. Nature. 2004;427(6970):171–175. doi: 10.1038/nature02281. [DOI] [PubMed] [Google Scholar]
- 42.Bakolitsa C, et al. Structural basis for vinculin activation at sites of cell adhesion. Nature. 2004;430(6999):583–586. doi: 10.1038/nature02610. [DOI] [PubMed] [Google Scholar]
- 43.Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science. 2009;323(5914):642–644. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- 44.Kong F, García AJ, Mould AP, Humphries MJ, Zhu C. Demonstration of catch bonds between an integrin and its ligand. J Cell Biol. 2009;185(7):1275–1284. doi: 10.1083/jcb.200810002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.