

Abstract
A cardinal symptom of major depressive disorder (MDD) is the disruption of circadian patterns. However, to date, there is no direct evidence of circadian clock dysregulation in the brains of patients who have MDD. Circadian rhythmicity of gene expression has been observed in animals and peripheral human tissues, but its presence and variability in the human brain were difficult to characterize. Here, we applied time-of-death analysis to gene expression data from high-quality postmortem brains, examining 24-h cyclic patterns in six cortical and limbic regions of 55 subjects with no history of psychiatric or neurological illnesses (“controls”) and 34 patients with MDD. Our dataset covered ∼12,000 transcripts in the dorsolateral prefrontal cortex, anterior cingulate cortex, hippocampus, amygdala, nucleus accumbens, and cerebellum. Several hundred transcripts in each region showed 24-h cyclic patterns in controls, and >100 transcripts exhibited consistent rhythmicity and phase synchrony across regions. Among the top-ranked rhythmic genes were the canonical clock genes BMAL1(ARNTL), PER1-2-3, NR1D1(REV-ERBa), DBP, BHLHE40 (DEC1), and BHLHE41(DEC2). The phasing of known circadian genes was consistent with data derived from other diurnal mammals. Cyclic patterns were much weaker in the brains of patients with MDD due to shifted peak timing and potentially disrupted phase relationships between individual circadian genes. This transcriptome-wide analysis of the human brain demonstrates a rhythmic rise and fall of gene expression in regions outside of the suprachiasmatic nucleus in control subjects. The description of its breakdown in MDD suggests potentially important molecular targets for treatment of mood disorders.
Keywords: circadian rhythms, depression, microarray
Circadian patterns are 24-h rhythms in physiology and behavior sustained by a biological timekeeping capability that has evolved in most life on earth (1). In mammals, these rhythms are controlled by a hierarchy of cellular oscillators, at the top of which are pacemaker cells in the suprachiasmatic nucleus (SCN) in the hypothalamus (2). Local oscillators throughout the body coordinate daily cycles by integrating signals from the SCN with other internal and external time cues. Within cells, rhythmicity is maintained by transcriptional and posttranslational feedback loops involving a set of “clock genes” (a brief overview is provided in SI Summaries and Discussions, Mammalian Circadian Molecular Machinery). Recently, transcriptome-wide analyses from animal tissues, such as blood, brain, liver, kidney, skeletal muscle, and heart (3–6), have revealed that many genes beyond the core clock genes undergo daily variations in expression levels. The engagement of these additional circadian genes likely reflects tissue-specific functional needs. Genetic and epidemiological evidence suggests that disruption of circadian rhythms in humans can lead to many pathological conditions, including depression, metabolic syndrome, and cancer (7, 8).
Circadian control in the human brain is generally presumed based on parallels with other mammalian brains. Indeed, sleep, along with other cyclic events is among the most fundamental processes regulated by the CNS and provides the backdrop for all aspects of its function and dysfunction. Mood disorders represent a compelling example of dysregulation of circadian function, with many studies describing abnormal circadian rhythms in hormonal, body temperature, sleep, and behavioral patterns in major depressive disorder (MDD) (9). For example, patients who have MDD show persistent shortening of rapid eye movement (REM) latency (10), increased REM density, and decreases in total sleep time and sleep efficiency (11). In addition, chronotherapeutic interventions can often alleviate depressive symptoms (9, 12, 13).
However, direct demonstration of the molecular basis of circadian control in the human brain presents many unique challenges. Compared with in vitro systems or animal models, human studies lack control of genetic or environmental variables, and they pose major difficulties in collecting biologically relevant samples. Previous analyses of human tissues involved easily accessible oral mucosa (14), skin biopsies (15), hair follicle cells (16), and cultured cell lines (17, 18). Some human postmortem brain studies have focused on a limited number of candidate clock genes (19–21), but the overall orchestration of circadian regulation of gene expression in the human brain and its potential dysregulation in major depression remained unknown.
We addressed this problem by analyzing postmortem brain tissues from subjects ordered around a 24-h cycle based on their time of death (TOD), effectively treating the independently sampled data points, one for each subject, as a pseudo-time series spanning one cycle (Fig. 1A and Fig. S1). Our dataset covers ∼12,000 transcripts for each of six brain areas for 55 carefully screened normal “controls” and 34 patients with MDD (diagnosed in accordance with the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition).
Fig. 1.
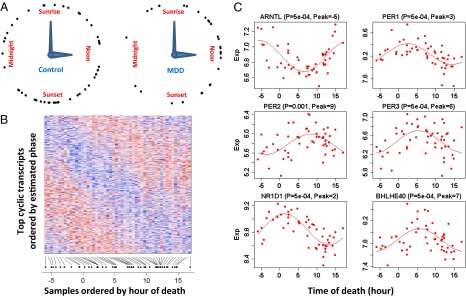
Discovery of cyclic gene expression in the human brain: examples from the DLPFC. (A) TOD distribution in the controls (n = 52) and patients with MDD (n = 33 in the DLPFC). TODs (zeitgeber time, ZT) were individually adjusted by sunrise time. (B) Heat map of expression levels for top (P < 0.05) cyclic genes (n = 922) in DLPFC samples of 52 control subjects. Genes are shown in the vertical direction and ordered by inferred phase, and samples are shown along the horizontal direction and ordered by ZT across the 24-h day, where sunrise time is ZT = 0. Expression levels for each gene are rescaled by its observed SD. The color scale represents 0.25-fold to fourfold of SD. Red indicates higher expression, and blue indicates lower expression. (C) Expression (Exp) levels of six known circadian genes in samples ordered by TOD. P values and peak times are indicated above each panel. The red lines depict the best-fitting sinusoidal curves.
Results
We first characterized circadian gene expression in the control human brain. Experimental procedures are described in Materials and Methods. At P < 0.05, there were 922 transcripts in the dorsolateral prefrontal cortex (DLPFC), 417 in the amygdala (AMY), 444 in the cerebellum (CB), 565 in the nucleus accumbens (NAcc), 566 in the anterior cingulate cortex (AnCg), and 659 in the hippocampus (HC). Fig. 1B shows a heat map of the 922 cyclic genes in the DLPFC, with the genes ordered by peak time and the samples ordered by TOD. For each gene, the pattern across samples (rows) has a characteristic phase. Meanwhile, for each sample, the pattern across genes (columns) has a rise-and-fall phase relationship typical of the subject’s TOD. Such a TOD-specific pattern across cyclic genes can serve as the basis of expression-based prediction of TOD for samples of unknown TOD.
Many core clock genes, including aryl hydrocarbon receptor nuclear translocator-like (brain and muscle Arnt-like protein-1) [ARNTL (BMAL1)]; three Period homolog (PER1–3) genes; nuclear receptor subfamily 1, group D, member 1 [NR1D1(REV-ERBα)]; D-site of albumin promoter binding protein (DBP); and basic helix–loop–helix family gene member e40 (deleted in esophageal cancer 1) [BHLHE40 (DEC1)] and member e41 [BHLHE41(DEC2)], were among those showing the strongest cyclic patterns (six examples are shown in Fig. 1C). They accounted for the 5 highest ranked cyclic genes summarized over six regions and 11 of the top 50 (highlighted in yellow in Fig. 2A). Notably, the top-ranked gene across all six brain regions was ARNTL (BMAL1), a central component in the clock gene machinery (Fig. S2). Pathway analyses using several databases consistently identified “circadian patterns” or “biological rhythms” as the top pathways enriched among top cyclic genes (SI Summaries and Discussions, Pathway Analysis, and Table S1).
Fig. 2.

Characterization of the top cyclic genes in the human brain. (A) Comparison of statistical significance for the top cyclic genes across regions. Shown are P values of the top 50 genes across six regions, with the genes ordered by the average logged P value across the six regions. The 11 gene symbols that are highlighted in yellow were annotated as being part of the circadian rhythm pathway in the Kyoto Encyclopedia of Genes and Genomes (KEGG) or the Protein Information Resource (PIR). Among the 41 “core circadian genes” reviewed by Yan et al. (5), 38 were on the microarray platform used in our study and 8 (marked by *) overlapped with the 50 genes shown here. In addition, 5 genes among the 38 (TFRC, NAMPT, USP2, NR1D2, and CRY1) ranked among the top 5% in our study (ranked at 0.7%, 0.7%, 1.3%, 1.6%, and 4.2%, respectively). (B) Peak time of expression for PER genes in our study follows what might be predicted by the animal literature. PER1 expression peaks 0–2 h after sunrise, PER2 peaks in the afternoon, and PER3 peaks in the interval between PER1 and PER2 in all six brain regions.
Our data uncovered a staggered phase relationship between the three Period genes, with PER1 peaking soon after sunrise, PER3 peaking during midday, and PER2 peaking in the afternoon (Fig. 2B). This stagger is highly characteristic of Period genes in the SCN of rodents (Fig. S3) [e.g., mice (22), Arvicanthis ansorgei (23), Octodon degus (24)], but it has not been demonstrated in brain regions outside of the SCN, although it has long been predicted (25). The detection of small phase differences in this study was enabled by the sampling density of our pseudo-time series data, because such subtle shifts may not be evident when samples are collected at fixed, multihour intervals.
The strength of cyclic variation was consistent across brain regions: P values for top genes were largely similar across the six brain regions (Fig. 2A) and were quantitatively correlated (SI Summaries and Discussions, Correlation of Statistical Significance Across Regions and Fig. S4). To identify genes with consistent cyclic patterns in six regions, we combined the P values across regions using Fisher’s method (Materials and Methods). The resulting “meta”-P values of the top 100–200 genes were smaller P values than those expected under a uniform distribution, with 169 genes having a Benjamini–Hochberg false discovery rate of <0.5 (Fig. 3A). Peak times (acrophase) for 445 transcripts with evidence of rhythmicity (P < 0.05) in at least two regions were similar across regions (Fig. 3B). The estimated peak hours for the top 50 genes are provided in Fig. S5. Similarly, the amplitude of the cyclic pattern, defined as the difference between the highest and the lowest points in the fitted sinusoidal curves, was consistent across regions (Fig. 3C and Fig. S6).
Fig. 3.

Top cyclic genes show consistent rhythmicity, phasing, and amplitude across brain regions. (A) More than 100 genes exhibit consistently significant rhythmicity. The quantile–quantile plot compares the distribution of the combined P values across the six brain regions (using Fisher’s method) and a uniform distribution, showing that 100–200 genes had smaller combined P values than expected. The top 100 genes were colored in red, and the next 100 genes were colored in green. Gray lines indicate the sorted original P values in the six individual brain regions. The dotted red line indicates uniformly distributed P values. (B) Phasing of the top cyclic genes is consistent across brain regions, as indicated by a heat map of peak times. Genes are ordered from top to bottom by mean peak time. Genes of nonsignificant (P > 0.1) cyclic patterns in a given region were shown as missing (gray) because their peak times could not be accurately determined. (C) Amplitude of rhythms is similarly consistent across brain regions, as indicated by a heat map of the amplitude for 445 transcripts with P < 0.05 in at least two of six regions. Genes are ordered from top to bottom by mean amplitude. (D) Phasing of the top cyclic genes differs between species with different chronotypes (day-active human vs. night-active mouse). Shown is a comparison of peak times for genes that overlapped between a metaanalysis of circadian gene expression in the mouse (5) and our study (P < 0.01 in controls). The y axis shows the peak time in the mouse prefrontal cortex (PFR) or whole brain (WB). The line in the plot models a linear relationship using the 7 top genes (highlighted in red). When fit with robust linear modeling, they revealed a shift of 6.51 h and a slope of 1.18 (r = 0.88).
Our dataset represents the largest transcriptome-wide resource to date for studying brain circadian patterns in any diurnal (day-active) species. We therefore compared our results with those previously reported in animal studies, especially on the nocturnal mouse. Yan et al. (5) performed a metaanalysis of gene expression data from 14 mouse tissues and identified 41 common circadian genes. Among the 27 of these genes that were found to be rhythmic in the mouse brain outside of the SCN (5) and that were analyzed in our study, 8 (30%) overlapped with the top 50 genes shown in Fig. 2 (marked with an asterisk). Four more genes, TFRC (transferrin receptor), USP2 (ubiquitin specific peptidase 2), NR1D2 (nuclear receptor subfamily 1, group D, member 2), and CLOCK (circadian locomotor output cycles kaput), ranked among the top 7% in our study. In all, 17 (63%) of the 27 genes were rhythmic (P < 0.05) in at least one human brain region. A comparison of P values across the >5,000 genes that overlapped between our data on human subjects and the mouse data from 14 tissues (5) showed that the greatest level of concordance was found in canonical clock genes (SI Summaries and Discussions, Comparison with Results from Animal Models and Fig. S7). To identify human-mouse differences in phasing of circadian genes, we compared peak times for genes reported as rhythmic in mouse prefrontal cortex or in the whole brain by Yan et al. (5) with those that had P < 0.01 in our study. The 7 top genes showed a linear relationship (Pearson’s r = 0.88, circular correlation coefficient = 0.61) between the human and mouse data, but the phase in the mouse was delayed by ∼6.5 h (Fig. 3D), consistent with the idea that clock genes in non-SCN regions (“local oscillators”) reflect the behavioral chronotype of the species.
The identification of cyclic genes in controls allowed us to ask whether these genes were also cyclic in patients with MDD. We found that most of the top cyclic genes in controls were not significant in MDD. Indeed, among the top 16 genes, 11 had P < 0.05 in four or more regions in controls (Fig. 2A), yet only 2 had P < 0.05 in more than one region in patients with MDD and none had P < 0.05 in more than three regions (Fig. 4A). In a Fisher’s metaanalysis, P values in MDD were not appreciably different from a uniform distribution (Fig. 4B), in contrast to the increased significance of the top 100–200 genes seen in controls due to between-region consistency. According to Fisher’s P values, the top 5 ranked genes in controls, ARNTL (BMAL1), PER2, PER3, NR1D1, and DBP, ranked the 171st, 532nd, 10,191st, 27th, and 684th, respectively, in patients with MDD. The decrease in significance was paralleled by the reduction of amplitude of the best-fitting sinusoidal curves (Fig. S8 A and B), even though the overall variance for these genes was similar between the MDD and control groups (Fig. S8C). By testing a subset of controls that (i) have an equivalent sample size to the MDD group for each brain region and (ii) have TODs that were matched as closely as possible between the MDDs and the selected controls, we confirmed that the weaker signal observed in the MDD group was not due to its smaller sample size than the control group (SI Summaries and Discussions, Effect of Sample Size in Comparison of Controls and MDD Cases and Fig. S8 D and E).
Fig. 4.
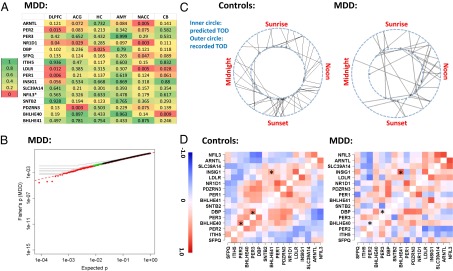
Disruption of cyclic pattern in patients with MDD. (A) Top 16 cyclic genes from controls are not rhythmic in the MDD group. The P values for the genes are formatted similar to Fig. 2A (ranked by the average logged P value across the six regions in controls). (B) Genes in patients with MDD do not exhibit consistently significant rhythmicity, as illustrated by a quantile–quantile plot comparing the combined P values across the six brain regions in MDD (using Fisher’s method) vs. the expected P values in a uniform distribution using the same style as in Fig. 3A. (C) Rhythms of patients with MDD are less synchronized with the solar day compared with controls. The predicted TOD in 55 controls (Left) and 34 patients with MDD (Right) are shown on the inner circle of a 24-h clock, and their documented TODs are shown on the outer circle. The deviations were smaller in controls than in patients with MDD (P = 0.012, Mann–Whitney nonparametric test). (D) Patterns of gene-gene correlations seen in controls (in-phase = positive correlation, out-of-phase = negative correlation) are only partially present in patients with MDD. Depicted are the correlation coefficients across the top 16 genes, calculated using DLPFC data for 52 controls (Left) and 33 MDD cases (Right). Genes are ordered by the peak time derived from the control dataset. Examples of gene pairs with significant differences between controls and patients with MDD are marked with an asterisk.
The weaker cyclic patterns in MDD group could be due to (i) a flattened or disrupted rhythmicity of the circadian genes in patients with MDD or (ii) large time shifts of the rhythms in many patients. In the latter scenario, patients with MDD could still carry robust cyclic patterns (just as in controls) but their actual phase at death might have deviated from what is expected according to their recorded TOD. To test these hypotheses, we first used the top cyclic genes (n = 108) to calculate sample-sample correlations in the DLPFC and found a clear pattern of positive correlations among control samples with similar TODs and negative correlations between those with opposing TODs (e.g., noon vs. midnight). This pattern was much weaker between patients with MDD and controls or among MDD cases (SI Summaries and Discussions, Sample–Sample Correlations Suggest Phase Shift in MDD Cases and Fig. S9), suggesting that biological cycles for many MDD cases may have fallen out of synchronization with the solar day. Next, we applied the concerted rise and fall of the top 100 cyclic genes in a training set of 60 randomly selected subjects, containing both cases and controls (Fig. 1), to predict the likely TOD for each subject in the remaining test set (Materials and Methods). The absolute deviations of the predicted TOD from the recorded TOD were smaller for controls than for patients with MDD (Fig. 4C; P = 0.012, Mann–Whitney test), further suggesting that the circadian rhythms of MDD cases were not synchronized (“entrained”) normally to the solar day. Finally, if the cyclic patterns had persisted in patients with MDD, we would expect in-phase genes to be positively correlated with each other and out-of-phase genes to be negatively correlated. Importantly, this analysis of gene-gene correlations across samples should be unaffected by how the samples were ordered and immune to any desynchronization between the “internal time” of the patients and the solar day. In controls, we found that the top cyclic genes showed positive correlations between genes with similar phases and negative correlations between genes of opposing phases (an example for the top 16 genes is shown in Fig. 4D). This pattern was partially preserved in patients with MDD (Mantel statistic based on Kendall’s rank correlation: 0.38, P < 0.001), albeit with notable alterations (Fig. 4D). Some normally in-phase gene pairs (e.g., BHLHE40-PER2, DBP-PER3, with large correlations shown in red) were out-of-phase in patients with MDD, whereas some normally out-of-phase genes were in-phase in patients with MDD [e.g., insulin-induced gene 1 (INSIG1)-BHLHE41]. These results suggest that both scenarios may be in play in patients with MDD: a disrupted regulatory relationship among portions of the cyclic genes and shifted timing in many patients.
The apparent disruption of the circadian clock could be due to a number of biological causes, including the mood disorder itself, the use of antidepressant drugs, or the presence of other nontherapeutic drugs taken by the subject as ascertained by the toxicology screen of the brains (Table S2). We explored several variables and found that the TOD deviations of MDD cases were not significantly different between suicide (n = 20) and nonsuicide (n = 14) cases, with P = 0.62, or between the witnessed (n = 7) and nonwitnessed (n = 27) deaths, with P = 0.72. We also examined a group of patients (n = 10) who were highly homogeneous: They had all died of suicide, had no known history of antidepressant treatment (i.e., newly diagnosed for MDD), and had negative findings on the postmortem toxicology screen. Thus, these patients represent a “clean” group in which the primary difference from controls is the diagnosis of MDD with suicide. Because members of this group all died during the daytime, we compared them not only with the entire group of controls but with the subset of controls who died during the same daytime period. The average TOD deviation for the 10 suicide/toxicology screen-negative MDD cases is 3.3 h, which is larger than the average deviation for the entire control group (1.9 h; P = 0.068, Kolmogorov–Smirnov test) and from the average deviation of the daytime-only controls (n = 30, 2.1 h; P = 0.038, Kolmogorov–Smirnov test). These findings support the view that the circadian disruption observed in this work is partially linked to the disease process itself rather than being exclusively due to the impact of psychoactive drugs. Meanwhile, the average deviation between predicted and recorded TOD in this group (3.3 h) is lower than in the entire MDD group (3.9 h, n = 34), suggesting that other factors, including prescription and nonprescription drugs, may contribute to the observed circadian dysregulation.
Discussion
Cumulatively, these results provide convincing evidence that there exists a rhythmic rise and fall in the transcriptional activity of hundreds of genes in the control human brain, initiating or responding to the regulation of 24-h behavioral and hormonal cycles. The data presented here are notable for their transcriptome-wide coverage (∼12,000 transcripts) and large sample size, encompassing 365 RNA samples from controls isolated from six brain regions with sample sizes of 29–55 per region and covering the daily cycle, with an average of 1.2–2.3 data points per hour. Despite these strengths, it was conceivable that no consistently cyclic gene would emerge in our analysis due to the numerous sources of noise in the independent subjects design, both biological and technical. Indeed, even though there was no clinical record regarding the state of consciousness of control subjects at the TOD, many subjects might have been awake or experiencing disrupted sleep. Despite these challenges, over 100 genes showed consistent cyclic patterns across the six regions (Fig. 3), reflecting the robust, slow-changing nature of circadian rhythms in extra-SCN regions even in the presence of environmental disturbances (2). The two regions with the smallest sample size, the CB and AMY, showed the weakest significance, suggesting that a larger sample size (≥55) could reveal additional cyclic genes.
Two lines of evidence support the validity of our observations in the normal human brain. First, several core circadian genes essential to the clock machinery ranked as top cyclic genes in each of the six brain areas, including ARNTL (BMAL1), PER1–3, NR1D1 (REV-ERBα), DBP, and BHLHE40–41 (DEC1–2). Second, the phase relationships between core circadian genes resembled those found in model organisms. Indeed, the order of PER peak expression (i.e., PER1, PER3, PER2) matched the pattern of PER expression in the SCN of rodents, demonstrating a consistency in phase relationships across mammalian species.
In addition to confirming the cyclic patterns of most known circadian genes, this study revealed additional cyclic genes, including, for example, LDLR (low-density lipoprotein receptor) and INSIG1, which are known to be involved in lipid synthesis and metabolism (26), and the hypocretin receptor, HCRTR2, which is important for sleep/wake regulation (27). Because DNA variations in several circadian genes underlie seasonal affective disorder (28) and familial advanced sleep phase syndrome (29), the cyclic genes described here may also serve as candidates for genetic analyses of inherited disorders that involve dysfunction of the circadian system. Moreover, this study provides the most complete transcriptomic description to date for the brain of a diurnal species, and it could serve as the knowledge base for future efforts to define signaling pathways underlying basic chronotype generation, a long-standing question in the field of chronobiology.
The present findings also offer empirical evidence of molecular dysregulation of circadian rhythmicity across six brain regions of clinically depressed individuals. Our analysis indicates that patients with MDD exhibit abnormal phasing of circadian gene expression and potentially disrupted phase relationships between individual circadian genes. This disruption may have an impact on the functional regulation of numerous neural processes and behaviors, consistent with the broad range of symptoms seen in MDD. A caveat in this analysis is that gene pairs that appeared significantly disrupted in one region (e.g., DLPFC as shown in Fig. 4D) are not necessarily disrupted in another region of the brain of patients with MDD. Rather, some other gene pairs appear disrupted in that different region. This complexity could arise from region-specific biological factors, with MDD conferring distinct patterns of transcriptional dysregulation in different brain areas. However, the differential effects could also result from technical factors (e.g., sample processing and microarray experiments conducted separately by region). Thus, it is possible that few gene pairs in the core machinery of circadian regulation were truly uncoupled and that phase shifts played a primary role in giving rise to the apparently dampened cyclic pattern in MDD cases. Finally, the observed effect may also be due to clinical heterogeneity among the subjects with MDD, with some patients exhibiting faulty entrainment of an otherwise normally functioning circadian machinery, whereas others have a more fundamental disruption of circadian regulation. As such, we can glimpse the likelihood of multiple patterns of dysregulation within the depressed group. Future studies, with larger MDD sample sizes, are required to unravel the complex interplay of these factors fully. Emerging approaches to mimic the biology of human neural cells, such as induced pluripotent stem cells, together with appropriate animal models (e.g. refs. 30, 31), may also prove useful for uncovering molecular cascades associated with mood dysregulation.
In sum, the current study identifies hundreds of genes in the human brain that are likely involved in important daily rhythmic events, including the sleep/wake cycle and metabolism. Using this knowledge, we discovered that daily rhythms in these genes are profoundly dysregulated in MDD. Although this disruption can result from numerous factors, including the disease itself and the patient’s drug history, we show that the dysregulation can exist in the absence of any drug exposure. These results pave the way for the identification of novel biomarkers and treatment targets for mood disorders.
Materials and Methods
Sample collection, including human subject recruitment and characterization, tissue dissection, and RNA extraction, was described previously (32, 33). RNA samples for different regions came from the same set of brains from 55 control subjects and 34 patients with MDD for whom the recorded hour of death was available. Sample size varied by region: AnCg (n = 55 controls), DLPFC (n = 52), CB (n = 34), AMY (n = 29), HC (n = 48), and NAcc (n = 51) (Table S3). Tables S2 and S4 provide demographic and medical details for the study subjects, including sex, age at death, ethnicity, agonal factor scores, brain tissue pH, cause of death, and TOD. The brain tissues were of high quality: All subjects died rapidly and had an agonal factor score of 0 (34), with an average pH of 6.87 (SD = 0.23). We ran each sample on at least two microarrays using Affymetrix U133-A or U133Plus-v2 GeneChips. We applied robust multiarray analysis (35, 36) to summarize probe set expression levels, using custom chip definition files, resulting in expression data for 11,912 ENTREZ transcripts. Microarray data for each region were analyzed separately. All downstream analyses were performed in R (37). Details of the data processing, including data cleaning and normalization, are provided in SI Materials and Methods. After data filtering, 1,424 microarrays remained, corresponding to 776 unique RNA samples in six regions. The raw data and processed data for the complete set of controls were deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (accession no. GSE45642) and on our Web site (www.pritzkerneuropsych.org/?page_id=1196).
We adjusted the recorded TOD for each subject by the sunrise time of his/her date and place of death, and we used this zeitgeber time (ZT) scale for downstream analysis. In the adjusted scale, sunrise time is ZT = 0, noon is approximately ZT = 6, and midnight is approximately ZT = 18 (18 h after sunrise) or −6 (6 h before sunrise). To detect potential cyclic patterns for a given gene, we fit its TOD-ordered expression values to a sinusoidal function with a 24-h period, with phase and amplitude as free parameters, and calculated the percentage of variance explained (PVE) as a goodness-of-fit index. By comparing the observed PVE for each gene with its null PVE distribution in 1,000 TOD-randomized datasets, we assigned empirical P values and identified transcripts with small P values as candidate cyclic genes. To quantify the overall rhythmicity across regions, we combined the P values from six regions using Fisher’s method (SI Materials and Methods, Fisher’s P, Phase, and Pathway Analysis). To identify phase, or peak time, we calculated the correlation coefficient of the actual data series for each gene with a family of 24 sinusoidal functions that are shifted by 1 h. The maximal correlation coefficient indicates the estimated peak time. For functional analyses, we referred to “known circadian genes” as those documented by KEGG (38) and PIR (39) databases.
Enrichment analysis relied on online tools at the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (40) and Pathway Analysis Using Logistic Regression (LRpath) (41). Prediction of TOD is described in SI Materials and Methods, Prediction.
Supplementary Material
Acknowledgments
We thank Dr. Kerby Shedden and John Basler for statistical advice, Dr. Jennifer Mohawk for reviewing clock gene regulatory circuitry, and Hanna Larcinese for assistance in enrichment analysis. This work was supported, in part, by the Pritzker Neuropsychiatric Disorders Research Fund, National Institute of Mental Health (NIMH) Conte Center Grant P50 MH60398, the William Lion Penzner Foundation (W.E.B.), the Della Martin Foundation (W.E.B.), NIMH R01MH085801 (M.P.V.), and Office of Naval Research Grants N00014-09-1-059 and N00014-12-1-0366 (to H.A. and S.J.W.). J.Z.L. is supported by a National Alliance for Research on Schizophrenia and Depression Abramson Family Foundation Investigator Award and an International Mental Health Research Organization–Johnson & Johnson Rising Star Translational Research Award.
Footnotes
Conflict of interest statement: The authors are members of the Pritzker Neuropsychiatric Disorders Research Consortium, which is supported by Pritzker Neuropsychiatric Disorders Research Fund, LLC. A shared intellectual property agreement exists between the academic and philanthropic entities of the consortium. The Pritzker Neuropsychiatric Disorders Research Fund had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data deposition: The raw and processed data for this complete set of controls have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE45642) and on our Web site, www.pritzkerneuropsych.org/?page_id=1196.
2Deceased June 6, 2011.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305814110/-/DCSupplemental.
References
- 1.DeCoursey PJ. The behavioral ecology and evolution of biological timing systems. In: Dunlap JC, Loros JJ, Decoursey PJ, editors. Chronobiology: Biological Timekeeping. Sunderland, MA: Sinauer; 2004. pp. 26–65. [Google Scholar]
- 2.Yamazaki S, et al. Resetting central and peripheral circadian oscillators in transgenic rats. Science. 2000;288(5466):682–685. doi: 10.1126/science.288.5466.682. [DOI] [PubMed] [Google Scholar]
- 3.Akhtar RA, et al. Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr Biol. 2002;12(7):540–550. doi: 10.1016/s0960-9822(02)00759-5. [DOI] [PubMed] [Google Scholar]
- 4.Panda S, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109(3):307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- 5.Yan J, Wang H, Liu Y, Shao C. Analysis of gene regulatory networks in the mammalian circadian rhythm. PLOS Comput Biol. 2008;4(10):e1000193. doi: 10.1371/journal.pcbi.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang S, Wang K, Valladares O, Hannenhalli S, Bucan M. Genome-wide expression profiling and bioinformatics analysis of diurnally regulated genes in the mouse prefrontal cortex. Genome Biol. 2007;8(11):R247. doi: 10.1186/gb-2007-8-11-r247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahar S, Sassone-Corsi P. Regulation of metabolism: The circadian clock dictates the time. Trends Endocrinol Metab. 2012;23(1):1–8. doi: 10.1016/j.tem.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat Rev Genet. 2008;9(10):764–775. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kronfeld-Schor N, Einat H. Circadian rhythms and depression: Human psychopathology and animal models. Neuropharmacology. 2012;62(1):101–114. doi: 10.1016/j.neuropharm.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 10.Kupfer DJ. REM latency: A psychobiologic marker for primary depressive disease. Biol Psychiatry. 1976;11(2):159–174. [PubMed] [Google Scholar]
- 11.Mendlewicz J, Kerkhofs M. Sleep electroencephalography in depressive illness. A collaborative study by the World Health Organization. Br J Psychiatry. 1991;159:505–509. doi: 10.1192/bjp.159.4.505. [DOI] [PubMed] [Google Scholar]
- 12.Berger M, van Calker D, Riemann D. Sleep and manipulations of the sleep-wake rhythm in depression. Acta Psychiatr Scand Suppl. 2003;(418):83–91. doi: 10.1034/j.1600-0447.108.s418.17.x. [DOI] [PubMed] [Google Scholar]
- 13.Bunney BG, Bunney WE. Rapid-acting antidepressant strategies: Mechanisms of action. Int J Neuropsychopharmacol. 2012;15(5):695–713. doi: 10.1017/S1461145711000927. [DOI] [PubMed] [Google Scholar]
- 14.Zieker D, et al. Circadian expression of clock- and tumor suppressor genes in human oral mucosa. Cell Physiol Biochem. 2010;26(2):155–166. doi: 10.1159/000320547. [DOI] [PubMed] [Google Scholar]
- 15.Brown SA, et al. The period length of fibroblast circadian gene expression varies widely among human individuals. PLoS Biol. 2005;3(10):e338. doi: 10.1371/journal.pbio.0030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akashi M, et al. Noninvasive method for assessing the human circadian clock using hair follicle cells. Proc Natl Acad Sci USA. 2010;107(35):15643–15648. doi: 10.1073/pnas.1003878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffman AE, et al. Phenotypic effects of the circadian gene Cryptochrome 2 on cancer-related pathways. BMC Cancer. 2010;10:110. doi: 10.1186/1471-2407-10-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hughes ME, et al. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5(4):e1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cermakian N, Lamont EW, Boudreau P, Boivin DB. Circadian clock gene expression in brain regions of Alzheimer’s disease patients and control subjects. J Biol Rhythms. 2011;26(2):160–170. doi: 10.1177/0748730410395732. [DOI] [PubMed] [Google Scholar]
- 20.Ackermann K, Dehghani F, Bux R, Kauert G, Stehle JH. Day-night expression patterns of clock genes in the human pineal gland. J Pineal Res. 2007;43(2):185–194. doi: 10.1111/j.1600-079X.2007.00461.x. [DOI] [PubMed] [Google Scholar]
- 21.Wu YH, et al. Pineal clock gene oscillation is disturbed in Alzheimer’s disease, due to functional disconnection from the “master clock”. FASEB J. 2006;20(11):1874–1876. doi: 10.1096/fj.05-4446fje. [DOI] [PubMed] [Google Scholar]
- 22.Takumi T, et al. A light-independent oscillatory gene mPer3 in mouse SCN and OVLT. EMBO J. 1998;17(16):4753–4759. doi: 10.1093/emboj/17.16.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caldelas I, Poirel VJ, Sicard B, Pévet P, Challet E. Circadian profile and photic regulation of clock genes in the suprachiasmatic nucleus of a diurnal mammal Arvicanthis ansorgei. Neuroscience. 2003;116(2):583–591. doi: 10.1016/s0306-4522(02)00654-1. [DOI] [PubMed] [Google Scholar]
- 24.Vosko AM, Hagenauer MH, Hummer DL, Lee TM. Period gene expression in the diurnal degu (Octodon degus) differs from the nocturnal laboratory rat (Rattus norvegicus) Am J Physiol Regul Integr Comp Physiol. 2009;296(2):R353–R361. doi: 10.1152/ajpregu.90392.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96(2):271–290. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- 26.Javitt NB. Oxysterols: Novel biologic roles for the 21st century. Steroids. 2008;73(2):149–157. doi: 10.1016/j.steroids.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Taheri S, Zeitzer JM, Mignot E. The role of hypocretins (orexins) in sleep regulation and narcolepsy. Annu Rev Neurosci. 2002;25:283–313. doi: 10.1146/annurev.neuro.25.112701.142826. [DOI] [PubMed] [Google Scholar]
- 28.Partonen T, et al. Three circadian clock genes Per2, Arntl, and Npas2 contribute to winter depression. Ann Med. 2007;39(3):229–238. doi: 10.1080/07853890701278795. [DOI] [PubMed] [Google Scholar]
- 29.Vanselow K, et al. Differential effects of PER2 phosphorylation: Molecular basis for the human familial advanced sleep phase syndrome (FASPS) Genes Dev. 2006;20(19):2660–2672. doi: 10.1101/gad.397006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roybal K, et al. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci USA. 2007;104(15):6406–6411. doi: 10.1073/pnas.0609625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang WG, et al. Chronic unpredictable stress induces a reversible change of PER2 rhythm in the suprachiasmatic nucleus. Brain Res. 2011;1399:25–32. doi: 10.1016/j.brainres.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Evans SJ, et al. DNA microarray analysis of functionally discrete human brain regions reveals divergent transcriptional profiles. Neurobiol Dis. 2003;14(2):240–250. doi: 10.1016/s0969-9961(03)00126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li JZ, et al. Systematic changes in gene expression in postmortem human brains associated with tissue pH and terminal medical conditions. Hum Mol Genet. 2004;13(6):609–616. doi: 10.1093/hmg/ddh065. [DOI] [PubMed] [Google Scholar]
- 34.Tomita H, et al. Effect of agonal and postmortem factors on gene expression profile: Quality control in microarray analyses of postmortem human brain. Biol Psychiatry. 2004;55(4):346–352. doi: 10.1016/j.biopsych.2003.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 36.Irizarry RA, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.R Development Core Team . R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2005. [Google Scholar]
- 38.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38(Database issue):D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu CH, et al. The Protein Information Resource. Nucleic Acids Res. 2003;31(1):345–347. doi: 10.1093/nar/gkg040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 41.Sartor MA, Leikauf GD, Medvedovic M. LRpath: A logistic regression approach for identifying enriched biological groups in gene expression data. Bioinformatics. 2009;25(2):211–217. doi: 10.1093/bioinformatics/btn592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.