

Abstract
After four decades of research aimed at understanding tRNA selection on the ribosome, the mechanism by which ribosomal ambiguity (ram) mutations promote miscoding remains unclear. Here, we present two X-ray crystal structures of the Thermus thermophilus 70S ribosome containing 16S rRNA ram mutations, G347U and G299A. Each of these mutations causes miscoding in vivo and stimulates elongation factor thermo unstable (EF-Tu)-dependent GTP hydrolysis in vitro. Mutation G299A is located near the interface of ribosomal proteins S4 and S5 on the solvent side of the subunit, whereas G347U is located 77 Å distant, at intersubunit bridge B8, close to where EF-Tu engages the ribosome. Despite these disparate locations, both mutations induce almost identical structural rearrangements that disrupt the B8 bridge—namely, the interaction of h8/h14 with L14 and L19. This conformation most closely resembles that seen upon EF-Tu⋅GTP⋅aminoacyl-tRNA binding to the 70S ribosome. These data provide evidence that disruption and/or distortion of B8 is an important aspect of GTPase activation. We propose that, by destabilizing B8, G299A and G347U reduce the energetic cost of attaining the GTPase-activated state and thereby decrease the stringency of decoding. This previously unappreciated role for B8 in controlling the decoding process may hold relevance for many other ribosomal mutations known to influence translational fidelity.
The molecular mechanisms controlling the fidelity of DNA replication, transcription, and translation have been areas of intense interest since the discovery of the genetic code. Thermodynamic differences between standard Watson–Crick and alternative (e.g., wobble) base pairs in solution are insufficient to explain the high fidelity for any of the three polymerase reactions of the central dogma (1), indicating an active role for the enzymes in substrate selectivity (1–3). Mechanistic studies of polymerases have revealed some common themes, such as the specific recognition of Watson–Crick base pair geometry, larger forward rate constants for correct substrates (induced fit), separate opportunities for incorrect substrate rejection (kinetic proofreading), and postincorporation correction mechanisms (1–5).
During translation, the ribosome must select aminoacyl-tRNA (aa-tRNA) substrates based on the mRNA sequence. Extensive biochemical studies have shed light on the kinetics of this decoding process (reviewed in ref. 6). The aa-tRNA is delivered to the ribosome as part of a ternary complex (TC) with elongation factor thermo unstable (EF-Tu) and GTP. Initial binding of TC, mediated primarily by L7/L12 of the 50S subunit, is followed by the sampling of codon–anticodon interactions in the 30S A site. Codon–anticodon pairing leads to GTPase activation and GTP hydrolysis, which allows release of the acceptor end of aa-tRNA from EF-Tu. The aa-tRNA then either moves completely into the ribosomal A site (a step termed accommodation), where it can participate in peptide bond formation, or is rejected and released into solution (7, 8).
For each decoding event, the ribosome selects cognate aa-tRNA from a large pool of non- and near-cognate aa-tRNAs with high speed (>20 s−1) and fidelity (error rate ∼10−4) (1). High fidelity can be explained in part by a kinetic proofreading mechanism, in which differences in substrate binding affinity are exploited twice to increase the overall level of discrimination (7–9). Basically, the functionally irreversible GTP hydrolysis step of the pathway provides a second independent opportunity for rejection of near-cognate aa-tRNA. It is clear, however, that kinetic proofreading is not maximally exploited for fidelity (6, 10). Instead, the ribosome additionally employs an induced-fit mechanism to achieve both high speed and fidelity in decoding. Indicative of this mechanism is that cognate codon recognition increases the forward rate constants for GTPase activation/GTP hydrolysis and accommodation (11–14), which allows rapid incorporation of cognate aa-tRNA specifically, effectively obviating the need for substrate binding equilibria to be approached.
An important question is how cognate codon recognition stimulates GTPase activation by EF-Tu. Cognate codon–anticodon pairing results in docking of A-site rRNA nucleotides A1492, A1493, and G530 into the minor groove of the first two base pairs of the codon–anticodon helix (13). Presumably, these changes in the decoding center are transmitted via conformational signaling ∼80 Å to the GTPase domain of EF-Tu, although the molecular basis of such signaling remains unclear. One potential conduit for signaling is the tRNA itself, which is known to adopt a distorted or bent conformation in the GTPase-activated state (15, 16). Another possibility is that conformational signaling occurs through the 30S subunit. Crystallographic studies of the 30S subunit suggest that cognate A-site codon recognition is accompanied by a global conformational change in the subunit termed domain closure (13). Domain closure involves an inward rotation of the 30S shoulder domain, which may facilitate productive engagement of the TC and GTPase activation (5).
Most chromosomal mutations affecting the fidelity of decoding have mapped to ribosomal protein genes rather than rRNA genes, presumably because the latter typically exist in multiple copies. Recently, a specialized ribosome system was used to isolate a number of 16S rRNA mutations that increase miscoding (referred to as ribosomal ambiguity, or ram mutations) (17). Many of these ram mutations clustered along interfaces between the 30S shoulder domain and other portions of the ribosome, generally consistent with a role for shoulder movement in aa-tRNA selection. Nearly half the mutations mapped to helices 8 (h8) and 14 (h14), which interact with each other and with the 50S subunit proteins L14 and L19, forming intersubunit bridge B8. Ribosomes with truncation of either h8 or h14 retain activity but are error-prone and exhibit elevated rates of EF-Tu–dependent GTP hydrolysis (17). These data indicate that B8 acts in some way to negatively regulate GTP hydrolysis by EF-Tu.
Another cluster of 16S ram mutations mapped to h12 (17), close to the interface of ribosomal proteins S4 and S5. In earlier studies, numerous mutations affecting decoding fidelity were mapped to the S4/S5 interface (18–20). These mutations were isolated based on their ability to suppress certain S12 mutations, which confer streptomycin dependence and hyperaccurate decoding. Unlike S5, S4 is part of the shoulder domain, and hence domain closure results in slight separation of these two proteins. It has been proposed that the S4/S5 mutations destabilize the open state of the 30S subunit and thereby influence decoding fidelity (5). However, several of these S4 suppressor mutations have been found to confer a restrictive (hyperaccurate) phenotype rather than the expected ram phenotype (21). Moreover, analysis of S4/S5 mutations in a yeast two-hybrid system showed no correlation between S4/S5 interaction and decoding fidelity (22). These observations cannot be easily reconciled with the domain closure model and hint that another mechanism may be at play.
Here, we present two X-ray crystal structures of the Thermus thermophilus 70S ribosome, containing ram mutations G299A or G347U. These mutations lie 77 Å apart, in h12 and h14, respectively (Fig. 1). Despite this, both structures show similar rearrangements of the B8 bridge that mimic rearrangements resulting from TC binding (16, 23). These data provide evidence that GTPase activation involves B8 disruption and reveal long-distance conformational signaling across the 30S.
Fig. 1.
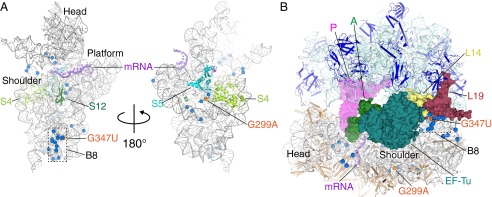
Location of ram mutations in 16S rRNA. (A) Positions of 16S rRNA mutations that increase miscoding (blue), mapped onto the 30S ribosomal subunit (PDB ID code 2WDG), viewed from the subunit interface (Left) and the solvent side (Right). The 16S rRNA, mRNA, and r-proteins are shown as cartoon. The positions of the mutations G299A and G347U (orange), and the head, shoulder, and platform domains and intersubunit bridge B8 of the subunit are indicated. The 30S r-proteins are removed for clarity. (B) Most of the 16S rRNA ram mutations cluster to regions distant from the tRNA binding sites, nearly half of which map to h8/14, which contacts L14 (yellow) and L19 (maroon) to form B8 (boxed). Although mutation G347U is proximal to B8, G299A resides ∼80 Å away.
Results
Mutations G299A and G347U Promote Activation of EF-Tu.
It was shown previously that mutations predicted to disrupt the interaction between h8 and h14 cause defects in the initial selection phase of decoding (17). These mutations (e.g., G347U and truncations of either h8 or h14) stimulate EF-Tu–dependent GTP hydrolysis, particularly in the near-cognate case. To test whether G299A similarly affects initial selection, control and G299A ribosomes were purified and used to assemble 70S initiation complexes programmed with either a cognate 5′-UUU-3′ or a near-cognate 5′-CUU-3′ codon in the A site (Materials and Methods). Each complex (at various concentrations ≥0.5 μM) was rapidly mixed with EF-Tu⋅[γ-32P]⋅GTP⋅Phe-tRNAPhe (<0.3 μM), and the rate of GTP hydrolysis was measured. Apparent rates were plotted as a function of ribosome concentration, and the data were fit to a hyperbolic function to obtain the maximal rate (kGTPmax) and the concentration at which half-maximal rate is observed (K1/2; Table 1; Fig. S1). In the cognate case, the G299A mutation increased kGTPmax and K1/2 by ∼twofold, whereas in the near-cognate case, G299A increased kGTPmax by ninefold with little or no effect on K1/2. These effects of G299A are similar to those seen for G347U, h8Δ3, and h14Δ2 (17). Previous studies of EF-Tu–dependent GTP hydrolysis by Rodnina and coworkers (11, 12, 24) have demonstrated that a conformational change attributed to GTPase activation is rate-limiting in the near-cognate case and partially rate-limiting in the cognate case. Hence, each of these 16S ram mutations acts, at least in part, by accelerating the GTPase activation step (17). Indeed, in the framework of the Rodnina model, an increase of the forward rate constant for GTPase activation of the same magnitude in both the cognate and near-cognate cases would be sufficient to reduce the selectivity of the reaction, as we observe.
Table 1.
Kinetic parameters for EF-Tu–dependent GTP hydrolysis on control and mutant ribosomes
| Ribosomes | Cognate | Near cognate | Selectivity* | Source | ||
| kGTPmax, s−1 | K1/2, μM | kGTPmax, s−1 | K1/2, μM | |||
| Control | 51 ± 3 | 1.1 ± 0.2 | 1.8 ± 0.1 | 1.5 ± 0.2 | 39 | This study and ref. 17 |
| G299A | 110 ± 9 | 2.3 ± 0.4 | 17 ± 1 | 1.0 ± 0.2 | 2.7 | This study |
| G347U | 130 ± 10 | 2.8 ± 0.5 | 15 ± 1 | 2.5 ± 0.4 | 7.7 | Ref. 17 |
(Cognate kGTPmax/K1/2)/(Near-cognate kGTPmax/K1/2).
Crystallization and Structural Analysis of T. thermophilus G299A and G347U Ribosomes.
To understand the structural basis of the effects of ram mutations G299A and G347U, T. thermophilus strains containing homogeneous populations of mutant ribosomes were genetically engineered. T. thermophilus ribosomes were used because of their ability to produce crystals that are amenable for X-ray structure determination (25). One of the two 16S rRNA genes (rrsB) was first replaced with the mutant allele, and then the other (rrsA) was replaced with the null allele ΔrrsA::htk1, marked with the high-temperature kanamycin resistance 1 gene (26). In both Escherichia coli (strain ∆7 prrn) and T. thermophilus, G299A conferred a larger growth defect than G347U. Mutation G299A slowed the growth of E. coli by 40% and T. thermophilus by 37%, whereas G347U slowed the growth of E. coli by 6% and T. thermophilus by 7% (Table S1).
From these T. thermophilus strains, 70S ribosomes carrying either the G299A or G347U mutation were purified, pretranslocation complexes were formed with P-site tRNAfMet and A-site tRNAPhe, and these complexes were crystallized using established conditions (25). The structures were solved using molecular replacement with the most complete available model of the 70S ribosome (27). The 70S G299A and G347U structures were refined at 3.5 and 3.9 Å resolution, respectively (Materials and Methods; Table S2).
In both structures, P-site tRNAfMet, A-site ASLPhe, and mRNA were clearly visible in the original, unbiased difference Fo–Fc density. Regions that had noticeably shifted compared with the start model and required rebuilding included h8 and h14 (Fig. S2) and h12 for the G299A structure only (Fig. S3). The codon–anticodon interactions in the A and P sites, the placement of the tRNAs in the E, P, and A sites, and the mRNA path all look identical to those of the wild-type 70S structure (27).
G347U Directly Alters h14, Thereby Disrupting Bridge B8.
The 16S rRNA nucleotide G347 is located in h14, which, together with h8, contacts L14 and L19 of the large subunit to form intersubunit bridge B8 (Figs. 1 and 2A) (28). The G347U mutation is 87 Å from where tRNA initially engages the ribosome at the 30S A site, and is one of several ram mutations that cluster to the B8 region (17, 29). G347 is normally involved in a base triple interaction with C342 of h14 and A160 of h8 (Fig. 3A), forming a Watson–Crick and Hoogsteen interaction, respectively (30). Our structure reveals that the G347U mutation disrupts this base triple, resulting in a widening of the entire h14 (Fig. 3B). This widening also causes movement away from the large ribosomal subunit, thereby preventing interactions that form B8 (Fig. 2B). From original, unbiased Fo–Fc difference density, the entire phosphate backbone of h14 moves away from the large subunit between 2 Å and 6 Å (Fig. S2). In wild-type 70S structures, interactions are observed between the E36 side chain of L19 and the phosphate oxygens of 16S rRNA h14 nucleotides 345 and 346 via a Mg2+ ion; the K35 side chain of L19 and the phosphate oxygen of 16S rRNA nucleotide 346; and the main chain of residues V116/A118 of L14 and the phosphate oxygen of nucleotide 347 via a Mg2+ ion (Fig. 2A). The movement of the 16S rRNA h14 backbone away from the 50S subunit results in distances that are too large to maintain this hydrogen bonding network with L14 and L19. Not all of the side chains of L14 and L19 were interpretable, and therefore these residues were kept modeled as in the wild-type 70S structure. Only one specific B8 interaction remains between the R97 side chain of L14 and the 16S rRNA backbone phosphate oxygen of C339.
Fig. 2.

The 16S rRNA mutations G299A and G347U cause similar conformational rearrangements that disrupt intersubunit bridge B8. (A) B8 is formed by contacts between h14 of the 30S subunit and L14/L19 of the 50S subunit (PDB ID code 2WDG), many of which are lost in the mutant structures. Color scheme is the same as in Fig. 1B. (B) 70S G347U (green) and (C) G299A (blue) mutations cause h14 displacement away from L14 and L19, disrupting the hydrogen bonding network between the subunits. The rRNA backbone of the wild-type 70S structure is shown for comparison (gray).
Fig. 3.
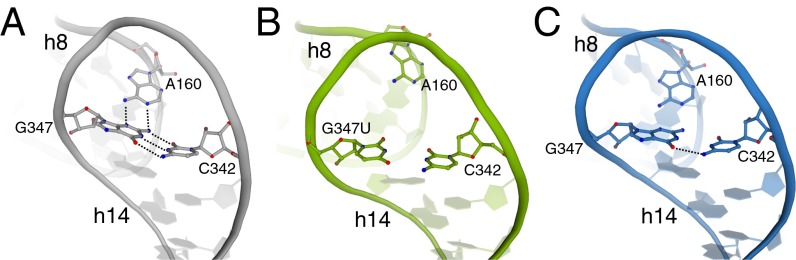
Mutations G347U and G299A similarly alter the conformation of h14 and its interaction with h8. (A) The structural integrity of h14 is normally maintained by the base triple G347⋅A160⋅C342, as shown in the wild-type 70S structure (gray; PDB ID code 2WDG). In the presence of either G347U (B, green) or G299A mutations (C, blue), h14 widens, resulting in disruption of this base triple and the interaction network of B8.
G299A Indirectly Alters h14, also Disrupting Bridge B8.
The 16S rRNA G299A mutation is located in h12, near previously identified ram mutations at the interface of ribosomal proteins S4 and S5 (18, 19) (Fig. 1; Fig. S3A). G299 normally interacts with the conserved 16S rRNA 560 loop, which contains several sharp backbone turns and links the 5′ and central domains of 16S rRNA (Fig. S3B). The Watson–Crick face of G299A makes hydrogen bonds with the Hoogsteen face of G566 and coordinates a Mg2+ ion with G558, appearing to stabilize the 560 loop (Fig. S3B) (25). The G299A mutation results in a loss of coordination to the Mg2+ ion, presumably due to the predicted electrostatic repulsion between the N6 of adenosine and the metal (17). In addition to the loss of the Mg2+ ion, the mutation to adenosine precludes interaction with G566 (Fig. S3B). The loss of these two interactions does not, however, disrupt the compressed fold of the 560 loop.
Despite the modest nature of the local changes around the G299A mutation, significant structural changes occur ∼80 Å away, at B8 (Fig. S2B). As in the G347U structure, contacts between h14 and L14 are abrogated as h8 and h14 shift away from the 50S subunit (Fig. 2C); this indicates an important role for RNA tertiary interactions between h12 and the 560 loop, which have long-range implications for h14 positioning. These data provide direct evidence that 16S rRNA helices are capable of transmitting conformational signals across the ribosome subunits. Moreover, the similar structural effects of G299A and G347U imply that they influence decoding via a common mechanism.
Comparison of G347U and G299A Structures with Previous 70S Structures.
We compared our two 70S ram structures with other ribosome structures using the program Theseus (31, 32). This program uses maximum-likelihood superpositioning of the phosphate backbone and is a powerful method to look at conformational changes between two similar structures. Consistent with the original difference maps (Fig. S2), the most striking backbone differences between our two 70S ram structures with that of an analogous wild-type 70S structure (27) occurs in the 16S rRNA at h8 and h14 (Fig. 4B and Figs. S4B and S5). These helices, along with h5, move upon binding of TC binding to the 70S (stabilized with a nonhydrolyzable GTP analog or the antibiotic kirromycin) (16, 23). When we compare the two 70S ram structures to either TC-bound structures, we see little to no backbone changes at h8/h14 for the G299A structure (Fig. 4C) and reduced changes for the G347U structure (Fig. S4C) [The G347U mutation causes additional distortions in h14 not observed in either the G299A or TC-bound 70S structures (Fig. S4C), presumably because the mutation is located nearby.] These data indicate that G347U and G299A mutations partially recapitulate the conformational changes that occur upon TC binding, in the absence of this factor (Fig. 5). Overall, there were minimal differences between the 5S and 23S rRNA except for previously seen L1 stalk movements that depend upon which tRNA is bound at the E site (nucleotides 2116–2165) (33) and 5S rRNA changes (nucleotides 83–94) that appear to arise from either different crystal packing interactions (16, 23) or the lack of A-site ligands (34).
Fig. 4.
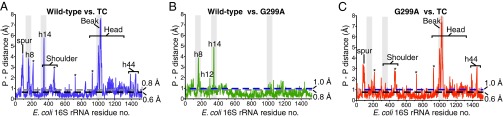
Comparison of the 70S G299A structure with other 70S structures. (A) Comparison of the 16S rRNA phosphate–phosphate backbone differences between wild-type 70S (PDB ID code 2WDG) and 70S TC (PDB ID code 2XQD). The coordinate error for the wild-type 70S (0.8 Å) and the 70S TC structures (0.6 Å) are shown as gray and black dashed lines, respectively. The asterisks denotes rRNA adjacent to the mobile spur regions that moves presumably because of different crystal forms used in the 2WDG and 2XQD structures (as not seen in B). (B) Same comparison as in A of wild-type 70S and 70S G299A. Major differences are almost exclusively in the h8 and h14 regions. The coordinate error for the wild-type 70S (0.8 Å) and 70S G299A structures (1.0 Å) are shown as gray and blue dashed lines, respectively. (C) Same comparison as in A of 70S TC and 70S G299A. Differences in h8 and h14 as seen in B are absent. The coordinate errors for the 70S G299A (1.0 Å) and 70S TC structures (0.6 Å) are shown as blue and black dashed lines, respectively.
Fig. 5.

Conformational changes in h14 are propagated to h5. (A) An overview of the 5′ domain of 16S rRNA (Left) with the secondary structure diagram of the junction between h5 and h14 of 16S rRNA (E. coli numbering; Right). Nucleotides conserved at >90% in all cytoplasmic ribosomes are colored red (36). The base triple between C352 and the U56⋅A356 base pair is illustrated with a broken line. (B) A comparison of 70S G299A and either wild-type 70S (Left) or 70S TC (Right) reveals how the mutant ribosomes are poised for productive interaction of h5 with EF-Tu, whereas wild-type 70S are less organized for interactions with EF-Tu.
The mutant ribosomes were crystallized in the presence of the miscoding antibiotic paromomycin, which might raise concern that the observed conformational changes are due to the antibiotic. However, all of the structures compared above derive from complexes containing paromomycin, and thus the changes seen in h8/h14 can be specifically attributed to the 16S rRNA mutations. Moreover, there is no evidence for paromomycin-dependent conformational changes in h8/h14 from earlier structural studies (25, 27).
Discussion
In this study, we report X-ray crystal structures of 70S ribosomes carrying mutation G299A or G347U in 16S rRNA. Each of these mutations confers a strong miscoding phenotype in vivo and accelerates the GTPase activation/GTP hydrolysis step of aa-tRNA selection in vitro (17). The most prominent structural effects of these mutations are remarkably similar—each widens h14, resulting in a loss of the base triple involving nucleotides 342, 347, and 160 and disruption of many of the specific contacts contributing to B8. Comparison of the mutant ribosome structures with other previously determined ribosome structures shows that the altered conformation of h8/h14 is most similar to that seen in the TC-bound structures (16, 23), which are believed to closely resemble the GTPase-activated state of decoding. Together, these observations provide compelling evidence that disruption of B8 is an important aspect of GTPase activation. Consistent with this interpretation, ram mutations are found in B8 on both subunits, in h8/h14 on the 30S and in L14 and L19 on the 50S (17, 29). Moreover, truncations of either h8 or h14, which undoubtedly disrupt B8, cause miscoding and accelerate GTPase activation/GTP hydrolysis by EF-Tu (17). We propose that all of these ram mutations, which compromise (or effectively “predisrupt”) B8, reduce the energetic cost of attaining the GTPase-activated state and thereby increase miscoding.
The combination of biochemical and structural results reported herein indicate that B8 puts a “brake” on, or negatively regulates GTPase activation of TC. How does this occur, because B8 components h8 and h14 do not physically interact with TC (23)? Helix 5 of 16S rRNA, which is physically adjacent to h14 (Fig. 5A), interacts with domain 2 of EF-Tu and the acceptor end of aa-tRNA in the TC-bound ribosome structures. Data from EF-Tu mutants confirms this interaction is crucial for GTPase activation (35). Comparison of structures of wild-type 70S with and without bound TC suggests that movement of h8/h14 away from the 50S subunit is accompanied by a movement of h5. Despite h8/h14 clearly moving in TC-bound ribosome structures (16, 23), its significance was not noted. Our structures reported here show that ram mutants can disrupt B8 in the absence of TC, positioning both h5 and h14 so that they mimic the GTPase-activated conformation (Fig. 5B). The basis for the coupling of h5 and h14 movement is likely the base triple formed by U56⋅A356⋅C352 (Fig. 5B). As h5 and h14 adopt their GTPase productive conformation, this tertiary interaction between them is maintained rather than broken, causing the helices to move as a unit. This observation establishes a link between B8 disruption and GTPase activation.
Because the general features of aa-tRNA delivery to the ribosome are conserved in all organisms, one would expect the details of GTPase activation of TC to also be conserved. In fact, most h5 and h14 nucleotides are greater than 90% conserved in cytoplasmic ribosomes across all three domains of life, making these helices a “hotspot” for rRNA sequence conservation similar to other known functional regions, such as the decoding center (36). Structurally, the U56⋅A356⋅C352 base triple is present in all of the X-ray crystallographic models of prokaryotic ribosomes as well as the eukaryotic ribosome structure from Saccharomyces cerevisiae (25, 37, 38). This conservation provides additional support for the functional importance of the role for h5 and h14 in GTPase activation.
An unexpected and remarkable finding of our study is that h12 mutation G299A causes the largest structural changes ∼80 Å away, in h8/h14 at B8. How such long-range signaling is mediated remains unclear, although we imagine that G299A acts in a manner analogous to well-studied allosteric mutations in protein enzymes (39, 40). In this view, enzymes are inherently dynamic, interconverting between multiple distinct (but similar) conformational states at timescales relevant for catalysis. Intermediates along the reaction coordinate are similarly well described as dynamic ensembles of related conformational states, and hence the reaction proceeds through a combinatorial multitude of interrelated parallel pathways. These dynamics, which largely govern catalysis, entail coordinated motions of elements across the whole enzyme. By perturbing the overall conformational equilibria of the enzyme, an allosteric mutation impacts the active site from a distance. In the case of G299A, its long-range effect on h8/h14 presumably shifts the conformational equilibria of the ribosome forward along the reaction coordinate of decoding, enabling the GTPase-activated state to be more readily attained.
Mutation G299A lies near the interface of ribosomal proteins S4 and S5, where a number of mutations affecting decoding fidelity have been mapped (18–20). It was hypothesized that these mutations act by destabilizing the S4/S5 interface to promote domain closure (5, 17). However, our current findings suggest that G299A acts by disrupting B8 and raises the possibility that nearby S4/S5 mutations also act by influencing B8. Intriguingly, the majority of ribosomal mutations that affect decoding fidelity cluster to either B8 or the h12/S4/S5 region (Fig. 1) (17, 19, 29). It is tempting to speculate that nearly all ram mutations either directly or indirectly destabilize B8, similarly altering the conformational equilibria of the ribosome. Further experiments will be necessary to investigate this possibility.
In summary, our results provide evidence for a long-range interaction network across the 30S subunit to communicate with the GTPase center of EF-Tu. It is unknown whether this network is normally used for signaling by wild-type ribosomes, but such a scenario seems plausible. For example, cognate codon recognition in the 30S A site might promote conformational signaling to h8/h14 via the same interaction network, facilitating disruption of B8 and productive interaction of EF-Tu within the intersubunit space. Undoubtedly, GTPase activation is controlled by several other events, such as distortion (bending) of the tRNA and conformational changes resulting from interaction between h5 of the 30S shoulder and domain 2 of EF-Tu. Further biochemical analyses are necessary to elucidate the relative contribution of each of these events to GTPase activation and other aspects of the decoding process.
Materials and Methods
Bacterial Strains.
E. coli Δ7 prrn strains harboring derivatives of plasmid p278MS2 (41) were made as described previously (42). T. thermophilus strains expressing homogeneous populations of mutant ribosomes were constructed as follows. A ∼1,500-bp DNA fragment that includes the 5′ two-thirds of rrsB and adjacent DNA upstream was amplified from the T. thermophilus genome and cloned into pUC18-htk1, a vector encoding a thermostable kanamycin adenyltransferase (26, 43). The resulting plasmid pMD3 was subjected to site-directed mutagenesis to produce the derivatives pMD5 and pMD6, with mutations corresponding to 16S rRNA substitutions G347U and G299A, respectively. These plasmids were each transformed into T. thermophilus HB8 (44), selecting for kanamycin resistance (KanR). Transformants were screened by PCR to identify those in which the plasmid integrated into the rrsB locus, as opposed to rrsA. Such isolates were cultured for several days in the absence of kanamycin and then plated for single colonies. These colonies were screened for kanamycin sensitivity (KanS), due to loss of the integrated plasmid through a second recombination event, and such KanS isolates were further screened by PCR and sequencing to identify those in which rrsB was replaced with the mutant allele. Finally, these mutant strains were transformed with chromosomal DNA from HG286 (ΔrrsA::htk1) to delete the rrsA gene (26), producing strains KLF1211 and KLF1212.
Kinetic Assays.
Single-turnover GTP hydrolysis experiments were performed with ribosomes purified from E. coli Δ7 prrn strains as described previously (17).
T. thermophilus 70S Purification and Crystallization.
T. thermophilus ribosomes containing either the G299A or G347U mutations were purified as described previously (25). E. coli tRNAfMet and tRNAPhe were purchased from Chemical Block. The mRNA oligonucleotide was chemically synthesized (Dharmacon) with a sequence of 5′ GGCAAGGAGGUAAAAAUGUUCAAAA 3′, where the underlined AUG and UUC represent the P- and A-site codons, respectively. Ribosome complexes were formed, and crystals were grown and cryoprotected using previously established procedures (25). The crystals were soaked in a final cryo-solution containing 200 μM paromomycin for 4 h before being harvested and flash frozen in liquid nitrogen for data collection.
Data Collection and Refinement.
X-ray diffraction data were collected on three crystals for each structure at the Southeast Regional Collaborative Access Team (SER-CAT) and Northeastern Collaborative Access Team (NE-CAT) beamlines at the Advanced Photon Source (Argonne, IL). Data were integrated and scaled using the XDS software package (45). The structures were solved by molecular replacement using the PHENIX software suite using Protein Data Bank (PDB) ID codes 2WDG, 2WDH, 2WDI, and 2WDJ as a search model with all ligands removed (46). Coordinate refinement was performed with each ribosomal subunit defined as a rigid group followed by additional rigid and Translation, Liberation, Screw-movement (TLS) refinement with groups defined by the head, body, platform, and 3′ minor domain of the 30S subunit, along with mobile elements of the 50S subunit: 5S rRNA, L1 arm, protein L9, A-site finger, and the central protuberance. Modeling of conformational changes in rRNA and ribosomal proteins along with the placement of mRNA, tRNA, and Mg2+ ions was performed using Coot guided by MolProbity identification of all-atom contacts (47, 48). Iterative rounds of model-building were followed by positional and atomic displacement parameter (ADP) refinement in PHENIX, yielding a final model with the statistics reported in Table S2. Figures were generated using PyMOL (www.pymol.org).
Global Phosphate Backbone Changes.
The 23S, 5S, or 16S platform domains (residues 560–912) were superpositioned using the program Theseus to generate maximum-likelihood covariance weighting for the wild-type, TC-bound, or 70S ram mutant structures (32). Individual rRNA was pairwise aligned against the wild-type tRNA 70S structure as a reference (27). Nucleotide movement was analyzed for each pairwise alignment by determining the phosphate-to-phosphate distance (Å) between the two structures.
Supplementary Material
Acknowledgments
We thank G. L. Conn for helpful discussions throughout the project and critical reading of the manuscript; S. Gregory for plasmid pUC18-htk, strain HG286, and helpful suggestions; R. Gonzalez and S. McClory for useful discussions; and F. Murphy and staff members at the NE-CAT beamline for assistance during data collection. This work is based on research conducted at the Advanced Photon Source on the NE-CAT beamlines (supported by National Center for Research Resources [National Institutes of Health (NIH)] Award RR-15301) and SER-CAT beamline. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by US DOE Contract DE-AC02-06CH11357. Support for this work was provided by the Department of Defense through the National Defense Science and Engineering Graduate Fellowship Program and NIH Training Grant T32 GM8367 (to C.E.F.), and National Institute of General Medical Sciences of the NIH Awards R01GM093279 (to C.M.D.) and R01GM072528 (to K.F.). C.M.D. is a Pew Scholar in the Biomedical Sciences.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3V6U–3V6X (G347U) and 4EJ9, 4EJA, 4EJB, and 4EJC (G299A).
See Commentary on page 9627.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1301585110/-/DCSupplemental.
References
- 1.Zaher HS, Green R. Fidelity at the molecular level: Lessons from protein synthesis. Cell. 2009;136(4):746–762. doi: 10.1016/j.cell.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 3.Sydow JF, Cramer P. RNA polymerase fidelity and transcriptional proofreading. Curr Opin Struct Biol. 2009;19(6):732–739. doi: 10.1016/j.sbi.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Francklyn CS. DNA polymerases and aminoacyl-tRNA synthetases: shared mechanisms for ensuring the fidelity of gene expression. Biochemistry. 2008;47(45):11695–11703. doi: 10.1021/bi801500z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu Rev Biochem. 2005;74:129–177. doi: 10.1146/annurev.biochem.74.061903.155440. [DOI] [PubMed] [Google Scholar]
- 6.Wohlgemuth I, Pohl C, Mittelstaet J, Konevega AL, Rodnina MV. Evolutionary optimization of speed and accuracy of decoding on the ribosome. Philos Trans R Soc Lond B Biol Sci. 2011;366(1580):2979–2986. doi: 10.1098/rstb.2011.0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hopfield JJ. Kinetic proofreading: A new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc Natl Acad Sci USA. 1974;71(10):4135–4139. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ninio J. Kinetic amplification of enzyme discrimination. Biochimie. 1975;57(5):587–595. doi: 10.1016/s0300-9084(75)80139-8. [DOI] [PubMed] [Google Scholar]
- 9.Ruusala T, Ehrenberg M, Kurland CG. Is there proofreading during polypeptide synthesis? EMBO J. 1982;1(6):741–745. doi: 10.1002/j.1460-2075.1982.tb01240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansson M, Zhang J, Ehrenberg M. Genetic code translation displays a linear trade-off between efficiency and accuracy of tRNA selection. Proc Natl Acad Sci USA. 2012;109(1):131–136. doi: 10.1073/pnas.1116480109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gromadski KB, Daviter T, Rodnina MV. A uniform response to mismatches in codon-anticodon complexes ensures ribosomal fidelity. Mol Cell. 2006;21(3):369–377. doi: 10.1016/j.molcel.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 12.Gromadski KB, Rodnina MV. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol Cell. 2004;13(2):191–200. doi: 10.1016/s1097-2765(04)00005-x. [DOI] [PubMed] [Google Scholar]
- 13.Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002;111(5):721–732. doi: 10.1016/s0092-8674(02)01086-3. [DOI] [PubMed] [Google Scholar]
- 14.Pape T, Wintermeyer W, Rodnina M. Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. EMBO J. 1999;18(13):3800–3807. doi: 10.1093/emboj/18.13.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valle M, et al. Cryo-EM reveals an active role for aminoacyl-tRNA in the accommodation process. EMBO J. 2002;21(13):3557–3567. doi: 10.1093/emboj/cdf326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voorhees RM, Schmeing TM, Kelley AC, Ramakrishnan V. The mechanism for activation of GTP hydrolysis on the ribosome. Science. 2010;330(6005):835–838. doi: 10.1126/science.1194460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McClory SP, Leisring JM, Qin D, Fredrick K. Missense suppressor mutations in 16S rRNA reveal the importance of helices h8 and h14 in aminoacyl-tRNA selection. RNA. 2010;16(10):1925–1934. doi: 10.1261/rna.2228510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahlgren A, Rydén-Aulin M. A novel mutation in ribosomal protein S4 that affects the function of a mutated RF1. Biochimie. 2000;82(8):683–691. doi: 10.1016/s0300-9084(00)01160-3. [DOI] [PubMed] [Google Scholar]
- 19.Maisnier-Patin S, Berg OG, Liljas L, Andersson DI. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol Microbiol. 2002;46(2):355–366. doi: 10.1046/j.1365-2958.2002.03173.x. [DOI] [PubMed] [Google Scholar]
- 20.Olsson MO, Isaksson LA. Analysis of rpsD mutations in Escherichia coli. I. Comparison of mutants with various alterations in ribosomal protein S4. Mol Gen Genet. 1979;169(3):251–257. doi: 10.1007/BF00382271. [DOI] [PubMed] [Google Scholar]
- 21.Björkman J, Samuelsson P, Andersson DI, Hughes D. Novel ribosomal mutations affecting translational accuracy, antibiotic resistance and virulence of Salmonella typhimurium. Mol Microbiol. 1999;31(1):53–58. doi: 10.1046/j.1365-2958.1999.01142.x. [DOI] [PubMed] [Google Scholar]
- 22.Vallabhaneni H, Farabaugh PJ. Accuracy modulating mutations of the ribosomal protein S4-S5 interface do not necessarily destabilize the rps4-rps5 protein-protein interaction. RNA. 2009;15(6):1100–1109. doi: 10.1261/rna.1530509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmeing TM, et al. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326(5953):688–694. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodnina MV, Wintermeyer W. GTP consumption of elongation factor Tu during translation of heteropolymeric mRNAs. Proc Natl Acad Sci USA. 1995;92(6):1945–1949. doi: 10.1073/pnas.92.6.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selmer M, et al. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313(5795):1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 26.Gregory ST, Dahlberg AE. Genetic and structural analysis of base substitutions in the central pseudoknot of Thermus thermophilus 16S ribosomal RNA. RNA. 2009;15(2):215–223. doi: 10.1261/rna.1374809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voorhees RM, Weixlbaumer A, Loakes D, Kelley AC, Ramakrishnan V. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat Struct Mol Biol. 2009;16(5):528–533. doi: 10.1038/nsmb.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yusupov MM, et al. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292(5518):883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 29.Maisnier-Patin S, Paulander W, Pennhag A, Andersson DI. Compensatory evolution reveals functional interactions between ribosomal proteins S12, L14 and L19. J Mol Biol. 2007;366(1):207–215. doi: 10.1016/j.jmb.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 30.Firdaus-Raih M, Harrison AM, Willett P, Artymiuk PJ. Novel base triples in RNA structures revealed by graph theoretical searching methods. BMC Bioinformatics. 2011;12(Suppl 13):S2. doi: 10.1186/1471-2105-12-S13-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Theobald DL, Steindel PA. Optimal simultaneous superpositioning of multiple structures with missing data. Bioinformatics. 2012;28(15):1972–1979. doi: 10.1093/bioinformatics/bts243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Theobald DL, Wuttke DS. Accurate structural correlations from maximum likelihood superpositions. PLOS Comput Biol. 2008;4(2):e43. doi: 10.1371/journal.pcbi.0040043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fei J, Richard AC, Bronson JE, Gonzalez RL., Jr Transfer RNA-mediated regulation of ribosome dynamics during protein synthesis. Nat Struct Mol Biol. 2011;18(9):1043–1051. doi: 10.1038/nsmb.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jenner LB, Demeshkina N, Yusupova G, Yusupov M. Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat Struct Mol Biol. 2010;17(5):555–560. doi: 10.1038/nsmb.1790. [DOI] [PubMed] [Google Scholar]
- 35.Vorstenbosch E, Pape T, Rodnina MV, Kraal B, Wintermeyer W. The G222D mutation in elongation factor Tu inhibits the codon-induced conformational changes leading to GTPase activation on the ribosome. EMBO J. 1996;15(23):6766–6774. [PMC free article] [PubMed] [Google Scholar]
- 36.Cannone JJ, et al. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-Shem A, Jenner L, Yusupova G, Yusupov M. Crystal structure of the eukaryotic ribosome. Science. 2010;330(6008):1203–1209. doi: 10.1126/science.1194294. [DOI] [PubMed] [Google Scholar]
- 38.Schuwirth BS, et al. Structures of the bacterial ribosome at 3.5 A resolution. Science. 2005;310(5749):827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 39.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4(8):474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 40.Benkovic SJ, Hammes GG, Hammes-Schiffer S. Free-energy landscape of enzyme catalysis. Biochemistry. 2008;47(11):3317–3321. doi: 10.1021/bi800049z. [DOI] [PubMed] [Google Scholar]
- 41.Youngman EM, Green R. Affinity purification of in vivo-assembled ribosomes for in vitro biochemical analysis. Methods. 2005;36(3):305–312. doi: 10.1016/j.ymeth.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 42.Qin D, Abdi NM, Fredrick K. Characterization of 16S rRNA mutations that decrease the fidelity of translation initiation. RNA. 2007;13(12):2348–2355. doi: 10.1261/rna.715307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hashimoto Y, Yano T, Kuramitsu S, Kagamiyama H. Disruption of Thermus thermophilus genes by homologous recombination using a thermostable kanamycin-resistant marker. FEBS Lett. 2001;506(3):231–234. doi: 10.1016/s0014-5793(01)02926-x. [DOI] [PubMed] [Google Scholar]
- 44.Koyama Y, Hoshino T, Tomizuka N, Furukawa K. Genetic transformation of the extreme thermophile Thermus thermophilus and of other Thermus spp. J Bacteriol. 1986;166(1):338–340. doi: 10.1128/jb.166.1.338-340.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabsch W. Xds. Acta Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.