

Abstract
Pirin is a nuclear nonheme Fe protein of unknown function present in all human tissues. Here we describe that pirin may act as a redox sensor for the nuclear factor κB (NF-κB) transcription factor, a critical mediator of intracellular signaling that has been linked to cellular responses to proinflammatory signals and controls the expression of a vast array of genes involved in immune and stress responses. Pirin’s regulatory effect was tested with several metals and at different oxidations states, and our spectroscopic results show that only the ferric form of pirin substantially facilitates binding of NF-κB proteins to target κB genes, a finding that suggests that pirin performs a redox-sensing role in NF-κB regulation. The molecular mechanism of such a metal identity- and redox state-dependent regulation is revealed by our structural studies of pirin. The ferrous and ferric pirin proteins differ only by one electron, yet they have distinct conformations. The Fe center is shown to play an allosteric role on an R-shaped surface area that has two distinct conformations based on the identity and the formal redox state of the metal. We show that the R-shaped area composes the interface for pirin-NF-κB binding that is responsible for modulation of NF-κB’s DNA-binding properties. The nonheme Fe protein pirin is proposed to serve as a reversible functional switch that enables NF-κB to respond to changes in the redox levels of the cell nucleus.
Keywords: metalloprotein, coregulator, reactive oxygen species (ROS), oxidative stress, signal transduction activation
The transcription factor, nuclear factor κB (NF-κB), discovered in 1986 (1, 2), is a ubiquitous cellular regulator widely recognized as a critical mediator of intracellular signaling for immune/inflammatory response including oxidative stress (3–5). NF-κB is a family of structurally related and functionally conserved dimeric proteins that consist of RelA (p65), RelB, c-Rel, p50, and p52. All members can form both homo- and heterodimers with different physiological functions and gene targets (6–8). NF-κB proteins bind and activate several hundred κB genes in response to activating stimuli (9–11). The activity of NF-κB is tightly controlled by regulatory proteins and cofactors collectively known as coregulators (12, 13). Complex regulatory networks exist to ensure that NF-κB is activated only by appropriate stimuli and is then inactivated when no longer needed (14–16). NF-κB typically resides in the cytoplasm in an apparently inhibited state, bound with an inhibitory protein known as IκBα. Previous studies have established that NF-κB activation is first achieved by phosphorylation of the inhibitory protein IκBα to liberate NF-κB proteins to their free forms. Next, NF-κB proteins are translocated to the nucleus where, with the help of coregulators, they will orchestrate the cascade of signaling responses to the external stimuli. At present, the precise roles played by the coregulators in the cell nucleus are poorly understood.
Winnacker and coworkers in 1997 identified a highly conserved human protein by using nuclear factor I as a “bait protein” and named it pirin (17). This previously unknown protein is expressed in all human tissues. Biochemical data suggest that pirin is a nuclear protein composed of 290 aa, ca. 32 kDa (17). Experiments using genomic Southern and Western blots have demonstrated the presence of pirin genes and their expression in dot-like subnuclear structures that represent loci of specific nuclear processes, including DNA replication and/or RNA polymerase II transcription (17). Pirin has been found to form tight complexes with the inducible antiapoptotic transcription factor NF-κB p50 homodimer and the B-cell lymphoma 3-encoded protein Bcl-3 (18). In a gel mobility shift assay using cell lysates, pirin was shown to enhance the DNA-binding activity of the p50–Bcl-3 complex (18). These observations led to the initial suggestion that pirin functions as a coregulator in the NF-κB transcription complex (18). However, such a proposed regulation has not been confirmed and the mechanism of it is completely unknown.
In the subsequent studies by various laboratories, pirin was found to be a nonheme iron-binding protein that belongs to an ancient redox-sensitive subclass of proteins in the cupin superfamily (19–21). Pirin has additionally been linked to the metastasis of melanoma cells (22, 23). It has been shown that pirin expression is significantly up-regulated in the spleen and kidney of cytosolic superoxide dismutase (SOD)-deficient mice (24). Similarly, up-regulation of pirin expression by chronic cigarette smoking has been associated with bronchial epithelial cell apoptosis (25). The crystal structure of pirin at 2.1-Å resolution reveals a ferrous site that is octahedrally coordinated with three histidines, one glutamate, and two water molecules (19). In the present work, we describe that pirin substantially facilitates the DNA binding of NF-κB p65 to the IgκB gene when the metal center is a ferric ion. An unprecedented redox regulation mechanism is proposed from the previous studies and the results are described below.
Results
Human Pirin Facilitates the DNA Binding of p65 to the κB Gene in a Redox-State-Dependent Manner.
We studied the effect of pirin on the binding affinity of p65 (residues 21–325) (26) homodimers to the Ig κB target sequence (IgκB) by testing isolated protein, using surface plasmon resonance (SPR) spectroscopy. The first significant observation is that Fe(III)-pirin, generated by oxidizing Fe(II)-pirin with ferricyanide, or O2 in a relatively slower oxidation process, and verified by electronic paramagnetic resonance (EPR) spectroscopy substantially enhanced p65 binding to the IgκB target site (Fig. 1A). In the presence of 0–25 µM Fe(III)-pirin, the binding was a two-phase process: a fast phase in the first 60 s with kon1′ = 1.1 × 106 M−1⋅s−1 and a slow association phase after 60 s with kon2′ = 0.02 M−1⋅s−1. The buffer rinse resulted in the loss of only a small amount of mass presumably due to washing away of nonspecifically bound proteins observed in the slower binding phase. The binding is very tight, with virtually no dissociation after removing nonspecifically bound proteins. This is unique experimental evidence of pirin interacting with p65 and dramatically enhancing the binding affinity for p65 to the κB site. In contrast, Fe(II)-pirin does not affect p65 binding to the IgκB site. The dose response with the truncated p65 is shown in Fig. 1B. The sequence specificity of pirin’s effect on p65 was further established by performing the same SPR experiment using a nonspecific DNA sequence.
Fig. 1.

The ferric, not ferrous, form of pirin substantially facilitates binding of p65 to the IgκB gene in SPR spectroscopy. (A) Detection of apo-, Fe(II)-, and Fe(III)-pirin effects on p65 binding to the IgκB site. (B) Response unit as a function of the pirin (1–290):p65 (19–291) ratio. (C) SPR spectroscopy with Fe(III)- and metal-substituted pirin proteins. (D) Effect of exogenous pirin on native NF-κB of the nuclear extract of HeLa cells. (E) QCM-D spectroscopy of p65 alone (red trace) and along with Fe(III)-pirin in 1:1–1:10 ratio (indicated by 1×, 3×, 5×, and 10×). (F) Comparison of the ΔD/Δf slope in a QCM-D study that indicates a conformational difference of the protein–DNA complex. The SPR traces are color coded with p65 only (red), p65 with Fe(II)-pirin (black), Fe(III)-pirin (blue), metal-striped pirin (purple), Co(II)-pirin (pink), Mn(II)-pirin (dark yellow), Mn(III)-pirin (wine), and Ni(II)-pirin (olive). Fe(III)-pirin alone (cyan) is shown in A and HeLa nuclear extract only (navy) is shown in D as controls, respectively.
To determine whether other transition metal ions can function in place of the iron cofactor, apo-pirin was prepared from Fe-depleted minimal medium and charged with divalent metal ions. Co-, Mn-, and Ni-reconstituted proteins were stable and contained about 0.6 metal ions per molecule of pirin as determined by inductively coupled plasma optical emission spectrometry (ICP-OES) analysis (Table S1 and SI Materials and Methods). These metal-substituted pirin proteins had minimal effects on p65 and were unable to achieve the effect observed for NF-κB with ferric pirin (Fig. 1C). We wondered whether a metal, other than iron, with correct valence state (i.e., trivalent) would make pirin active. We oxidized Mn(II)-pirin protein, using potassium ferricyanide. After an overnight incubation, the protein was desalted to remove the oxidant and crystallized. As the oxidation state rises, each metal ligand increases its bond length in the Mn-substituted pirin metal center, a scenario similar to what is observed in Fe-pirin structures. The change of the oxidation state of pirin was validated by low-temperature EPR spectroscopy measured before and after the oxidation reaction by the disappearance of the Mn(II) EPR signal. This is the same method we used to generate Fe(III)-pirin from Fe(II)-pirin, with the exception of a shorter incubation time (4 h) after which the colorless Fe(II)-pirin turned into a pale yellow-colored solution and a high-spin (S = 5/2) ferric EPR signal was fully developed from the EPR-silent state. As shown in Fig. 1C, the Co(II)-, Mn(II)-, and Mn(III)-substituted pirin proteins have little or no activity toward NF-κB. A similar effect was also observed by fluorescence spectroscopy. When the 5′-FAM-labeled κB DNA was titrated by p65 in the absence of pirin, the 520-nm emission intensity increased as previously reported (27). In contrast, the titration using p65 and Fe(III)-pirin induced an increase in emission signal intensity that was nearly fourfold larger than that using p65 alone, an effect not seen when pirin is complexed with other metals such as Co(II) and Cu(II) (Fig. S1 and SI Materials and Methods). The change in fluorescence intensity as a function of p65 concentration resulted in a sigmoidal curve, indicative of the binding cooperativity previously reported in studies of NF-κB and DNA interactions (27). All of the available reducing agents affect the fluorescein in FAM and resulted in less excitation energy. Thus, the fluorescence assays with Fe(II)-pirin cannot be performed in the current stage.
Pirin’s effect on NF-κB was further investigated with native NF-κB proteins in cell lines. Fig. 1D shows the effect of isolated pirin on the nuclear extract from HeLa cells, which were not under redox stress; i.e., the NF-κB proteins were not yet induced for activation. The freshly prepared nuclear extract (at a total protein concentration of 0.08 µg/mL) was injected into the IgκB-coated SPR sensor chip simultaneously with exogenous Fe(III)-pirin. The inclusion of isolated Fe(III)-pirin protein with the nuclear extract increased binding by about 25-fold compared with that with the extract alone. Fe(II)-pirin and metal-substituted pirin were unable to induce the same effect.
Protein–DNA Supercomplex in the Presence of Pirin Is Compact and Rigid.
We also tested whether NF-κB and the DNA change their structures after forming a complex. Quartz crystal microbalance with dissipation (QCM-D) was used to monitor pirin’s effect on the conformation of the p65–DNA complex (Fig. 1E). The plot of the change in dissipation (ΔD) vs. the shift in frequency (Δf) displays a more moderate slope when pirin is present (Fig. 1F). A less steep ΔD vs. Δf slope indicates a more compact, rigid layer formed upon the sensor (28). This result suggests that the conformation of the supercomplex is altered with respect to the NF-κB and DNA mixture. This observation is also consistent with previous studies showing that κB sequences are bent to various degrees upon NF-κB binding (6, 29). DNA bending is a common prerequisite to the formation of a fully active transcriptional complex (30).
Pirin–NF-κB–DNA Supercomplex Dissociates by Added Reducing Agent.
We determined the midpoint reduction potential (Em) for the ferrous/ferric couple of pirin by cyclic voltammetry. The resulting curve shown in Fig. 2A resembles published data on the nonheme FeB of nitric oxide reductase (31). At pH 7.4, the Em value is +80 mV vs. an Ag/AgCl electrode. This is 140 mV lower than what was determined for a structurally related protein, Fe-superoxide dismutase (32), which is similarly coordinated by a 3-His-1-carboxylate protein ligand motif. EPR spectroscopic characterization shown in Fig. 2B provides further support that the high-spin ferric ion (S = 5/2) in pirin is slowly reduced by l-ascorbate (+58 mV) in a physiologically meaningful context (33). The reduced ferrous ion in pirin is readily oxidized by O2.
Fig. 2.

The function of pirin on p65 is reversible depending on the redox state of the iron center. (A) Determination of the reduction potential of pirin by film cyclic voltammetry in an anaerobic chamber. (B) X-band EPR spectroscopy shows reversibility of the Fe(II)/Fe(III) couple in response to l-ascorbate. Shown are Fe(II)-pirin (250 µM, trace 1) after oxidation by O2 for 30 min (or 1 equivalent of H2O2 for 1 min, trace 2) and addition of 1 mM l-ascorbate for 5 min (trace 3) and for 60 min (trace 4). (C) l-ascorbate was introduced to the system, after forming the supercomplex from initial injection of p65/Fe(III)-pirin, at concentrations of 0 (black), 0.25 (navy), 2.5 (magenta), and 25 mM (green) at 980 s.
If pirin is indeed a regulatory component in the NF-κB signaling transduction, it must be able to dissociate after the redox levels resume to normal. We designed a continuous SPR experiment to test whether l-ascorbate would be able to dissociate the pirin•p65•IgκB supercomplex (Fig. 2C). First, the supercomplex was formed and the nonspecifically bound proteins were washed away by buffer. At this point, l-ascorbate was introduced to stimulate a shift in intracellular redox conditions to a less oxidative environment. Fig. 2C shows that l-ascorbate caused the protein supercomplex to dissociate in a dose-dependent manner, and buffer wash without the l-ascorbate immediately halted the dissociation.
Redox State of the Fe in Pirin Affects the Conformation of a Specific R-Shaped Surface Area As Revealed by X-Ray Protein Crystallography.
To further our understanding of the structural impact of pirin metal center, especially the differences between the active Fe(III)- and inactive Fe(II)-pirin and the metal-substituted pirin variants, we determined multiple X-ray crystal structures of the active form of Fe(III)-pirin at 1.80- to 2.50-Å resolutions (Table S2). All of the pirin crystals, including ones we analyzed and previously published (19), belong to the P212121 space group with one molecule per asymmetric unit.
An apparent structural change is observed when the active ferric form of pirin is aligned with the inactive resting state of ferrous pirin. Fig. 3A shows that the deviations are primarily located on one face of the protein. The residues involved collectively account for ca. 20% of 290 total residues. Notable in our structural study, a specific, R-shaped surface loop area is identified that is substantially altered due to switching of the metal oxidation state. This area consists of residues 7–41 and 53–62, which includes the surface area surrounding the metal-binding cavity at the N terminus as well as the interface between the two cupin domains. Although the root-mean-square deviation (rmsd) of shift in the protein backbone of this R-shaped region between the Fe(II) and Fe(III) states is modest, the widespread coverage and especially the cumulative effect of the changes in the amino acid residues are extensive for a protein of this size. Both sheet-to-coil transition and twist of orientations are observed in the R-shaped region. Four charged residues (Arg14, Arg23, Glu32, and Lys34) in the R-shaped region show two distinct conformations, depending on the oxidation state of Fe (Fig. 3B). Arg14, for example, shifts 5.1 Å when comparing the two structures (Fig. S2).
Fig. 3.

(A) Structural alignment of ferric (active toward p65) and ferrous (inactive) pirin. Colored regions indicate areas of deviation (≥1 Å), whereas gray regions are identical. The structural differences of Fe(III)-pirin are shown in yellow and those of Fe(II)-pirin are in green. (B) the zoom-in view highlights the deviation of a special R-shaped area with distinct conformations. (C) Superimposed Fe center structure of ferric (yellow), ferrous (green), and l-ascorbate–reduced (cyan) pirin structures.
The structural data also provide clues for the cause of the extensive conformational changes. His56, His58, His101, and Glu103 are the coordination ligands at 2.1, 2.1, 2.3, and 2.3 Å distance from the Fe(II) ion, respectively. Whereas the histidine residues remain coordinated to the Fe ion at 2.1, 2.1, and 2.2 Å, the negatively charged ligand, Glu103, moves away from the Fe ion in the oxidized protein (Fig. 3C). The Fe-Glu103 distance lies in 2.5–3.2 Å in multiple ferric pirin structures determined (Fig. S3). The variation may be caused by metal reduction during data collections at 100 K. Even though the overall protein structure still maintains at the oxidized state, the metal ligand distances may vary. Nonetheless, this is an unexpected observation and the positively charged Fe ion still sits in a highly negatively charged cavity. We noticed that Glu103 connects the R-shaped surface through a well-defined H-bond network (Fig. S4). It is highly possible that redox state change-induced R-shape loop deviations are mainly triggered by the iron ion through Glu103.
Metal Identity Affects the Conformation of the R-Shaped Surface Area.
To understand the structural basis for the functional incapability of metal-substituted pirin proteins, we solved the X-ray crystal structures of Co- and Mn-substituted pirin (Fig. 4 and Table S3). A similar R-shaped conformational difference is observed when comparing the active form of ferric pirin with functionally inactive pirin variants charged with Co(II) and Mn(II) (Fig. S5). The changes in the R-shaped region are different from one transition metal to the next, but the location and direction of the conformation changes show the same general trend. To understand why oxidized Mn-pirin is not active, which has an oxidation state of +3, we determined its crystal structure. The structure of Mn(III)-pirin (at 2.15 Å resolution) indeed differs from that of Fe(III)-pirin, but the conformation differences are not the same as those for Mn(II)-pirin (1.56 Å resolution).
Fig. 4.
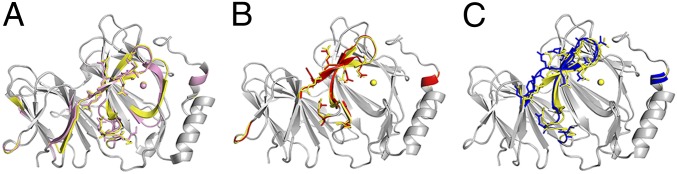
(A–C) Structural differences shown in the alignment of ferric pirin (yellow) (A) with Co(II)-substituted pirin (pink), (B) with Mn(II)-pirin (red), and (C) with Mn(III)-pirin (blue). Colored regions indicate areas of deviation (≥1 Å), and gray regions indicate identical structural features.
R-Shaped Region on Pirin Interacts with the C-Terminal Domain of NF-κB Proteins.
The structural data led us to hypothesize that the R-shaped region is the key protein surface in pirin that directly interacts with p65. A docking model study was subsequently conducted using ZDOCK (34) to further investigate how pirin makes contacts with p65 (26). The result suggests that the R-shaped surface region is the pirin·p65 interface (Fig. 5A). The R-shaped region forms much of the central binding surface between the two proteins (Fig. 5B). The binding takes place at the C-terminal Rel homology domain. The most interesting finding from the docking study is the presence of four ion pairs interacting between pirin and p65. The R-shaped surface residues Arg14, Arg23, Glu32, and Lys34 of ferric pirin are electrostatically complemented by p65 residues Glu279, Glu282, Arg273, and Glu234 at distances of 3.2, 2.7, 3.0, and 3.5 Å, respectively, forming four ion pairs (Fig. 5B). Interestingly, these four residues contribute to the major changes of the R-shaped region as shown in Fig. 3. In contrast, these ion pair interactions are missing when the Fe(II)-pirin structure is used for docking.
Fig. 5.

(A) In silico docking model of pirin (green/yellow)–p65 (blue)–IgκB (golden/blue) supercomplex built from the corresponding crystal structures (Materials and Methods). The R-shaped region is highlighted in yellow and the Fe ion is represented in the sphere (ferric in yellow and ferrous in light green). (B) Zoomed-in view of the pirin (upper, green)–p65 (lower, cyan) interface region shows multiple complementary ion-pair interactions, including K34-E234, E32-R273, R14-E279, and R23-E282 (pirin-p65).
The R-shaped region was tested by site-directed mutagenesis. R23E, E32V, K34V, and double-mutant S13R/R14W of the R-shaped region mutants were generated and analyzed. These pirin variants were reconstituted with Fe(II) and oxidized before SPR analysis in the same manner as for the wild-type protein. Fig. 6A shows that these mutations have a significant adverse effect on the binding of pirin to p65 compared with the wild-type protein, reducing the binding efficiency to one- to two-thirds of the wild type. Although there was less DNA binding in these mutants, the resulting supercomplex was still tightly bound and showed a similar lack of dissociation to that of the wild type. Q115N pirin was generated and used as a control along with wild-type pirin. Q115 is not part of the R-shaped region. It is located in the interior of the protein. Not surprisingly, Q115N essentially behaved the same as wild-type pirin. These results demonstrate that site-directed mutation of the surface loop residues dramatically changes the ability of pirin to facilitate p65 binding to the κB site and thereby supports the docking model.
Fig. 6.
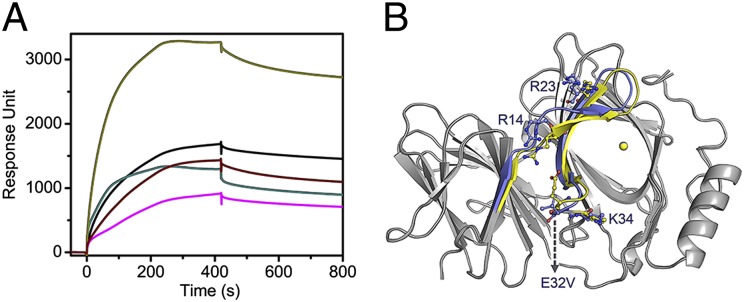
Mutation at the R-shaped surface impairs the ability of pirin to enhance the DNA-binding ability of p65 to the IgκB site. (A) SPR assay using p65 and immobilized IgκB with R23E (black trace), E32V (cyan), K34V (wine), and S13R/R14W (magenta). The nonsurface mutant Q115N (dark yellow) was used as a positive control. (B) Crystal structure of E32V aligned with native pirin structure. Colored regions indicate areas of deviation (≥1 Å), and gray regions indicate identical structural features.
A 1.70-Å resolution crystal structure of the E32V mutant was obtained in its oxidized form (4HLT, Table S4). The structure shows that mutation of Glu32 disrupts the electrostatic interaction between Glu32 and Arg14 (Fig. 6B). The two residues are within 3.2 Å in the ferric form of native pirin, but Val32 and Arg14 are 9.4 Å apart in E32V. Consequently, the position of the guanidinium group of Arg23 is also shifted. Among the four charged residues, Lys34 is the only residue that is nearly not affected by mutation. This structure provides a molecular rationale for the negative impact of the mutation. Together, these data validate the R-shaped region as the functionally important area of pirin for interaction with p65.
Discussion
On the basis of previous work and this study, it is concluded that human pirin is a redox sensor in the cell nucleus that functions as a coregulator of NF-κB. Fig. 7 presents an iron-dependent bioinorganic scheme for how NF-κB could respond to redox changes within the nucleus. Previous studies have established that NF-κB activation is first achieved by phosphorylation of the inhibitory IκB proteins in the cytoplasm to liberate NF-κB. Upon translocation of NF-κB to the nucleus, several layers of activation take place, involving different coregulators and mechanisms. In the nucleus, under normal reducing conditions, pirin resides in its inactive ferrous state. The results presented here show that, when the redox environment shifts to more oxidizing conditions, pirin responds by changing to its functionally active ferric form and turns on the transcription factor. This is a reversible redox process, depending on the oxidation state of the iron. The dependence of pirin on its iron cofactor is likely an additional contributing factor to the link between iron and NF-κB in the nucleus.
Fig. 7.

Proposed function of human pirin. This model depicts a nonheme iron- and oxidation state-dependent regulation mechanism of NF-κB in the cell nucleus.
It remains to be determined whether the proposed nonheme iron protein-based redox switch operates in vivo. However, our findings are consistent with recent biological studies showing increased expression of pirin in response to oxidative stress (24, 35, 36). Interestingly, pirin-like proteins in plants and prokaryotes have also been linked to stress-induced responses and programmed cell death (37). Up-regulation of pirin expression by chronic cigarette smoking has been associated with bronchial epithelial cell apoptosis, presumably due to increased oxidative stress that triggers Nrf2-modulated genes including pirin (35). The conclusion that the oxidation state and activity of pirin is regulated by cellular redox conditions is consistent with those previous findings proposing a role as a protector against oxidative stress. Thus, oxidative stress elevates both the expression of pirin and its activity. When redox levels are restored to normal reducing conditions in the cell nucleus, the transcriptional response induced by pirin may be ablated by reducing its expression and by losing its ability to form a complex with NF-κB and DNA. However, the present work does not exclude the possibility that there is another protein responsible for oxidizing pirin in the nucleus as a part of a signaling cascade.
Pirin has been proposed to be an important factor in cell differentiation (38). In progenitor cells, the activation of pirin is an important event that alters the transcription profile of the cell to one that primes the cell for differentiation (38). Reduced pirin expression is a common feature of acute myelogenous leukemia, which results in the proliferation of immature myeloid cells. Elevation in the concentration of reactive oxygen species (ROS) has been reported to facilitate differentiation in hematopoetic progenitor cells (39).
Pirin uses a structure-based conformational switch to quickly, reversibly, and efficiently inform the transcription factor of the positive/negative shift of redox levels in the nucleus. The data presented here suggest that the Fe center in pirin is not its active site. Instead, the role of the Fe center is allosteric control. The R-shaped surface loop region is the site that interacts with p65. The two distinct conformational states on the R-shaped surface loop regions endow pirin with the ability to regulate NF-κB with full reversibility. This unique property distinguishes pirin from any other redox regulators of transcription factors, including the H2O2 sensor PerR found in a bacterial source (40). PerR and pirin are both redox sensors with a similar dependence on nonheme iron. Unlike human pirin, PerR itself is a transcription factor. The Fe ion of PerR participates in a Fenton-type reaction with H2O2. The result of this reaction is the formation of a hydroxyl radical that irreversibly oxidizes one of its histidine metal-binding ligands, resulting in a drastically altered protein conformation that is no longer able to bind to DNA (40, 41). In contrast, the regulatory mechanism of redox sensing illustrated in Fig. 7 uses a reversible Fe(II)/Fe(III) oxidation state coupling and a consequent reversible conformational switch. The conformational variation of the R-shaped loop areas of pirin can respond accordingly to the oxidation level in the cell nucleus, which allows pirin to respond to redox potential shifts in both directions.
Materials and Methods
Protein Materials.
The full-length (1–290 aa) untagged form of human pirin was cloned and expressed in Escherichia coli strain BL21(DE3) (Invitrogen). The purification of pirin protein consisted of ammonium sulfate fractionation and three major chromatographic steps including SP Sepharose, Superdex 75, and MonoS on an ÅKTA FPLC system. The metal-free apo-protein of pirin was isolated from iron-depleted minimum medium (42) and metal reconstitution details are given in SI Materials and Methods. p65 (19–291) was obtained from Jim Maher, III of Mayo Clinic College of Medicine (Rochester, MN) (43, 44) and isolated as described in the SI Materials and Methods. The purified p65 was treated with 10 equivalents of EDTA overnight and then desalted with 20 mM Tris⋅HCl buffer (pH 7.4) containing 50 mM NaCl and 5% (vol/vol) glycerol.
Metal Analysis.
Metal contents of each pirin sample were determined by inductively coupled plasma optical emission spectrometry, using a Spectro Genesis spectrometer (Spectro Analytical Instruments GmbH). Calibration curves were obtained by measuring the standards at the concentrations of each element at 0, 0.1, 0.3, 0.5, 1.0, and 10 ppm (mg/L).
Nuclear Extract Containing Native NF-κB Proteins.
A nuclear extract of HeLa cells was prepared by using the Subcellular Protein Fractionation Kit for Cultured Cells from Pierce Protein Research Products (Thermo Fisher Scientific), according to the manufacturer’s protocols. Before injection, the extract was diluted to 0.08 µg/mL by the SPR binding buffer described above. The binding measurements were taken with the extract alone and together with added pirin (10 µM) protein.
DNA.
The κB-site DNA used was synthetic oligonucleotide 5′-biotin-GAGTTGAGGGGACTTTCCCAGGC-3′ (κB site is shown in boldface type). The single-strand DNA was annealed to its complementary strand before use. A nonspecific DNA sequence of 5′-CCTATATGCGGCGTATATCC-3′ was used as a negative control.
Spectroscopy.
SPR measurements were carried out on a Biacore T200 instrument (GE Healthcare). Biotinylated double-stranded IgκB DNA was immobilized to streptavidin-coated sensor chips, using standard procedures. The protein–DNA binding was carried out at 20 °C. The effect of pirin upon NF-κB binding the IgκB sequence was studied by flowing the protein over the sensor chip and comparing the levels of DNA binding in the absence and presence of the putative coregulator protein. The final concentration of p65 was 50 nM in the SPR injection buffer whereas pirin concentration was varied from 0 to 25 µM. After injecting protein samples for 420 s, buffer was injected from 420 to 1,220 s to rinse the weakly bound proteins from the chip surface to monitor the dissociation of the protein or protein complexes. Vitamin C was injected at 980 s in the reversibility testing experiments. The sensor chip surface was regenerated with 0.1% SDS and 3 mM EDTA between each measurement. The anaerobic SPR experiments were performed using O2-free, argon-saturated buffer.
QCM-D measurements were performed on an E4 instrument (Q-Sense) with biotin-functionalized sensors. Streptavidin was flowed over the biotinylated surface, followed by biotin-labeled IgκB-site DNA for immobilization. The equilibrium buffer was 20 mM Tris⋅HCl (pH 7.4) containing 50 mM NaCl. In each measurement, a total volume of 500 µL was continuously injected at a flow rate of 150 µL/min followed by buffer injection for an additional 300 s. The net change in frequency and dissipation was calculated by subtracting the values recorded at the sample injection time from those recorded at the end of each rinsing step. This eliminated the influence of nonspecific, loosely bound proteins on the DNA-immobilized surface. The surface was regenerated between each sample injection by flowing through 0.05% SDS followed by the equilibrium buffer until the baseline was restored. The concentration of p65 was maintained at 0.5 µM, whereas pirin concentration varied between 0, 0.5, 1.5, 2.5, and 5 µM for each run of the sample application.
Fluorescence spectroscopy was performed on a Cary Eclipse fluorescence spectrophotometer from Agilent Technologies. Purified p65 was titrated into the fluorescence cuvette containing FAM-labeled double-stranded DNA with IgκB sequence in the presence or absence of Fe(III)-pirin or pirin reconstituted with other metal ions in 20 mM Tris⋅HCl buffer (pH 7.4) and 50 mM NaCl.
The midpoint redox potential of pirin was determined by using a single-wall carbon nanotube-modified electrode in a spectroelectrochemical cell hosted in a N2-filled, O2-free plastic bag. Pirin (500 µM) in 50 mM Tris⋅HCl (pH 7.4) was used in this study. The formal potential was obtained from the recorded cyclic voltammetry curves.
X-band EPR spectra were obtained in perpendicular mode on a Bruker E200 spectrometer at 100-kHz modulation frequency, using a high-sensitivity resonator ER4119HS at 10 K, maintained by an ESR910 liquid helium cryostat and an Oxford ITC503S temperature controller. Pirin (160 µL per sample, 200–450 µM) was prepared in a quartz EPR tube with 20 mM Tris⋅HCl buffer (pH 7.4) containing 50 mM NaCl.
Crystallization and X-Ray Structure Determination.
Pirin was crystallized through optimization of the conditions previously established (19) by hanging-drop vapor diffusion in VDX plates (Hampton Research). Single crystals suitable for X-ray data collection were obtained from drops assembled with 1 μL protein solution layered with 1 μL reservoir solution containing 0.1 M MOPS buffer (pH 6.5), 15% PEG 20,000. Crystal growth took place in a vibration-free crystal growth refrigerator from Molecular Dimensions at 16 °C. The crystals were mounted in small loops of fine rayon fiber and flash-cooled directly in liquid nitrogen after being dipped into the cryoprotectant solution that contained 0.1 M MOPS (pH 6.5), 17% PEG 20,000 and 30% ethylene glycol. Initial screening was carried out using a Rigaku MicroMax 007 HF Cu-rotating anode X-ray generator, operated at 40 V and 30 mA, and using a Saturn 944 CCD area detector at Emory University (Atlanta). X-ray diffraction data of Fe(III)-pirin, putative Fe(II)-bound peroxo pirin, Mn(III)-pirin, and Co(II)-pirin were collected by the Rigaku X-ray facility. X-ray diffraction data of Mn(II)-pirin were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22-ID at the Advanced Photon Source (APS), Argonne National Laboratory, Argonne, IL. All data collection was performed at 100 K. The diffraction data were processed and analyzed as described in the SI Materials and Methods.
In Silico Docking of the Supercomplex.
The docking model of pirin with p65 and κB DNA was constructed by using the ZDOCK utility and server (http://zdock.umassmed.edu) (34). The PDB files of ferric pirin (PDB ID: 4GUL) and p65 (PDB ID: 1RAM) (26) were used in model building. The top-ranked result shown in Fig. 5 involves the R-shaped area of pirin, which has been shown in the structural study to differ in conformation when the metal oxidation state changes or metal substitution occurs.
Experimental Validation of the Docking Study.
The modeling results were further tested by site-directed mutagenesis experiments. The point mutants of pirin were generated by using the QuikChange II kit purchased from Qiagen (for details, see SI Materials and Methods). The sequences of the mutants were verified by DNA sequencing before expressing and purifying proteins. The procedures used for the expression, purification, metal reconstitution, and oxidation of the pirin variants were the same as described previously for the wild-type protein.
Accession Code.
Atomic coordinates and structure factors have been deposited with the Protein Data Bank under accession codes (resolution) 4GUL (1.80 Å), 4EWA (2.47 Å), 4EWE (1.56 Å), 4ERO (2.65 Å), 4EWD (2.15 Å), and 4HLT (1.70 Å).
Supplementary Material
Acknowledgments
We thank Michael Hebert and Howard Xu for assistance in pirin cloning, Jim Maher for providing us with a plasmid of p65, Jonathan Hosler for metal content analyses, W. David Wilson for helping us with SPR data acquisition and for providing us with a nonspecific DNA sequence, Binghe Wang for providing access to QCM-D instrumentation, Zhi-Ren Liu for providing the HeLa cells, Hiroshi Tachikawa for electrochemical experiments, and Xiaodong Chen for allowing access to the Saturn 944 X-ray facility through a fee-based contract. X-ray data were collected at Emory University and the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. We thank Bi-Cheng Wang and the SER-CAT staff as well as John Horton for assistance of X-ray data collections. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-Eng-38. We are grateful to John D. Lipscomb for helpful discussions. This work was supported in part by the Georgia Cancer Coalition Distinguished Cancer Scholar Program (A.L.) and National Science Foundation Grant MCB-0843537 (to A.L.). F.L. acknowledges a Georgia State University fellowship administrated by the Center for Diagnostics and Therapeutics.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. C.H. is a guest editor invited by the Editorial Board.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4GUL, 4EWA, 4EWE, 4ERO, 4EWD, and 4HLT).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1221743110/-/DCSupplemental.
References
- 1.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46(5):705–716. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 2.Sen R, Baltimore D. Inducibility of κ immunoglobulin enhancer-binding protein Nf-κB by a posttranslational mechanism. Cell. 1986;47(6):921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 3.Romashkova JA, Makarov SS. NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401(6748):86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 4.Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 5.Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. 2002;2(10):725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 6.Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature. 1998;391(6665):410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh S, May MJ, Kopp EB. NF-κB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh G, van Duyne G, Ghosh S, Sigler PB. Structure of NF-κB p50 homodimer bound to a κB site. Nature. 1995;373(6512):303–310. doi: 10.1038/373303a0. [DOI] [PubMed] [Google Scholar]
- 9.Pahl HL. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18(49):6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 10. Gilmore TD (2008) NF-κB target genes. Available at http://www.bu.edu/nf-kb/gene-resources/target-genes/
- 11. Gosselin K (2004) NF-κB target genes. Available at http://bioinfo.lifl.fr/NF-KB/
- 12.Jeong KW, et al. Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat Struct Mol Biol. 2011;18(12):1358–1365. doi: 10.1038/nsmb.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merson TD, et al. The transcriptional coactivator Querkopf controls adult neurogenesis. J Neurosci. 2006;26(44):11359–11370. doi: 10.1523/JNEUROSCI.2247-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Näär AM, Lemon BD, Tjian R. Transcriptional coactivator complexes. Annu Rev Biochem. 2001;70:475–501. doi: 10.1146/annurev.biochem.70.1.475. [DOI] [PubMed] [Google Scholar]
- 15.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12(8):695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 16.Chen FE, Ghosh G. Regulation of DNA binding by Rel/NF-κB transcription factors: Structural views. Oncogene. 1999;18(49):6845–6852. doi: 10.1038/sj.onc.1203224. [DOI] [PubMed] [Google Scholar]
- 17.Wendler WMF, Kremmer E, Förster R, Winnacker E-L. Identification of pirin, a novel highly conserved nuclear protein. J Biol Chem. 1997;272(13):8482–8489. doi: 10.1074/jbc.272.13.8482. [DOI] [PubMed] [Google Scholar]
- 18.Dechend R, et al. The Bcl-3 oncoprotein acts as a bridging factor between NF-κB/Rel and nuclear co-regulators. Oncogene. 1999;18(22):3316–3323. doi: 10.1038/sj.onc.1202717. [DOI] [PubMed] [Google Scholar]
- 19.Pang H, et al. Crystal structure of human pirin: An iron-binding nuclear protein and transcription cofactor. J Biol Chem. 2004;279(2):1491–1498. doi: 10.1074/jbc.M310022200. [DOI] [PubMed] [Google Scholar]
- 20.Gurmu D, et al. The crystal structure of the protein YhaK from Escherichia coli reveals a new subclass of redox sensitive enterobacterial bicupins. Proteins. 2009;74(1):18–31. doi: 10.1002/prot.22128. [DOI] [PubMed] [Google Scholar]
- 21.Adams M, Jia Z. Structural and biochemical analysis reveal pirins to possess quercetinase activity. J Biol Chem. 2005;280(31):28675–28682. doi: 10.1074/jbc.M501034200. [DOI] [PubMed] [Google Scholar]
- 22.Licciulli S, et al. Pirin inhibits cellular senescence in melanocytic cells. Am J Pathol. 2011;178(5):2397–2406. doi: 10.1016/j.ajpath.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyazaki I, Simizu S, Okumura H, Takagi S, Osada H. A small-molecule inhibitor shows that pirin regulates migration of melanoma cells. Nat Chem Biol. 2010;6(9):667–673. doi: 10.1038/nchembio.423. [DOI] [PubMed] [Google Scholar]
- 24.Brzóska K, Stępkowski TM, Kruszewski M. Putative proto-oncogene Pir expression is significantly up-regulated in the spleen and kidney of cytosolic superoxide dismutase-deficient mice. Redox Rep. 2011;16(3):129–133. doi: 10.1179/1351000211Y.0000000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelbman BD, Heguy A, O’Connor TP, Zabner J, Crystal RG. Upregulation of pirin expression by chronic cigarette smoking is associated with bronchial epithelial cell apoptosis. Respir Res. 2007;8:10. doi: 10.1186/1465-9921-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YQ, Ghosh S, Ghosh G. A novel DNA recognition mode by the NF-κB p65 homodimer. Nat Struct Biol. 1998;5(1):67–73. doi: 10.1038/nsb0198-67. [DOI] [PubMed] [Google Scholar]
- 27.Phelps CB, Sengchanthalangsy LL, Malek S, Ghosh G. Mechanism of κB DNA binding by Rel/NF-κB dimers. J Biol Chem. 2000;275(32):24392–24399. doi: 10.1074/jbc.M003784200. [DOI] [PubMed] [Google Scholar]
- 28.Peh WYX, Reimhult E, Teh HF, Thomsen JS, Su X. Understanding ligand binding effects on the conformation of estrogen receptor α-DNA complexes: A combinational quartz crystal microbalance with dissipation and surface plasmon resonance study. Biophys J. 2007;92(12):4415–4423. doi: 10.1529/biophysj.106.099382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schreck R, Zorbas H, Winnacker E-L, Baeuerle PA. The NF-κB transcription factor induces DNA bending which is modulated by its 65-kD subunit. Nucleic Acids Res. 1990;18(22):6497–6502. doi: 10.1093/nar/18.22.6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horton NC, Perona JJ. Crystallographic snapshots along a protein-induced DNA-bending pathway. Proc Natl Acad Sci USA. 2000;97(11):5729–5734. doi: 10.1073/pnas.090370797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cordas CM, Duarte AG, Moura JJ, Moura I. Electrochemical behaviour of bacterial nitric oxide reductase-evidence of low redox potential non-heme FeB gives new perspectives on the catalytic mechanism. Biochim Biophys Acta. 2013;1827(3):233–238. doi: 10.1016/j.bbabio.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 32.Vance CK, Miller AF. A simple proposal that can explain the inactivity of metal-substituted superoxide dismutases. J Am Chem Soc. 1998;120(3):461–467. [Google Scholar]
- 33.Fasman GD. CRC Handbook of Biochemistry and Molecular Biology: Physical Chemical Data. 3rd Ed. Cleveland: CRC; 1976. [Google Scholar]
- 34.Chen R, Li L, Weng Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins. 2003;52(1):80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 35.Hübner RH, et al. Coordinate control of expression of Nrf2-modulated genes in the human small airway epithelium is highly responsive to cigarette smoking. Mol Med. 2009;15(7–8):203–219. doi: 10.2119/molmed.2008.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mercer BA, Lemaître V, Powell CA, D’Armiento J. The epithelial cell in lung health and emphysema pathogenesis. Curr Respir Med Rev. 2006;2(2):101–142. doi: 10.2174/157339806776843085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orzaez D, de Jong AJ, Woltering EJ. A tomato homologue of the human protein PIRIN is induced during programmed cell death. Plant Mol Biol. 2001;46(4):459–468. doi: 10.1023/a:1010618515051. [DOI] [PubMed] [Google Scholar]
- 38.Licciulli S, Cambiaghi V, Scafetta G, Gruszka AM, Alcalay M. Pirin downregulation is a feature of AML and leads to impairment of terminal myeloid differentiation. Leukemia. 2010;24(2):429–437. doi: 10.1038/leu.2009.247. [DOI] [PubMed] [Google Scholar]
- 39.Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461(7263):537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee J-W, Helmann JD. The PerR transcription factor senses H2O2 by metal-catalysed histidine oxidation. Nature. 2006;440(7082):363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- 41.Traoré DAK, et al. Structural and functional characterization of 2-oxo-histidine in oxidized PerR protein. Nat Chem Biol. 2009;5(1):53–59. doi: 10.1038/nchembio.133. [DOI] [PubMed] [Google Scholar]
- 42.Sjöberg BM, Reichard P, Gräslund A, Ehrenberg A. The tyrosine free radical in ribonucleotide reductase from Escherichia coli. J Biol Chem. 1978;253(19):6863–6865. [PubMed] [Google Scholar]
- 43.Wurster SE, Maher LJ., 3rd Selection and characterization of anti-NF-κB p65 RNA aptamers. RNA. 2008;14(6):1037–1047. doi: 10.1261/rna.878908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen YQ, Sengchanthalangsy LL, Hackett A, Ghosh G. NF-κB p65 (RelA) homodimer uses distinct mechanisms to recognize DNA targets. Structure. 2000;8(4):419–428. doi: 10.1016/s0969-2126(00)00123-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.