

Abstract
The generation of toxic oligomers during the aggregation of the amyloid-β (Aβ) peptide Aβ42 into amyloid fibrils and plaques has emerged as a central feature of the onset and progression of Alzheimer’s disease, but the molecular pathways that control pathological aggregation have proved challenging to identify. Here, we use a combination of kinetic studies, selective radiolabeling experiments, and cell viability assays to detect directly the rates of formation of both fibrils and oligomers and the resulting cytotoxic effects. Our results show that once a small but critical concentration of amyloid fibrils has accumulated, the toxic oligomeric species are predominantly formed from monomeric peptide molecules through a fibril-catalyzed secondary nucleation reaction, rather than through a classical mechanism of homogeneous primary nucleation. This catalytic mechanism couples together the growth of insoluble amyloid fibrils and the generation of diffusible oligomeric aggregates that are implicated as neurotoxic agents in Alzheimer’s disease. These results reveal that the aggregation of Aβ42 is promoted by a positive feedback loop that originates from the interactions between the monomeric and fibrillar forms of this peptide. Our findings bring together the main molecular species implicated in the Aβ aggregation cascade and suggest that perturbation of the secondary nucleation pathway identified in this study could be an effective strategy to control the proliferation of neurotoxic Aβ42 oligomers.
Keywords: chemical kinetics, molecular mechanisms, protein misfolding, neurodegeneration
The 42-residue amyloid-β (Aβ) peptide, Aβ42, has been identified as a central constituent in the molecular pathways that underlie Alzheimer’s disease (AD) (1–6) through the generation of low molecular weight oligomers from this normally soluble peptide (2, 7–9). The highly complex self-assembly behavior exhibited by this peptide has, however, prevented its mechanism of aggregation from being defined in terms of molecular events in the manner that has been possible for other biomolecular assemblies, such as actin (10) and some prions (11, 12). Particular attention has been devoted to the study of oligomers, which are small multimers that do not yet possess the ability to elongate at the same rate as fibrils and are commonly associated with neuronal death both in vivo and in vitro (2, 7–9). The relationship between low molecular weight toxic oligomers and the mature fibrils has remained elusive, with some studies suggesting that oligomers are generated predominantly as on-pathway intermediates in fibril formation and others indicating that these different species originate mainly from independent pathways (13). Here, we connect the characteristic macroscopic features of Aβ42 fibril formation to their microscopic determinants through the analysis of experimental kinetic data in terms of microscopic rate laws and use selective radiolabeling, size-exclusion chromatography, and cell viability studies to define the origin of the toxic oligomers and their relationship with fibrillar aggregates. We uncover a close connection between the monomeric peptide, the toxic oligomeric species, and the mature fibrils by showing that after a small but critical concentration of amyloid fibrils has formed, the toxic oligomers are predominantly generated from the monomeric peptide in a secondary nucleation reaction that is catalyzed very strongly by the larger fibrillar species.
To elucidate the dominant mechanisms of aggregate proliferation, we focus first on the fibril population detected in thioflavin T (ThT) fluorescence experiments (Figs. 1–3). Because the nucleation pathways proceed through oligomeric intermediates, the information that we obtain from this kinetic analysis can also be used to predict the mechanisms for on-pathway oligomer formation. In the second part of this paper, we verify these predictions explicitly through isolating oligomeric fractions by means of chromatography and show that the overall generation of oligomers is an integral part of the Aβ secondary nucleation mechanism (Figs. 4 and 5).
Fig. 1.
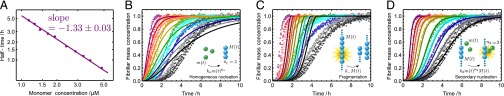
Experimental kinetics of Aβ42 aggregation under quiescent conditions for 10 initial monomer concentrations. (A) Power-law scaling of the time to half-completion with the initial monomer concentration. The slope gives the scaling exponent γ discussed in the text. (B–D) Global fits to the normalized experimental data, using the analytical solutions for systems where (B) the dominant nucleation mechanism is primary nucleation, and there are no secondary pathways (10, 17); (C) a (dominant) fragmentation process is active in addition to primary nucleation (16); and (D) secondary nucleation, in addition to primary nucleation, creates new aggregates, Eq. 1 (see SI Text for further discussion of these fits). The rate constants are (B) 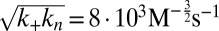 , with
, with 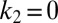 ,
, 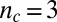 ; (C)
; (C) 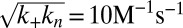 ,
, 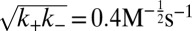 , with
, with 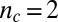 ; and (D)
; and (D) 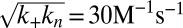 ,
, 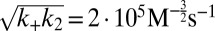 , with
, with 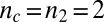 .
.
Fig. 3.

Shear alters the symmetry of the reaction profile. (A) Comparison of the shape of the kinetic profiles under quiescent and high-shear conditions (33), corresponding to concentrations marked B with a vertical dotted line. The solid lines are the theoretical rate laws with the rate constants identified in Fig. 2. (B) Power-law relationship for the monomer dependence at varying shear rates from Fig. 2 A–E (Lower). The weakening of the monomer dependence, given by the magnitude of the slope, occurs as fragmentation is gradually introduced as a molecular mechanism. Predicted deviations from the power law at high concentration are shown as open circles (SI Text).
Fig. 4.

Direct measurement of oligomer populations, using radioactive Aβ42 peptides. (A) Samples of monomer (light blue bar) or monomer mixed with 1% preformed fibrils (dark blue bar and right bar) with selective radiolabeling of monomer or fibrils, as indicated in red, were incubated followed by size-exclusion chromatography and liquid scintillation counting. The counts for the oligomer fractions are shown below the respective samples. The monomer counts are shown in Fig. S7. (B) Probing the chromatography fractions with the 6E10 antibody confirms the dramatically enhanced production of small oligomers in the presence of fibrils. Time 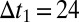 min. (C) Reduction in cell viability (MTS) for reactions without (light blue bars) and with (dark blue bars) a small concentration of added fibrils under the same conditions as in A and after filtration through a 200-nm filter. Values are averages over nine measurements at
min. (C) Reduction in cell viability (MTS) for reactions without (light blue bars) and with (dark blue bars) a small concentration of added fibrils under the same conditions as in A and after filtration through a 200-nm filter. Values are averages over nine measurements at 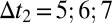 min. Gray bars are the initial (monomer) and end (fibril) reaction time points. (D) Normalized kinetic time courses without (light blue) and with (dark blue) added preformed fibrils that correspond to those in A–C. The rapid increase in the slope of the assay with preformed fibrils (dark blue) after ca. 10 min, before the matched reaction without preformed fibrils (light blue) has generated significant aggregate mass, indicates rapid creation of new aggregates through secondary nucleation (30) (SI Text). The concentration of monomeric Aβ42 was 4 μM and the mass concentration of added fibrils was 40 nM.
min. Gray bars are the initial (monomer) and end (fibril) reaction time points. (D) Normalized kinetic time courses without (light blue) and with (dark blue) added preformed fibrils that correspond to those in A–C. The rapid increase in the slope of the assay with preformed fibrils (dark blue) after ca. 10 min, before the matched reaction without preformed fibrils (light blue) has generated significant aggregate mass, indicates rapid creation of new aggregates through secondary nucleation (30) (SI Text). The concentration of monomeric Aβ42 was 4 μM and the mass concentration of added fibrils was 40 nM.
Fig. 5.
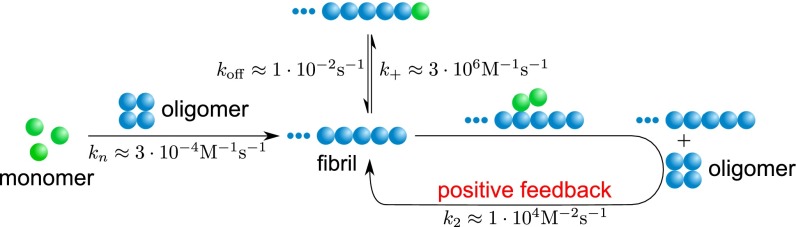
Schematic showing the overall reaction pathway and the corresponding rate constants identified in this paper. The approximate rates of the elongation-related processes have been identified in previous work (33, 35, 36).
Results and Discussion
Microscopic Mechanisms.
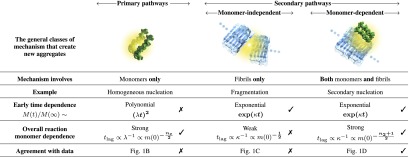
The general method underlying the kinetic analysis builds on earlier work (10, 14–17, 18, 19, 21, 22) and considers all of the possible sources of new aggregates, which consist of two or more monomers, from the species present in the system, as shown in Table 1, from both primary (10, 19, 23–25) and secondary (11, 12, 14, 26–28) pathways. Primary pathways, such as homogeneous nucleation (10, 19), generate new aggregates at a rate dependent on the concentration of monomers alone and independent of the concentration of existing fibrils. Secondary pathways are the complementary class of mechanisms that generate new aggregates at a rate dependent on the concentration of existing fibrils. The latter class can be subdivided into monomer-independent processes, such as fragmentation (11, 12, 18, 26), with a rate depending only upon the concentration of existing fibrils, and monomer-dependent processes, such as secondary nucleation (14, 22, 27, 28), where the surfaces of existing fibrils catalyze the nucleation of new aggregates from the monomeric state, with a rate dependent on both the concentration of monomers and that of existing fibrils. Together, these three classes of mechanism, shown in Table 1, form the basis of a general description of protein aggregation (15, 17), because they account for the generation of new aggregates from mechanisms that involve monomers alone, existing aggregates alone, or both monomers and existing aggregates. These pathways initially populate oligomeric intermediates (29), which lead to fibrillar forms that elongate at a rate that is independent of their length (10, 15, 17) and represent the bulk of the aggregate mass.
Table 1.
 |
Linear Theory.
These three classes of mechanism, summarized in Table 1, exhibit qualitatively different features, both as a function of time and as a function of the initial monomer concentration (14, 15, 18, 30). These differences are readily observed by considering the behavior of the system for early times before appreciable amounts of monomer have been sequestered into aggregates (15) when the rate equations can be linearized (15, 30). For cases where the mechanism that creates aggregates involves the preexisting fibrils, such as fragmentation or secondary nucleation, positive feedback for the increase of the fibril mass concentration, 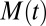 , results in the evolution of the form (14, 18)
, results in the evolution of the form (14, 18) 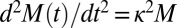 , and hence exponential growth
, and hence exponential growth 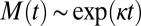 is observed, leading to a strong lag phase (14, 15, 17). The duration of the lag phase is commonly described by a lag time
is observed, leading to a strong lag phase (14, 15, 17). The duration of the lag phase is commonly described by a lag time 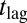 , defined as the time at which the aggregate concentration reaches a small fixed percentage of the total peptide concentration. Here,
, defined as the time at which the aggregate concentration reaches a small fixed percentage of the total peptide concentration. Here, 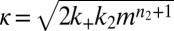 is the combined parameter that controls proliferation through secondary pathways,
is the combined parameter that controls proliferation through secondary pathways, 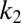 is the rate constant for the secondary process, m is the monomer concentration,
is the rate constant for the secondary process, m is the monomer concentration, 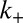 is the fibril elongation rate constant, and
is the fibril elongation rate constant, and 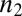 is the reaction order of the secondary pathway with respect to the monomer (15, 30). By contrast, for cases where nucleation is independent of the fibril concentration, such as for classical homogeneous nucleation (10), no feedback is generated,
is the reaction order of the secondary pathway with respect to the monomer (15, 30). By contrast, for cases where nucleation is independent of the fibril concentration, such as for classical homogeneous nucleation (10), no feedback is generated, 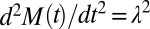 , and hence slow polynomial growth results,
, and hence slow polynomial growth results, 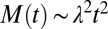 , with a weak lag phase (10, 15, 30), where
, with a weak lag phase (10, 15, 30), where 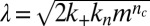 is the combined parameter controlling proliferation through primary nucleation,
is the combined parameter controlling proliferation through primary nucleation, 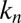 denotes the primary nucleation rate constant, and
denotes the primary nucleation rate constant, and 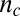 is the reaction order of the primary process (10, 15, 30). The reaction orders
is the reaction order of the primary process (10, 15, 30). The reaction orders 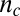 and
and 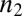 need not correspond to structural sizes of nuclei (30). The distinction between polynomial growth for primary processes and exponential-type growth for secondary processes is in general maintained for more complicated pathways, such as cascades through multiple intermediates (15, 30, 31).
need not correspond to structural sizes of nuclei (30). The distinction between polynomial growth for primary processes and exponential-type growth for secondary processes is in general maintained for more complicated pathways, such as cascades through multiple intermediates (15, 30, 31).
A second distinction between the basic mechanisms in Table 1 is whether or not the monomer concentration affects the process. If it does, a high concentration dependence of the lag time is possible, otherwise only a weak dependence emerges because a change in the monomer concentration has no direct effect on the nucleation pathway. It is convenient to describe the monomer dependence of the overall assembly reaction, including elongation-related processes, with a power-law relationship,  , which relates the lag time
, which relates the lag time 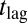 for the reaction to the initial peptide concentration
for the reaction to the initial peptide concentration 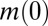 (15, 30). The exponent γ in the power law for the lag time (14, 15) is to a good approximation defined by the monomer dependence of the combined parameter λ in the case where primary pathways are dominant and from κ in the case of secondary pathways. In this manner, the exponent is given from λ as
(15, 30). The exponent γ in the power law for the lag time (14, 15) is to a good approximation defined by the monomer dependence of the combined parameter λ in the case where primary pathways are dominant and from κ in the case of secondary pathways. In this manner, the exponent is given from λ as 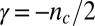 for processes where a classical homogeneous nucleation step is the major source of aggregates (10) and from κ as
for processes where a classical homogeneous nucleation step is the major source of aggregates (10) and from κ as 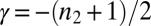 for phenomena where secondary nucleation processes dominate (14, 17). A strong monomer dependence,
for phenomena where secondary nucleation processes dominate (14, 17). A strong monomer dependence, 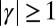 , can, therefore, always be captured by either primary or secondary nucleation through an appropriate value of
, can, therefore, always be captured by either primary or secondary nucleation through an appropriate value of 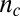 or
or 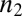 . By contrast, monomer-independent secondary processes, such as fragmentation, are associated with a weaker overall monomer scaling (16, 18) corresponding to a monomer reaction order
. By contrast, monomer-independent secondary processes, such as fragmentation, are associated with a weaker overall monomer scaling (16, 18) corresponding to a monomer reaction order 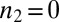 ,
, 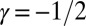 , with this remaining weak monomer dependence originating only from the fibril elongation step in the overall self-assembly pathway.
, with this remaining weak monomer dependence originating only from the fibril elongation step in the overall self-assembly pathway.
Nonlinear Theory.
Due to the complexity inherent in amyloid aggregation, it is challenging to acquire data in the regime where the linear solutions are fully valid, because in this early region the signals from bulk assays are low. To extend the applicability of kinetic analysis (14, 15) to amyloid systems, it is therefore desirable to consider the full reaction time course to maximize the constraints on the molecular mechanisms determined from the experimental data (22, 30). At later times in the reaction, as the monomer is consumed, the equations describing the overall assembly process become highly nonlinear and are challenging to integrate (15–17, 18, 22, 30). We have, however, recently derived self-consistent rate laws for the assembly process that are valid for the entire time course of the reaction (22). The full rate law reveals that the same two principal parameters κ and λ, which were identified in the early time behavior (Table 1), define much of the macroscopic behavior in the nonlinear regime also, although the rate laws themselves have a different form, Eq. 1. An analysis of the full time course, therefore, introduces additional constraints without introducing any additional freedom, resulting in a stringent test of the theory and robust mechanistic conclusions.
Secondary Nucleation Controls Aβ42 Fibril Formation.
To obtain a clear picture of the molecular mechanisms that give rise to Aβ42 fibrils, it is essential to generate highly reproducible experimental data reporting on this process. We have been able to collect these types of data at pH values and concentrations of the peptide that relate to physiological conditions (32) by controlling carefully the inertness of surfaces within which solutions of the peptide are contained and by purifying the recombinant monomeric peptide, using repeated applications of size-exclusion chromatography to ensure well-defined initial conditions before initiating kinetic assays (33) (Fig. S1). The kinetics of fibril formation are followed using ThT fluorescence measurements (33), which we have independently verified to be linearly related to the total mass of Aβ42 fibrils under our carefully controlled conditions (Fig. S2).
The value for the scaling exponent, which describes how the lag time or half-time of the reaction scales with the initial concentration of monomer, measured in Fig. 1A (and for preseeded growth in Fig. S3) for Aβ42 under quiescent conditions, is 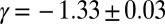 . The lag time and the time to half completion follow the same overall monomer scaling dependence (17, 22) (Table 1), and in this paper we use the half-time because it is available accurately from experimental data. It is interesting to note from Table 1 that this observation of the monomer dependence excludes aggregate fragmentation, believed to be vital, for example, in the propagation of prions (11, 12), as the dominant mechanism driving Aβ42 aggregation, because this process would result in an exponent of
. The lag time and the time to half completion follow the same overall monomer scaling dependence (17, 22) (Table 1), and in this paper we use the half-time because it is available accurately from experimental data. It is interesting to note from Table 1 that this observation of the monomer dependence excludes aggregate fragmentation, believed to be vital, for example, in the propagation of prions (11, 12), as the dominant mechanism driving Aβ42 aggregation, because this process would result in an exponent of 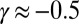 . The value of the scaling exponent is, however, consistent with a dominant secondary nucleation pathway characterized by a monomer dependence of
. The value of the scaling exponent is, however, consistent with a dominant secondary nucleation pathway characterized by a monomer dependence of 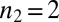 and a contribution from primary nucleation with a reaction order of
and a contribution from primary nucleation with a reaction order of 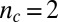 , the effect of which is to lower the scaling exponent (22) from the value
, the effect of which is to lower the scaling exponent (22) from the value 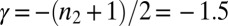 toward the value
toward the value 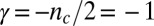 given for proliferation through primary nucleation only. We can now test this conclusion directly by checking explicitly the degree to which the experimental data determined for the full time course of the reaction are matched by the predictions from the rate law, Eq. 1, when all 10 initial peptide concentrations are used and the only two free parameters,
given for proliferation through primary nucleation only. We can now test this conclusion directly by checking explicitly the degree to which the experimental data determined for the full time course of the reaction are matched by the predictions from the rate law, Eq. 1, when all 10 initial peptide concentrations are used and the only two free parameters, 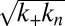 and
and 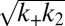 that enter κ and λ, are fixed globally to the same values for all 10 measured peptide concentrations to provide the best fit for the entire dataset consisting of 10 reaction profiles.
that enter κ and λ, are fixed globally to the same values for all 10 measured peptide concentrations to provide the best fit for the entire dataset consisting of 10 reaction profiles.
The results shown in Fig. 1D demonstrate the excellent agreement between the theoretical predictions from Eq. 1 and the experimental data over the full reaction time course under a wide range of concentrations. Moreover, the best fit when the secondary nucleation parameter κ is fixed to zero (Fig. 1B) shows that, whereas a description that lacks secondary pathways is able to account approximately for the scaling of the half-time of the reaction with the monomer concentration, it is not able to describe even qualitatively the full time courses observed in the experiments. In particular, the early-stage growth observed in the experimental data is much stronger than the polynomial form associated with primary nucleation and is instead described by the exponential growth associated with secondary pathways (Table 1). A fit to the case where most new aggregates are generated through fragmentation (Fig. 1C) is conversely able to account for the exponential growth at early times, but it is not able to match the more than linear monomer dependence of the reaction timescale (Table 1). By contrast, the fit including fibril-catalyzed secondary nucleation (Fig. 1D), where the surfaces of fibrils catalyze the nucleation of new aggregates from monomeric peptide (14, 27, 28), describes the entire set of time courses, including the characteristic exponential shape at early times and the monomer scaling, using only two global parameters that are fixed to the same value across all datasets. This result is particularly striking because it shows that the production of new Aβ42 fibrils does not occur predominantly through the classical mechanism of primary nucleation, which initially involves the coalescence of monomeric peptides into oligomers independently of existing fibrils, but occurs by secondary processes that are critically dependent on the latter species. It is interesting to note that the secondary nucleation mechanism identified here for the aggregation of the Aβ42 peptide is formally analogous to that originally identified for the polymerization of sickle hemoglobin (14). In the present case, however, electron microscopy indicates that fibrils are not generally attached to one another at the locations of secondary nucleation events (Fig. S4), implying that secondary nucleated aggregates detach from the fibril surface. Recent atomic force microscopy studies have captured the formation of such nuclei (34).
Rational Alteration of the Aggregation Pathway.
A factor that has contributed greatly to previous difficulties in developing a clear picture of Aβ42 aggregation is the high sensitivity of the kinetics of aggregation to even small changes in the reaction conditions. Here, we can use such differences in aggregation behavior in a systematic manner to reveal the underlying microscopic mechanisms. Thus, having shown that Aβ42 aggregation under quiescent conditions is controlled by a fibril-catalyzed secondary nucleation process, we sought to modify the dominant factors determining the aggregation pathway by introducing shear forces through shaking and to identify the signals in the kinetic data that report on this change.
The rate equations (16, 22) lead to the intriguing prediction for Aβ42 that, if such shear gradually introduces fibril fragmentation as a molecular mechanism (Fig. 2), the scaling exponent (16, 22, 30)—relating the lag time or half-time to the monomer concentration—will change monotonically from the quiescent value of 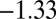 and approach the theoretical limit of
and approach the theoretical limit of 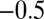 (Table 1) associated with fragmentation (16, 22, 30) at very high agitation rates (Fig. 2 A–E, Lower). Remarkably, the overall effect of fragmentation is incorporated in the rate equations through the introduction of a single additional parameter relative to the quiescent case (SI Text),
(Table 1) associated with fragmentation (16, 22, 30) at very high agitation rates (Fig. 2 A–E, Lower). Remarkably, the overall effect of fragmentation is incorporated in the rate equations through the introduction of a single additional parameter relative to the quiescent case (SI Text), 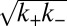 , by use of which we are able to fit very closely the kinetic traces at each agitation rate (Fig. 2 B–E, Upper). Furthermore, we verified using electron microscopy and seeding experiments that the morphology of the fibrils remained unchanged (Figs. S4 and S5). The global nature of the fit is equivalent to the ability to predict quantitatively the behavior of the system with changes in experimental conditions; such a situation is likely to be found only when the model captures correctly the molecular events taking place in the reaction.
, by use of which we are able to fit very closely the kinetic traces at each agitation rate (Fig. 2 B–E, Upper). Furthermore, we verified using electron microscopy and seeding experiments that the morphology of the fibrils remained unchanged (Figs. S4 and S5). The global nature of the fit is equivalent to the ability to predict quantitatively the behavior of the system with changes in experimental conditions; such a situation is likely to be found only when the model captures correctly the molecular events taking place in the reaction.
Fig. 2.
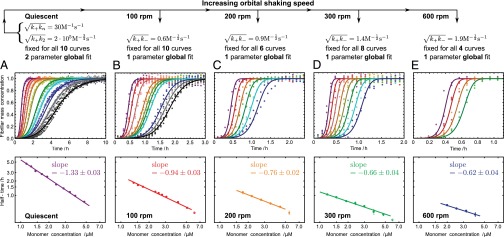
Experimental kinetics for Aβ42 aggregation under varying levels of shear generated by agitating the sample at different speeds (Upper) and the power-law relationships observed between the half-time and the initial monomer concentration (Lower). The slopes (Lower) give the scaling exponent γ discussed in the text. The two rate parameters determined from the global fit to the data under quiescent conditions (A, Upper) are held fixed and all of the normalized experimental profiles (B–E, Upper) are fitted with a single additional parameter for each shear rate. Note the different timescales (Upper). (Upper and Lower) The scale on the ordinate is the same. Predicted deviations from the power law at high concentration are shown as open circles (SI Text).
It is interesting to note from Fig. 2 that increasing levels of shear change not only the power law for the half-time, but also the characteristic form of the kinetic profiles at the late stages of the reaction (Fig. 3A) (30). A change occurs because fragmentation, unlike secondary nucleation, is not directly affected by the depletion of the monomeric peptide toward the end of the reaction. Although both fragmentation and secondary nucleation exhibit exponential growth, 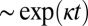 , in the early stages of the reaction (Table 1), an expansion of Eq. 1 for late times (22) predicts a sharper, double-exponential approach,
, in the early stages of the reaction (Table 1), an expansion of Eq. 1 for late times (22) predicts a sharper, double-exponential approach, 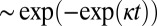 , to the plateau in the late stages of the reaction at high shear driven by fragmentation, in contrast to a less sharp,
, to the plateau in the late stages of the reaction at high shear driven by fragmentation, in contrast to a less sharp, 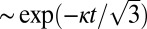 , exponential approach for the quiescent reaction driven by monomer-dependent secondary nucleation. In agreement with this prediction, a comparison of two traces at the same initial monomer concentration, under quiescent and high-shear conditions, is shown in Fig. 3A and reveals the predicted intricate transformation from an asymmetric curve under quiescent conditions that changes less rapidly toward the end of the reaction than at the beginning to a curve under high-shear conditions that has a sharper approach to the plateau and possesses the opposite asymmetry (30).
, exponential approach for the quiescent reaction driven by monomer-dependent secondary nucleation. In agreement with this prediction, a comparison of two traces at the same initial monomer concentration, under quiescent and high-shear conditions, is shown in Fig. 3A and reveals the predicted intricate transformation from an asymmetric curve under quiescent conditions that changes less rapidly toward the end of the reaction than at the beginning to a curve under high-shear conditions that has a sharper approach to the plateau and possesses the opposite asymmetry (30).
Confirming the Source of Oligomer Populations with Radioactive Peptides.
The analysis of the kinetic data indicates that a major and continuous source of new fibrils under quiescent conditions is a secondary nucleation mechanism that involves both the monomeric peptide and mature amyloid fibrils. Because a large number of peptides are required to form ordered fibrillar forms that are detected in ThT measurements, aggregates generated through the secondary pathway must initially be in prefibrillar, oligomeric states that can escape detection by this method (24, 35). To observe directly these ThT-invisible oligomer populations, which can ultimately convert to fibrils, and pinpoint their molecular origin, we studied a pair of samples with the same concentration of 35S-radiolabeled peptide, but one containing in addition to the soluble radioactive peptide a small concentration of unlabeled preformed fibrils. We measured the concentration of oligomers in both aggregating samples by quantifying through liquid scintillation assays the radioactivity in the oligomer fractions obtained from size-exclusion chromatography. This highly sensitive method of detecting oligomers has the advantage of not requiring any chemical labels and, therefore, leaves all of the chemical characteristics of the peptide intact.
The data in Fig. 4A show that the rate of generation of oligomers is dramatically enhanced in the solution that contains preformed fibrils even though the initial concentration of soluble peptide is kept constant. Crucially, because the added fibrils in these experiments are unlabeled, these results establish that oligomers are formed from the monomeric peptide, but in a reaction that is catalyzed very strongly by the presence of fibrils (Fig. S6). We also carried out the complementary experiments where unlabeled monomers were incubated with radiolabeled fibrils; the data in Fig. 4A show that no radioactivity is detectable in the oligomer fraction, confirming that the oligomers do not originate from the preformed fibrils themselves (e.g., through fragmentation or dissociation), but rather are formed from monomers through secondary nucleation. We verified these results using immunochemistry (Fig. 4B), by probing the amount of Aβ42 present in the oligomer fractions obtained from size-exclusion chomatography, confirming the formation of oligomers through secondary nucleation in a fibril-dependent manner.
The combination of the kinetic experiments and the detailed analysis of the chromatography fractions reveals that low molecular weight oligomers are formed in a pathway that involves both the monomer and fibrils. A key question, however, is whether the toxicity known to be associated with Aβ42 aggregation can originate from this same pathway. To address this issue, we measured the reduction in viability (Fig. 4C) and the increase in cytotoxicity (Fig. S8) of SH-SY5Y human neuroblastoma cells when exposed to oligomers formed as a result of secondary nucleation. We studied two solutions with an identical monomer concentration and, therefore, an identical population of oligomers generated by primary nucleation; marked differences in the resulting toxicity are evident, however, when a small concentration of preformed fibrils was added to one of these solutions to trigger the production of secondary oligomers as shown above. As the fibrils themselves are observed not to give rise to a high level of toxicity, these observations identify specifically that the major source of cytotoxic oligomers results from a process that involves both the monomeric peptide and the fibrils, i.e., secondary nucleation.
Significance and Conclusions
These results establish a general picture for the self-assembly of Aβ42 that brings together all of the species in the aggregation cascade (Fig. 5). Initially, in the absence of fibrils, all oligomers have to be generated through primary pathways because secondary nucleation requires the presence of fibrils. Once a critical concentration of amyloid fibrils has formed, however, secondary nucleation will overtake primary nucleation as the major source of new oligomers and further proliferation becomes exponential in nature (14, 16, 22) due to positive feedback (Fig. 5). The identification of secondary nucleation underlines the importance of elucidating the detailed structures of amyloid fibrils and their surfaces, information that will motivate molecular simulations to determine the origins of their surface-catalytic activity. The critical concentration of fibrils, above which secondary nucleation becomes the dominant mechanism generating new aggregates, is given from the ratio of the primary to secondary nucleation rate constants, 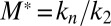 ; the parameters obtained in Fig. 1D define this concentration to be of the order of 10 nM. A survey of literature values (Tables S1 and S2) shows that the aggregate loads in the brains of patients suffering from AD are much greater than this critical concentration, and hence the results suggest that secondary nucleation is likely to be active under these conditions. It is therefore interesting to speculate that the secondary nucleation process identified in this in vitro study as the origin of the toxicity of Aβ42 aggregation could also play a major role in vivo, even accounting for the fact that differences in the morphological character and accessible surface area of the amyloid fibrils may cause variations in the rate of oligomer formation through secondary nucleation for different plaque loads.
; the parameters obtained in Fig. 1D define this concentration to be of the order of 10 nM. A survey of literature values (Tables S1 and S2) shows that the aggregate loads in the brains of patients suffering from AD are much greater than this critical concentration, and hence the results suggest that secondary nucleation is likely to be active under these conditions. It is therefore interesting to speculate that the secondary nucleation process identified in this in vitro study as the origin of the toxicity of Aβ42 aggregation could also play a major role in vivo, even accounting for the fact that differences in the morphological character and accessible surface area of the amyloid fibrils may cause variations in the rate of oligomer formation through secondary nucleation for different plaque loads.
In agreement with this idea, clear signatures of secondary nucleation are apparent in studies of living systems, as a halo of oligomeric Aβ42 aggregates (38) is found to emanate from amyloid plaques; close to plaques, the primary nucleation rate is unaffected, whereas the generation of oligomers through the secondary nucleation pathway is by definition very significantly enhanced. Furthermore, in the vicinity of plaques, dendritic spines have been found to be disrupted in a manner that depends on their distance from the plaques (39), an observation that suggests that the latter structures are not toxic by themselves in vivo but instead facilitate the generation of toxic oligomers by surface catalysis. The molecular picture that emerges from the present study, therefore, provides a mechanism by which the accumulation of amyloid fibrils is coupled to the generation of low molecular weight diffusive aggregates from monomeric peptide, thereby connecting together all of the main components in the Aβ cascade. This conclusion suggests that an important approach for suppressing the production of neurotoxic Aβ42 oligomers could be to focus on altering the secondary, rather than (or in addition to) the primary, nucleation pathway. Indeed, once the critical concentration of fibrils is exceeded, further perturbation of the primary nucleation pathway ceases to be effective in reducing the overall profileration of oligomers, as most new aggregates are not created via this mechanism.
Materials and Methods
Additional information can be found in SI Text.
Integrated Rate Law.
When both primary and secondary pathways are active, the integrated rate law describing the generation of total fibril mass, 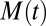 , over time as a function only of the initial conditions and the rate constants of the system is given as (16, 22)
, over time as a function only of the initial conditions and the rate constants of the system is given as (16, 22)
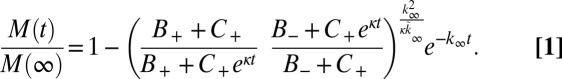 |
Although many distinct parameters, including microscopic rate constants for primary nucleation 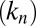 , elongation
, elongation 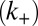 , depolymerization
, depolymerization 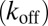 , fragmentation
, fragmentation 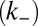 , and fibril-catalyzed secondary nucleation
, and fibril-catalyzed secondary nucleation 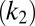 , are required to capture the complete assembly process (16, 22), only two particular combinations of the rate constants define much of the macroscopic behavior; these parameters are related to the rate of formation of new aggregates through primary pathways
, are required to capture the complete assembly process (16, 22), only two particular combinations of the rate constants define much of the macroscopic behavior; these parameters are related to the rate of formation of new aggregates through primary pathways 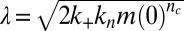 and through secondary pathways
and through secondary pathways 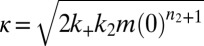 , where
, where 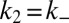 when
when 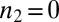 . Indeed, Eq. 1 depends on the rate constants through these two parameters, λ and κ, alone because
. Indeed, Eq. 1 depends on the rate constants through these two parameters, λ and κ, alone because 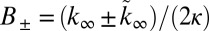 ,
, 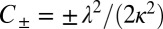 ,
,  , and
, and 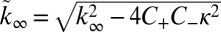 . The initial concentration of soluble monomers is
. The initial concentration of soluble monomers is 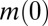 and the reaction orders describing the dependencies of the primary and secondary pathways on the monomer concentration are
and the reaction orders describing the dependencies of the primary and secondary pathways on the monomer concentration are 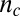 and
and 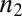 .
.
Materials.
We expressed in Escherichia coli and purified, as described previously (40), the Aβ(M1–42) peptide (MDAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA). Radiolabeled Aβ42 was expressed and purified in the same way, except that cells were grown in minimal medium supplemented with [35S]methionine 2 min before induction. Aliquots of purified Aβ42 were thawed and subjected to gel filtration on a Superdex 75 column in 20 mM sodium phosphate buffer, pH 8, 200 μM EDTA 0.02% NaN3.
Radiolabeling and Immunochemistry.
The peptide samples were taken from an ongoing seeded or unseeded aggregation reaction (Fig. 4) and immediately loaded into a 1 × 30-cm Superdex 75 column. Eluted fractions (2 mL) were diluted 1:4 in scintillation solution (Ready Safe liquid Scintillation Mixture; Beckman Coulter) and placed in a scintillator (Beckman LS6000IC) for counting for a total of 120 min per sample. The counts for fractions with average elution volumes of 6, 8, and 10 mL were binned as oligomer counts (sum of 3- to 20-mer, because the dominant monomer peak makes dimer quantification inaccurate) and counts for fractions eluting at 12, 14, and 16 mL were binned as monomer counts. The experiments were repeated with unlabeled species, and 1-mL eluted fractions were concentrated by lyophilization, dissolved in 8 M urea, and applied to a PVDF membrane for semiquantitative analysis using 6E10 primary antibody (Signet) and alkaline phosphatase-conjugated rabbit anti-mouse secondary antibody (Dako).
Cytotoxicity and Cell Viability Assays.
Assays were performed on SH-SY5Y human neuroblastoma cells cultured under standard conditions. The peptide samples were taken from ongoing seeded or unseeded reactions and subjected to filtration through a 200-nm filter (Anapour). Control peptide, monomer, and fibril were not filtrated. Buffer controls were both filtrated and unfiltrated. The cells were then cultured in the presence of the peptides, buffer, or media for a further 24 h before the cytotoxicity and viability assays were performed. Caspase-3/7 activity was measured using the Apo-ONE Homogeneous Caspase-3/7 assay (Promega). Cell viability was measured using the Cell Titer 96 Aqueous One MTS reagent (Promega).
Supplementary Material
Acknowledgments
We thank Dale Schenk, Daan Frenkel, and David Chandler for useful discussions. We acknowledge financial support from the Newman Foundation (T.P.J.K.), the Schiff Foundation (S.I.A.C.), the Kennedy Memorial Trust (S.I.A.C.), the Swedish Research Council (S.L.) and its Linneaus Centre Organizing Molecular Matter (S.L. and E.H.), the Crafoord Foundation (S.L.), the Royal Physiographic Society (E.H.), the Nanometer Structure Consortium at Lund University (S.L.), Alzheimerfonden (S.L.), Danish Research Foundation (D.E.O.), and the Wellcome Trust (M.V., C.M.D., and T.P.J.K.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1218402110/-/DCSupplemental.
References
- 1.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426(6968):900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 2.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 3.Aguzzi A, Haass C. Games played by rogue proteins in prion disorders and Alzheimer’s disease. Science. 2003;302(5646):814–818. doi: 10.1126/science.1087348. [DOI] [PubMed] [Google Scholar]
- 4.Dobson CM. Protein folding and misfolding. Nature. 2003;426(6968):884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 5.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell. 2005;120(4):545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Eisele YS, et al. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330(6006):980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kayed R, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300(5618):486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 8.Walsh DM, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 9.Bucciantini M, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416(6880):507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 10.Oosawa F, Asakura S. 1975. Thermodynamics of the Polymerization of Protein. (Academic, Waltham, MA)
- 11.Collins SR, Douglass A, Vale RD, Weissman JS. Mechanism of prion propagation: Amyloid growth occurs by monomer addition. PLoS Biol. 2004;2(10):e321. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442(7102):585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 13.Schnabel J. Amyloid: Little proteins, big clues. Nature. 2011;475(7355):S12–S14. doi: 10.1038/475S12a. [DOI] [PubMed] [Google Scholar]
- 14.Ferrone FA, Hofrichter J, Eaton WA. Kinetics of sickle hemoglobin polymerization. II. A double nucleation mechanism. J Mol Biol. 1985;183(4):611–631. doi: 10.1016/0022-2836(85)90175-5. [DOI] [PubMed] [Google Scholar]
- 15.Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- 16.Knowles TPJ, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326(5959):1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 17.Cohen SIA, et al. Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J Chem Phys. 2011;135(6):065105. doi: 10.1063/1.3608916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bishop MF, Ferrone FA. Kinetics of nucleation-controlled polymerization. A perturbation treatment for use with a secondary pathway. Biophys J. 1984;46(5):631–644. doi: 10.1016/S0006-3495(84)84062-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oosawa F, Kasai M. A theory of linear and helical aggregations of macromolecules. J Mol Biol. 1962;4:10–21. doi: 10.1016/s0022-2836(62)80112-0. [DOI] [PubMed] [Google Scholar]
- 20.Lührs T, et al. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc Natl Acad Sci USA. 2005;102(48):17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eaton WA, Hofrichter J. Sickle cell hemoglobin polymerization. Adv Protein Chem. 1990;40:63–279. doi: 10.1016/s0065-3233(08)60287-9. [DOI] [PubMed] [Google Scholar]
- 22.Cohen SIA, Vendruscolo M, Dobson CM, Knowles TPJ. Nucleated polymerization with secondary pathways. II. Determination of self-consistent solutions to growth processes described by non-linear master equations. J Chem Phys. 2011;135(6):065106. doi: 10.1063/1.3608917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73(6):1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 24.Serio TR, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289(5483):1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 25.Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat Struct Mol Biol. 2011;18(3):328–336. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wegner A. Spontaneous fragmentation of actin filaments in physiological conditions. Nature. 1982;296(5854):266–267. doi: 10.1038/296266a0. [DOI] [PubMed] [Google Scholar]
- 27.Ruschak AM, Miranker AD. Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc Natl Acad Sci USA. 2007;104(30):12341–12346. doi: 10.1073/pnas.0703306104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cacciuto A, Auer S, Frenkel D. Onset of heterogeneous crystal nucleation in colloidal suspensions. Nature. 2004;428(6981):404–406. doi: 10.1038/nature02397. [DOI] [PubMed] [Google Scholar]
- 29.Cremades N, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149(5):1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen SIA, Vendruscolo M, Dobson CM, Knowles TPJ. From macroscopic measurements to microscopic mechanisms of protein aggregation. J Mol Biol. 2012;421(2–3):160–171. doi: 10.1016/j.jmb.2012.02.031. [DOI] [PubMed] [Google Scholar]
- 31.Flyvbjerg H, Jobs E, Leibler S. Kinetics of self-assembling microtubules: An “inverse problem” in biochemistry. Proc Natl Acad Sci USA. 1996;93(12):5975–5979. doi: 10.1073/pnas.93.12.5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X, et al. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci USA. 2009;106(48):20324–20329. doi: 10.1073/pnas.0911281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hellstrand E, Boland B, Walsh DM, Linse S. Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem Neurosci. 2010;1(1):13–18. doi: 10.1021/cn900015v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeong JS, Ansaloni A, Mezzenga R, Lashuel HA, Dietler G. Novel Mechanistic Insight into the Molecular Basis of Amyloid Polymorphism and Secondary Nucleation during Amyloid Formation. J Mol Biol. 2013 doi: 10.1016/j.jmb.2013.02.005. 10.1016/j.jmb.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 35.Lee J, Culyba EK, Powers ET, Kelly JW. Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat Chem Biol. 2011;7(9):602–609. doi: 10.1038/nchembio.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buell AK, et al. Detailed analysis of the energy barriers for amyloid fibril growth. Angew Chem Int Ed Engl. 2012;51(21):5247–5251. doi: 10.1002/anie.201108040. [DOI] [PubMed] [Google Scholar]
- 37.Sánchez L, et al. Aβ40 and Aβ42 amyloid fibrils exhibit distinct molecular recycling properties. J Am Chem Soc. 2011;133(17):6505–6508. doi: 10.1021/ja1117123. [DOI] [PubMed] [Google Scholar]
- 38.Koffie RM, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA. 2009;106(10):4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spires-Jones TL, et al. Passive immunotherapy rapidly increases structural plasticity in a mouse model of Alzheimer disease. Neurobiol Dis. 2009;33(2):213–220. doi: 10.1016/j.nbd.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walsh DM, et al. A facile method for expression and purification of the Alzheimer’s disease-associated amyloid beta-peptide. FEBS J. 2009;276(5):1266–1281. doi: 10.1111/j.1742-4658.2008.06862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.