

Abstract
The lack of a robust pipeline of medical therapeutic agents for the treatment of heart disease may be partially attributed to the lack of in vitro models that recapitulate the essential structure–function relationships of healthy and diseased myocardium. We designed and built a system to mimic mechanical overload in vitro by applying cyclic stretch to engineered laminar ventricular tissue on a stretchable chip. To test our model, we quantified changes in gene expression, myocyte architecture, calcium handling, and contractile function and compared our results vs. several decades of animal studies and clinical observations. Cyclic stretch activated gene expression profiles characteristic of pathological remodeling, including decreased α- to β-myosin heavy chain ratios, and induced maladaptive changes to myocyte shape and sarcomere alignment. In stretched tissues, calcium transients resembled those reported in failing myocytes and peak systolic stress was significantly reduced. Our results suggest that failing myocardium, as defined genetically, structurally, and functionally, can be replicated in an in vitro microsystem by faithfully recapitulating the structural and mechanical microenvironment of the diseased heart.
Keywords: organs on chips, mechanotransduction, muscular thin films, microarray, contraction
Maladaptive cardiac hypertrophy occurs in response to a variety of stimuli, including myocardial infarction, genetic mutations, or hypertension (1). Initially, hypertrophic remodeling is compensatory, as myocytes counteract losses in cardiac output by adding myofibrils in parallel (2, 3). Over time, remodeling transitions from adaptive to maladaptive, which is characterized structurally by sarcomere disarray (4), fibrosis (5), myocyte elongation (2, 6, 7), and ventricular dilation (3). Gene expression reverts to an immature state, including down-regulation of α-myosin heavy chain (MHC) concurrent with up-regulation of β-MHC, reminiscent of the relative expression levels of these motor proteins in the embryonic heart (8, 9). Functionally, myocytes isolated from failing hearts show defective excitation–contraction coupling (10), including reduced calcium uptake into the sarcoplasmic reticulum (11). Contractile function is also reduced, as shown by decreased cardiac output in dogs subjected to chronic rapid pacing (12) and decreased tension in left ventricular strips isolated from failing human myocardium (13, 14). Thus, dilated and failing hearts are characterized by multiscale maladaptive remodeling, best studied in an experimental system capable of replicating and quantifying a broad range of these effects.
Drug discovery, safety pharmacology, and mechanistic studies have traditionally been performed in animal models. For example, cardiac hypertrophy and heart failure are reproduced in the rat by ligating coronary arteries or by using the spontaneously hypertensive rat strain (15). In vitro, heart disease has been modeled by exposing primary cardiac myocytes to chemical (16) or exogenous mechanical stimuli (17–21), which increases cell size, activates pathological gene expression, and remodels ion channel currents. However, cell culture systems often fail to predict the efficacy or toxicity of therapeutic candidates, potentially because they do not replicate the complex structure–function relationships of native cardiac tissue (22). Traditional cell culture systems also fail to quantify pathological changes in contractile function, instead focusing on endpoints such as gene expression or electrophysiology, which are difficult to justify as direct indicators of reduced cardiac output.
We reasoned that we could reproduce years of published animal studies on dilated and failing myocardium with a carefully designed in vitro microsystem. Our goal was to build failing myocardium “on a chip” by engineering arrays of laminar ventricular muscle on a substrate amenable to cyclic stretch to mimic mechanical overload. To test our system, we measured genetic, structural, and functional responses, including contractile stress generation. Cyclic stretch activated markers of pathological cardiac hypertrophy, disrupted myocyte shape and sarcomere alignment with directional specificity, remodeled calcium transients, and decreased stress generation in a manner similar to animal studies and clinical observations. These data suggest that we can recapitulate failing myocardium on a chip, which has potential value as a new in vitro platform for early stage drug discovery.
Results
Chip Design.
Cardiac myocytes are traditionally cultured on rigid substrates that prohibit mechanical intervention and contractility measurements and are limited to electrophysiology or gene expression studies of chemically induced pathologies. We reasoned that we could build a stretchable substrate amenable to microcontact printing and contractility studies in tissue constructs that mimic the aligned, laminar structure of ventricular tissue. We fabricated stretchable muscular thin film (MTF) substrates by selectively coating silicone membranes with a temperature-sensitive polymer and a thin layer of polydimethylsiloxane (PDMS; Fig. 1A) (23–25), building on previous efforts of Zhuang et al. (26) and Yamada et al. (27). Substrates micropatterned with fibronectin (FN) in a “brick wall” pattern (Fig. 1B) and seeded with neonatal rat ventricular myocytes promoted assembly of anisotropic cardiac monolayers (Fig. 1C) that recapitulate the laminar muscle architecture observed in the native ventricle (Fig. 1A) (28). Substrates were uniaxially and cyclically stretched at 10% strain and a frequency of 3 Hz by using a custom-built multiwell system (Movie S1) (23, 26). After conditioning the tissues, arrays of MTFs were cut from the PDMS layer so that, after the temperature-sensitive layer released, MTFs freely deflected away from the silicone membrane with each paced contraction while remaining fixed at one edge (Fig. 1A and Movie S2) (24). MTF deformation during systole could be tracked and translated to measurements of stress generation while diastolic tension was measured by the resting MTF curvature (24, 29). Silicone membranes could also be micropatterned directly and seeded with myocytes for optical measurements of calcium transients, staining of tissue structure, and collection of lysates for gene expression analysis, similar to our previous work (24). Thus, our chip recapitulates native tissue architecture and asynchronous mechanical stretch in the ventricular wall, and is amenable to measurements of contractile stress generation, calcium transients, cytoskeletal structures, and gene expression.
Fig. 1.

Failing myocardium on a chip design. (A, i) Schematic of the heart with right atrium (RA), left atrium (LA), right ventricle (RV), and left ventricle (LV) labeled. (A, ii) Ventricular myocardium consists of aligned, elongated cardiac myocytes. (A, iii) Schematic of failing myocardium on a chip system adapted for the MTF assay. (A, iv) Photograph of stretchable MTF experiment. (B) Elastomeric silicone membrane micropatterned with FN (white) in a brick wall pattern. (Scale bar: 10 µm.) (C) Micropatterned membrane seeded with neonatal rat ventricular myocytes. (D) Myocytes seeded onto membranes coated with isotropic FN, cultured for 1 h, and (E) cyclically stretched. (Scale bars: 50 µm) (F) Myocytes patterned and stretched in the longitudinal direction. (Scale bar: 50 µm.) (G) Myocytes patterned and stretched in the transverse direction. (Scale bar: 50 µm.)
Because many cardiomyopathies are associated with mechanical overload (4), we reasoned that cyclic stretch would induce pathological subcellular, cellular, and tissue remodeling. We seeded neonatal rat ventricular myocytes onto silicone membranes coated with isotropic FN and initiated cyclic stretch 1 h after seeding, when myocytes were adhered to the substrate but had not yet formed a tissue (Fig. 1D). Myocytes spontaneously aligned parallel to the axis of stretch (Fig. 1E). Myocytes seeded on micropatterned FN maintained uniaxial alignment when cyclic stretch was parallel to the direction of patterning (Fig. 1F). However, transverse stretch of patterned tissues disrupted tissue architecture observed in isolated regions where myocytes realigned with the exogenous stretch, creating a “parquet floor” pattern (Fig. 1G). This is reminiscent of clinical reports in which tissue sections taken from human hearts with hypertrophic cardiomyopathy displayed similar misalignment (4). Thus, our failing myocardium chip appears to recapitulate the form of laminar ventricular tissue in the healthy and diseased heart.
Cyclic Stretch Potentiates Pathological Gene Expression.
We asked if cyclic stretch would trigger known genetic indicators of pathological remodeling (30). We collected and amplified mRNA from conditioned tissues after 6, 24, and 96 h in culture and used Affymetrix whole-transcript microarrays to determine gene expression values. To visualize the dynamics of global gene expression, we used Gene Expression Dynamics Inspector (GEDI) bioinformatics software, which clusters genes with similar expression profiles and organizes the clusters into color-coded maps (31, 32). GEDI maps revealed that cyclic stretch-induced changes in gene expression became more prominent with time in culture (over 96 h; Fig. 2A). The microarray data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (33) and are accessible through Gene Expression Omnibus Series accession number GSE43846. Genes with significant differences at each time point are listed in Datasets S1, S2, and S3. Gene expression was relatively insensitive to the direction of stretch relative to tissue alignment, suggesting that stretch induces changes to gene expression that are not directionally dependent.
Fig. 2.
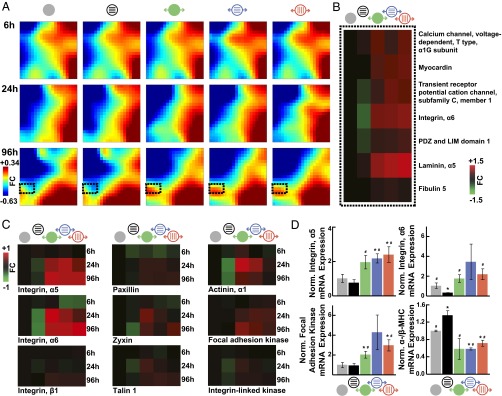
Cyclic stretch promotes pathological gene expression. (A) Time-course GEDI maps of microarray data filtered to include any gene that showed a significant fold change (FC) at any time point in any condition after 1 h plus the indicated hours in culture. Color bar indicates FC differences in gene expression relative to samples collected 1 h after seeding (n = 3). (B) Heat maps of microarray data for indicated genes, which are located in the dashed boxes in A. Color bar indicates FC differences relative to static, isotropic tissues at the same time point (n = 3). (C) Heat maps of microarray data for indicated genes. Color bar indicates FC differences relative to static, isotropic tissues at the same time point (n = 3). (D) Normalized mRNA expression as measured by RT-PCR for indicated genes (mean ± SE; n = 3 tissues; *P < 0.05 vs. static, isotropic tissues; #P < 0.05 vs. static, patterned tissues).
We asked if stretch-induced changes in tissue gene expression matched markers of pathological hypertrophy and/or heart failure. As shown in Fig. 2A, the region near the lower left corner of the mosaics (Fig. 2A, dashed black outline) was composed of genes most significantly up-regulated with stretch at 96 h. In this region, we found increased expression of genes encoding for T-type calcium channel α1G subunit, myocardin, and transient receptor potential cation channel 1 (TRPC1) in stretched tissues (Fig. 2B). Each of these genes has been reported to be up-regulated in pathological cardiac hypertrophy in several animal models (including rat) and human heart failure, except for TRPC1, in which, instead, TRPC5 and TRPC6 are up-regulated in human (34–38). We also observed stretch-dependent up-regulation of genes encoding for cytoskeletal proteins (Fig. S1), such as integrin α6 and PDZ and LIM domain 1, and ECM proteins, such as laminin α5 and fibulin 5 (Fig. 2B). These changes are also consistent with pathological remodeling, whereby cytoskeletal reorganization and increased ECM production are commonly reported in hypertrophy and failure in human and rat (39, 40). Thus, cyclic stretch activates expression of several genetic markers of pathological remodeling, including ion channels and components of the ECM and the cytoskeleton.
Focal adhesions are transmembrane, mechanosensitive protein complexes that couple the ECM to the cytoskeleton (41) and are commonly overexpressed in pathological hypertrophy (39, 42–44). Our microarray analysis revealed stretch-mediated up-regulation of integrin α6 mRNA at 96 h, so we asked if other focal adhesion genes were up-regulated with mechanical loading (Fig. S1). Integrins α5 and α6 mRNA were up-regulated as early as 24 h, and integrin β1 mRNA was up-regulated at 96 h after initiation of stretch. Genes encoding for paxillin, zyxin, talin 1, and α-actinin 1, which link integrins to the actin cytoskeleton, were up-regulated beginning at 24 h of stretch (Fig. 2C). mRNA for focal adhesion kinase 1 and integrin-linked kinase, which activate signaling in response to mechanical loads propagated through integrins, were also up-regulated in stretched tissues (Fig. 2C). RT-PCR results also indicated up-regulation of integrin α5, integrin α6, and focal adhesion kinase mRNA at 96 h in stretched relative to static tissues (Fig. 2D). Together, these results suggest that cyclic stretch up-regulates the expression of several genes essential to the structure and function of focal adhesions, analogous to pathological hypertrophy.
Heart failure is associated with increased expression of the slow MHC isoform β-MHC relative to the expression of the fast MHC isoform α-MHC, conserving energy but reducing contractility in rodents (8). We used RT-PCR to determine the ratio of α-/β-MHC mRNA at 96 h and found that this ratio was lowest for all stretched tissues (Fig. 2D). Interestingly, we also observed increased α-/β-MHC gene expression in patterned tissues relative to isotropic tissues, indicating that alignment alone potentially induces maturation of MHC expression, but cyclic stretch potentiates pathological MHC expression.
Cyclic Stretch Induces Orientation-Dependent Structural Remodeling.
Remodeling of myocyte shape, specifically length-to-width aspect ratio (AR), is a hallmark of cardiomyopathies across many species, including human and rat (2, 6, 7). Healthy ventricular myocytes have an average AR of 7:1, and myocytes isolated from hearts with eccentric and concentric hypertrophy have a higher (11:1) and lower (5:1) AR, respectively (3). We asked if cyclic stretch induces myocyte shape changes that correlate to pathological cellular remodeling. We stained cell membranes with di-8-ANEPPS, manually measured myocyte dimensions, and found that AR was higher in patterned tissues (7:1) compared with isotropic tissues (4.5:1) as a result of myocyte elongation on the FN pattern (Fig. 3 A and B and Fig. S2), which has been reported previously (45). Cyclic stretch elongated myocytes beyond 7:1, which was most prominent in patterned tissues stretched longitudinally (10:1; Fig. 3C and Fig. S2). Myocytes stretched transversely were longer and wider (Fig. S2), and thus did not have a change in overall AR relative to static, patterned tissues (Fig. 3D). Thus, longitudinal stretch increases the cellular AR of healthy tissues to values typically observed in dilated cardiomyopathy (2).
Fig. 3.

Cyclic stretch induces orientation-dependent structural remodeling. Cell membranes in (A) isotropic, (B) patterned, and (C) longitudinally stretched tissues stained with di-8-ANEPPS to indicate cell borders, as shown for representative cells outlined in yellow. (Scale bars: 10 µm.) (D) Average cell AR for each condition (mean ± SE; n ≥ 4 tissues; *P < 0.05 vs. static, isotropic tissues; #P < 0.05 vs. static, patterned tissues). (E) Isotropic, (F) patterned, and (G) transversely stretched cardiac tissues immunostained for α-actinin to detect sarcomeres. (H) Average sarcomere OOP for each condition (mean ± SE; n = 5 tissues; *P < 0.05 vs. static, isotropic tissues, #P < 0.05 vs. static, patterned tissues).
In mature cardiac tissue, ECM fibers are oriented parallel to the long axis of the cell and the direction of contraction (46). However, pathological hypertrophy is associated with fibrosis and ECM reorganization (47), which potentially disrupts the cooperative relationship between ECM alignment and the myocyte cytoskeleton (4). We asked how different orientations of cyclic stretch relative to ECM alignment alter sarcomere alignment. We immunostained for sarcomeric α-actinin in stretched and unstretched tissues, collected epifluorescent images, and used image processing techniques to threshold the immunosignal, detect pixel orientation angles (48), and calculate the orientational order parameter (OOP). The OOP ranges from zero for isotropic systems to one for perfectly aligned systems (24). Compared with isotropic tissues (Fig. 3E), tissues that were patterned (Fig. 3F), stretched on isotropic FN, or longitudinally stretched on patterned FN had higher OOPs. For transversely stretched tissues (Fig. 3G), the OOP was lower than in other aligned tissues (Fig. 3H) because ECM patterning and cyclic stretch competed for tissue alignment and induced the parquet-floor architecture described earlier (Fig. 1G). Together, these results demonstrate that sarcomere alignment is compromised when ECM is not aligned coincident to the direction of mechanical loading.
Cyclic Stretch Reduces Contractile Function.
The contractile strength of muscle is dependent on intracellular calcium levels (49). We asked if stretch alters calcium transient morphology, which could signify contractile dysfunction. Fluo-4 calcium transients in conditioned tissues paced at 2 Hz were imaged (Fig. 4A) and analyzed to determine peak fluorescence (F) relative to baseline fluorescence (F0) and time to peak. In all stretched tissues, transients showed decreased F/F0 (Fig. 4B) and increased time to peak (Fig. 4C), independent of the direction of stretch. These changes are reminiscent of those reported for hypertrophic and failing rat (10) and human (11) hearts. Thus, cyclic stretch induces remodeling of calcium transients that matches reports for failing myocytes.
Fig. 4.

Cyclic stretch reduces contractile function. (A) Average fluorescence intensity (marked as “F”) of Fluo-4 normalized to F0 and plotted for a cardiac cycle. (B) Peak F/F0 and (C) time to peak for each condition (mean ± SE; n ≥ 5; *P < 0.05 vs. static, isotropic tissues; #P < 0.05 vs. static, patterned tissues). (D) Schematic of stretchable MTFs at diastole and systole. Contraction of MTFs was recorded from above, as shown for representative (E) patterned and (F) transversely stretched films transitioning from (i) diastole to (ii) systole. (Scale bars: 1 mm.) (G) Displacement of films was used to determine stress generation over time, as shown for the films in E and F. (H) Average diastolic, systolic, and active stress for each condition (mean ± SE; n ≥ 16; *P < 0.05 vs. static, isotropic tissues; #P < 0.05 vs. static, patterned tissues).
We reasoned that, if our microsystem faithfully reproduced cardiac pathophysiology, we would measure contractile dysfunction in the stretched tissues. Thus, we measured stresses generated by conditioned MTFs paced at 2 Hz (Fig. 4D) (24, 29). Diastolic stresses were not statistically different between tissues, except between patterned tissues (Fig. 4E) and transversely stretched tissues (Fig. 4 F and G). As shown previously, patterned tissues generated more systolic and active stress than isotropic tissues (24, 48). However, all stretched tissues had lower systolic and twitch stresses than patterned tissues, independent of the direction of stretch (Fig. 4H), indicating that cyclic stretch reduces stress generation even if sarcomeres are highly aligned. Our results are similar to measurements made from myocytes isolated from failing and nonfailing human hearts, in which, at physiological frequencies, percent shortening in failing myocytes was approximately half that in nonfailing myocytes (13, 14). These data suggest that it is possible to recapitulate failing myocardium on a 2D chip that is amenable to comparison with animal and clinical studies.
Discussion
In this study, we modeled failing myocardium on a chip by applying cyclic stretch to engineered, anisotropic cardiac tissues. Gene expression in stretched tissues was consistent with pathological remodeling, including up-regulation of focal adhesion genes and a switch to the immature myosin isoform. Sarcomere alignment and cell shape were uniquely sensitive to the direction of stretch and matched clinical reports of pathological structural remodeling. Stretch-induced changes in calcium cycling and systolic stress generation were also comparable with measurements from patients with heart failure. Together, these results suggest that our in vitro microsystem can be used to study the genetic, structural, and functional aspects of failing myocardium.
Maladaptive gene expression is a commonly used test for experimental models of heart disease because it can be easily compared with clinical studies. Similarly, we observed a lower ratio of α- to β-MHC mRNA in stretched tissues, a commonly accepted indicator of heart disease (8, 9). We measured increased expression of genes encoding for several focal adhesion proteins, such as focal adhesion kinase and integrin α5, in response to stretch, which matches in vivo studies demonstrating up-regulation of focal adhesion expression and signaling during the hypertrophic response to mechanical overload (39, 42–44). Although our results do not identify molecular mechanisms, they do motivate future studies to discover and validate therapeutic targets, such as focal adhesion signaling. Thus, our chip matches the genetic criteria of a valid model of failing myocardium and could be used to further explore signaling and protein expression.
Heart failure is also characterized by significant remodeling of tissue architecture. Because disease progression often affects the distribution of biomechanical forces and ECM organization, we took advantage of our in vitro microsystem to address how these two extracellular cues coregulate structural remodeling. With transverse cyclic stretch, local regions of sarcomere misalignment resulted in disorganized tissue reminiscent of hypertrophic cardiomyopathy (4). Myocyte AR was also sensitive to the direction of stretch, as myocytes stretched longitudinally had an AR of 10:1, compared with patterned myocytes with an AR of 7:1. These values are similar to those reported for myocytes isolated from dilated and healthy hearts, respectively (2). Interestingly, transverse stretch did not affect AR, suggesting that structural remodeling is sensitive to the direction of stretch, similar to previous in vitro reports (18, 50). These data suggest that our in vitro microsystem replicates the maladaptive remodeling of the diseased heart.
Because heart failure is a broadly defined disease, it has been associated with diastolic and systolic dysfunction (51). Our stretched tissues exhibited reduced systolic stress, suggesting that excessive stretch affects myocyte shortening, but not relaxation. One potential explanation is that pathological hypertrophy and heart failure are also associated with increased fibrosis, which stiffens the cellular microenvironment (52). These pathological changes in compliance likely impede myocyte relaxation and could be essential to diastolic dysfunction. One limitation of our system is that we cannot measure the rate of diastolic relaxation, which could correlate to isovolumetric relaxation, because the material properties of PDMS make it difficult to separate the relaxation of our engineered tissues from the elastic recoil of the PDMS substrate. Thus, our system recapitulates the systolic dysfunction of failing myocardium, but does not reveal a mechanism for diastolic dysfunction in the heart.
Organs on chips have promising potential as substitutes for in vivo animal studies with improved predictability compared with current in vitro systems (53, 54). In this study, we used neonatal rat myocytes because rat is the pharmaceutical and biotechnology industry standard for the study of heart disease in vitro and in vivo. Our model could be further developed as failing myocardium on a chip technology with applications for testing therapeutic agents against specific diseases and patient populations in combination with in vitro models that focus on other cardiac diseases (55). The uniqueness of our model lies in its ability to recapitulate native tissue architecture and its capacity to quantify not only genetic and structural remodeling, but also functional remodeling, including contractile force generation. As the quality and consistency of human stem cell-derived cardiomyocytes improves, we hope to extend our model to these cells that will potentially be more predictive than rodent cells (56). We also appreciate the importance of incorporating other cell types found in the myocardium, such as fibroblasts and endothelial cells, which contribute to the multicellular architecture of the heart but are difficult to coculture with myocytes. Further development of organs on chips with specific physiological and pathological phenotypes has the potential to significantly reduce drug development costs while also improving the fidelity and predictability of efficacy and toxicity studies.
Materials and Methods
Experimental methods are described in detail in SI Materials and Methods; a brief description is included here.
Chip Fabrication.
Elastic silicone membranes (Specialty Manufacturing) were clamped into stainless steel brackets (23, 26) and affixed with 25-mm-diameter rings of silicon tubing. Membranes treated in a UV-ozone cleaner (Jelight) were micropatterned with PDMS stamps coated with 50 μg/mL FN (BD Biosciences) or coated uniformly with 50 μg/mL FN. To fabricate stretchable MTFs (23, 24), silicone membranes were temporarily cured to glass platforms with 200 proof ethanol to maintain baseline tension and provide rigid surfaces for spin-coating. Two pieces of tape were applied across the length or width of the membrane separated by 8 mm. Poly(N-isopropylacrylamide) (Polysciences) was spin-coated onto membranes followed by tape removal. PDMS cured at room temperature for 4 to 5 h was then spin-coated onto membranes, which were reclamped into brackets after fully curing.
Cell Culture.
All animal protocols were approved by the Harvard University Animal Care and Use Committee. Cardiac myocytes were isolated by enzymatically digesting ventricles from 2-d-old Sprague–Dawley rats by using previously published protocols (24, 48, 57). A custom bioreactor based on a previous design (26) was built to apply cyclic strain to tissues cultured on silicone membranes using a linear motor (LinMot) to stretch as many as 10 samples simultaneously in an incubator (23).
Gene Expression Analysis.
RNA was isolated by using Stratagene Absolutely RNA Miniprep Kit (Agilent Technologies). mRNA was amplified and hybridized to Affymetrix GeneChip Rat Gene 1.0 ST Arrays following manufacturer’s instructions, and scanned with Affymetrix GeneChip Scanner 3000 7G. Probe cell intensity data files were normalized using the robust multichip average method (58) in Affymetrix Expression Console Software. Signal values were log2-transformed and analyzed with Bioconductor open-source software and the limma package in R (59). Average expression values for each condition were fit to a linear model and compared using Bayes statistics. Gene expression values were analyzed with GEDI bioinformatics software package (31, 32). Differentially regulated genes were categorized by selected Gene Ontology terms by inputting filtered probe set IDs into the AmiGO! Slimmer from the Gene Ontology project (http://amigo.geneontology.org/cgi-bin/amigo/slimmer).
Sarcomere Alignment Quantification.
Tissues were fixed, immunostained for sarcomeric α-actinin (A7811; Sigma-Aldrich), and imaged on an inverted fluorescent microscope (DMI 6000B; Leica). Custom MATLAB software (MathWorks) was used to calculate the sarcomere OOP of each tissue (24, 48), which were statistically compared by Student t test.
Cell Shape Measurements.
Tissues were incubated with 10 µM di-8-ANEPPS (Invitrogen) for 10 min at 37 °C, rinsed with Tyrode solution (1.8 mM CaCl2, 5 mM glucose, 5 mM Hepes, 1 mM MgCl2, 5.4 mM KCl, 135 mM NaCl, 0.33 mM NaH2PO4, pH 7.4), moved to a confocal microscope maintained at 37 °C (LSM 510; Zeiss), and imaged with a 40× objective. Custom MATLAB software (MathWorks) was used to manually outline cell borders of multiple cells per tissue, which were fit to an ellipse. The average major (cell length) and minor (cell width) axes and ARs of fitted ellipses for each condition were statistically compared by Student t test.
Calcium Transient Measurements.
Tissues were incubated with 10 µg/mL Fluo-4 acetoxymethyl ester calcium indicator (Invitrogen) for 30 min at 37 °C, rinsed with Tyrode solution, and transferred to a confocal microscope maintained at 37 °C (Zeiss LSM 510). Tissues were paced with a point stimulation electrode operating at 2 to 5 V and 2 Hz for at least 5 min before data acquisition with a 40× objective. For multiple fields, 10 × 10-µm2 regions were selected in the cytoplasm of a cell and change in F over time for four beats was averaged to determine F over F0 and time to peak (60). Average F/F0 and time to peak for each condition were statistically compared by Student t test.
Contractile Stress Measurements.
Stretchable MTFs were incubated in Tyrode solution and moved to a stereomicroscope (Leica Microsystems). The PDMS layer was cut to release arrays of MTFs that remained attached to the substrate at one longitudinal end. As the temperature decreased to less than 32 °C, poly(N-isopropylacrylamide) transitioned to the aqueous phase, allowing MTF release (24). Temperature was restored to 37 °C, and MTFs were paced with a field stimulation electrode operating at 5 to 10 V and 2 Hz. Movies were acquired at 120 to 150 Hz. PDMS film thickness was measured with a profilometer (P-16+ Contact Stylus Profiler; KLA-Tencor). MATLAB software (MathWorks) was used to calculate stress generation based on the projection of the radius of curvature and film thickness (24, 29). Average diastolic, systolic, and twitch stresses for each condition were statistically compared by Student t test.
Supplementary Material
Acknowledgments
We thank André G. Kléber for design of the initial cyclic stretch system and the Harvard Center for Nanoscale Systems for use of cleanroom facilities. This work was funded by American Heart Association Predoctoral Fellowship 0815729D, National Institutes of Health Grants 1 R01 HL079126 and 1 UH2 TR000522-01, the Harvard Materials Research Science and Engineering Center as supported by National Science Foundation Division of Materials Research Grant DMR-0213805, the Harvard Stem Cell Institute and GlaxoSmithKline, and the Harvard School of Engineering and Applied Sciences.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE43846).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304913110/-/DCSupplemental.
References
- 1.Lorell BH, Carabello BA. Left ventricular hypertrophy: Pathogenesis, detection, and prognosis. Circulation. 2000;102(4):470–479. doi: 10.1161/01.cir.102.4.470. [DOI] [PubMed] [Google Scholar]
- 2.Gerdes AM. Cardiac myocyte remodeling in hypertrophy and progression to failure. J Card Fail. 2002;8(6, suppl):S264–S268. doi: 10.1054/jcaf.2002.129280. [DOI] [PubMed] [Google Scholar]
- 3.Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56(1):56–64. doi: 10.1172/JCI108079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho CY. Hypertrophic cardiomyopathy: Preclinical and early phenotype. J Cardiovasc Transl Res. 2009;2(4):462–470. doi: 10.1007/s12265-009-9124-7. [DOI] [PubMed] [Google Scholar]
- 5.Jane-Lise S, Corda S, Chassagne C, Rappaport L. The extracellular matrix and the cytoskeleton in heart hypertrophy and failure. Heart Fail Rev. 2000;5(3):239–250. doi: 10.1023/A:1009857403356. [DOI] [PubMed] [Google Scholar]
- 6.Gerdes AM, Onodera T, Wang X, McCune SA. Myocyte remodeling during the progression to failure in rats with hypertension. Hypertension. 1996;28(4):609–614. doi: 10.1161/01.hyp.28.4.609. [DOI] [PubMed] [Google Scholar]
- 7.Gerdes AM. Remodeling of ventricular myocytes during cardiac hypertrophy and heart failure. J Fla Med Assoc. 1992;79(4):253–255. [PubMed] [Google Scholar]
- 8.Gupta MP. Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J Mol Cell Cardiol. 2007;43(4):388–403. doi: 10.1016/j.yjmcc.2007.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86(4):386–390. doi: 10.1161/01.res.86.4.386. [DOI] [PubMed] [Google Scholar]
- 10.Gómez AM, et al. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276(5313):800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 11.Piacentino V, 3rd, et al. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92(6):651–658. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- 12.Wilson JR, et al. Experimental congestive heart failure produced by rapid ventricular pacing in the dog: cardiac effects. Circulation. 1987;75(4):857–867. doi: 10.1161/01.cir.75.4.857. [DOI] [PubMed] [Google Scholar]
- 13.Mulieri LA, Hasenfuss G, Leavitt B, Allen PD, Alpert NR. Altered myocardial force-frequency relation in human heart failure. Circulation. 1992;85(5):1743–1750. doi: 10.1161/01.cir.85.5.1743. [DOI] [PubMed] [Google Scholar]
- 14.Davies CH, et al. Reduced contraction and altered frequency response of isolated ventricular myocytes from patients with heart failure. Circulation. 1995;92(9):2540–2549. doi: 10.1161/01.cir.92.9.2540. [DOI] [PubMed] [Google Scholar]
- 15.Carll AP, Willis MS, Lust RM, Costa DL, Farraj AK. Merits of non-invasive rat models of left ventricular heart failure. Cardiovasc Toxicol. 2011;11(2):91–112. doi: 10.1007/s12012-011-9103-5. [DOI] [PubMed] [Google Scholar]
- 16.Kelso EJ, et al. Mechanical effects of ET-1 in cardiomyocytes isolated from normal and heart-failed rabbits. Mol Cell Biochem. 1996;157(1-2):149–155. doi: 10.1007/BF00227893. [DOI] [PubMed] [Google Scholar]
- 17.Frank D, et al. Gene expression pattern in biomechanically stretched cardiomyocytes: evidence for a stretch-specific gene program. Hypertension. 2008;51(2):309–318. doi: 10.1161/HYPERTENSIONAHA.107.098046. [DOI] [PubMed] [Google Scholar]
- 18.Gopalan SM, et al. Anisotropic stretch-induced hypertrophy in neonatal ventricular myocytes micropatterned on deformable elastomers. Biotechnol Bioeng. 2003;81(5):578–587. doi: 10.1002/bit.10506. [DOI] [PubMed] [Google Scholar]
- 19.Blaauw E, et al. Stretch-induced hypertrophy of isolated adult rabbit cardiomyocytes. Am J Physiol Heart Circ Physiol. 2010;299(3):H780–H787. doi: 10.1152/ajpheart.00822.2009. [DOI] [PubMed] [Google Scholar]
- 20.Kensah G, et al. A novel miniaturized multimodal bioreactor for continuous in situ assessment of bioartificial cardiac tissue during stimulation and maturation. Tissue Eng Part C Methods. 2011;17(4):463–473. doi: 10.1089/ten.tec.2010.0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rana OR, et al. A simple device to apply equibiaxial strain to cells cultured on flexible membranes. Am J Physiol Heart Circ Physiol. 2008;294(1):H532–H540. doi: 10.1152/ajpheart.00649.2007. [DOI] [PubMed] [Google Scholar]
- 22.Bhadriraju K, Chen CS. Engineering cellular microenvironments to improve cell-based drug testing. Drug Discov Today. 2002;7(11):612–620. doi: 10.1016/s1359-6446(02)02273-0. [DOI] [PubMed] [Google Scholar]
- 23.Balachandran K, et al. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci USA. 2011;108(50):19943–19948. doi: 10.1073/pnas.1106954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grosberg A, Alford PW, McCain ML, Parker KK. Ensembles of engineered cardiac tissues for physiological and pharmacological study: Heart on a chip. Lab Chip. 2011;11(24):4165–4173. doi: 10.1039/c1lc20557a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alford PW, et al. Blast-induced phenotypic switching in cerebral vasospasm. Proc Natl Acad Sci USA. 2011;108(31):12705–12710. doi: 10.1073/pnas.1105860108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhuang J, Yamada KA, Saffitz JE, Kléber AG. Pulsatile stretch remodels cell-to-cell communication in cultured myocytes. Circ Res. 2000;87(4):316–322. doi: 10.1161/01.res.87.4.316. [DOI] [PubMed] [Google Scholar]
- 27.Yamada K, Green KG, Samarel AM, Saffitz JE. Distinct pathways regulate expression of cardiac electrical and mechanical junction proteins in response to stretch. Circ Res. 2005;97(4):346–353. doi: 10.1161/01.RES.0000178788.76568.8a. [DOI] [PubMed] [Google Scholar]
- 28.Greenbaum RA, Ho SY, Gibson DG, Becker AE, Anderson RH. Left ventricular fibre architecture in man. Br Heart J. 1981;45(3):248–263. doi: 10.1136/hrt.45.3.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alford PW, Feinberg AW, Sheehy SP, Parker KK. Biohybrid thin films for measuring contractility in engineered cardiovascular muscle. Biomaterials. 2010;31(13):3613–3621. doi: 10.1016/j.biomaterials.2010.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barry SP, Davidson SM, Townsend PA. Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol. 2008;40(10):2023–2039. doi: 10.1016/j.biocel.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 31.Eichler GS, Huang S, Ingber DE. Gene Expression Dynamics Inspector (GEDI): For integrative analysis of expression profiles. Bioinformatics. 2003;19(17):2321–2322. doi: 10.1093/bioinformatics/btg307. [DOI] [PubMed] [Google Scholar]
- 32.Sheehy SP, Huang S, Parker KK. Time-warped comparison of gene expression in adaptive and maladaptive cardiac hypertrophy. Circ Cardiovasc Genet. 2009;2(2):116–124. doi: 10.1161/CIRCGENETICS.108.806935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eder P, Molkentin JD. TRPC channels as effectors of cardiac hypertrophy. Circ Res. 2011;108(2):265–272. doi: 10.1161/CIRCRESAHA.110.225888. [DOI] [PubMed] [Google Scholar]
- 35.Martínez ML, Heredia MP, Delgado C. Expression of T-type Ca(2+) channels in ventricular cells from hypertrophied rat hearts. J Mol Cell Cardiol. 1999;31(9):1617–1625. doi: 10.1006/jmcc.1999.0998. [DOI] [PubMed] [Google Scholar]
- 36.Xing W, et al. Myocardin induces cardiomyocyte hypertrophy. Circ Res. 2006;98(8):1089–1097. doi: 10.1161/01.RES.0000218781.23144.3e. [DOI] [PubMed] [Google Scholar]
- 37.Torrado M, et al. Myocardin mRNA is augmented in the failing myocardium: expression profiling in the porcine model and human dilated cardiomyopathy. J Mol Med (Berl) 2003;81(9):566–577. doi: 10.1007/s00109-003-0470-7. [DOI] [PubMed] [Google Scholar]
- 38.Cribbs L. T-type calcium channel expression and function in the diseased heart. Channels (Austin) 2010;4(6):447–452. doi: 10.4161/chan.4.6.12870. [DOI] [PubMed] [Google Scholar]
- 39.Heling A, et al. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ Res. 2000;86(8):846–853. doi: 10.1161/01.res.86.8.846. [DOI] [PubMed] [Google Scholar]
- 40.LeGrice IJ, et al. Progression of myocardial remodeling and mechanical dysfunction in the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol. 2012;303(11):H1353–H1365. doi: 10.1152/ajpheart.00748.2011. [DOI] [PubMed] [Google Scholar]
- 41.Parker KK, Ingber DE. Extracellular matrix, mechanotransduction and structural hierarchies in heart tissue engineering. Philos Trans R Soc Lond B Biol Sci. 2007;362(1484):1267–1279. doi: 10.1098/rstb.2007.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terracio L, et al. Expression of collagen binding integrins during cardiac development and hypertrophy. Circ Res. 1991;68(3):734–744. doi: 10.1161/01.res.68.3.734. [DOI] [PubMed] [Google Scholar]
- 43.Clemente CF, et al. Focal adhesion kinase governs cardiac concentric hypertrophic growth by activating the AKT and mTOR pathways. J Mol Cell Cardiol. 2012;52(2):493–501. doi: 10.1016/j.yjmcc.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 44.Lu H, et al. Integrin-linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice. Circulation. 2006;114(21):2271–2279. doi: 10.1161/CIRCULATIONAHA.106.642330. [DOI] [PubMed] [Google Scholar]
- 45.Pong T, et al. Hierarchical architecture influences calcium dynamics in engineered cardiac muscle. Exp Biol Med (Maywood) 2011;236(3):366–373. doi: 10.1258/ebm.2010.010239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pope AJ, Sands GB, Smaill BH, LeGrice IJ. Three-dimensional transmural organization of perimysial collagen in the heart. Am J Physiol Heart Circ Physiol. 2008;295(3):H1243–H1252. doi: 10.1152/ajpheart.00484.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strijkers GJ, et al. Diffusion tensor imaging of left ventricular remodeling in response to myocardial infarction in the mouse. NMR Biomed. 2009;22(2):182–190. doi: 10.1002/nbm.1299. [DOI] [PubMed] [Google Scholar]
- 48.Feinberg AW, et al. Controlling the contractile strength of engineered cardiac muscle by hierarchal tissue architecture. Biomaterials. 2012;33(23):5732–5741. doi: 10.1016/j.biomaterials.2012.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 50.Simpson DG, Majeski M, Borg TK, Terracio L. Regulation of cardiac myocyte protein turnover and myofibrillar structure in vitro by specific directions of stretch. Circ Res. 1999;85(10):e59–e69. doi: 10.1161/01.res.85.10.e59. [DOI] [PubMed] [Google Scholar]
- 51.Gaasch WH. Diagnosis and treatment of heart failure based on left ventricular systolic or diastolic dysfunction. JAMA. 1994;271(16):1276–1280. [PubMed] [Google Scholar]
- 52.Berry MF, et al. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol. 2006;290(6):H2196–H2203. doi: 10.1152/ajpheart.01017.2005. [DOI] [PubMed] [Google Scholar]
- 53.Huh D, et al. Reconstituting organ-level lung functions on a chip. Science. 2010;328(5986):1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huh D, Torisawa YS, Hamilton GA, Kim HJ, Ingber DE. Microengineered physiological biomimicry: Organs-on-chips. Lab Chip. 2012;12(12):2156–2164. doi: 10.1039/c2lc40089h. [DOI] [PubMed] [Google Scholar]
- 55.Hirt MN, et al. Increased afterload induces pathological cardiac hypertrophy: a new in vitro model. Basic Res Cardiol. 2012;107(6):307. doi: 10.1007/s00395-012-0307-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davis RP, van den Berg CW, Casini S, Braam SR, Mummery CL. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends Mol Med. 2011;17(9):475–484. doi: 10.1016/j.molmed.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 57.Bray MA, Sheehy SP, Parker KK. Sarcomere alignment is regulated by myocyte shape. Cell Motil Cytoskeleton. 2008;65(8):641–651. doi: 10.1002/cm.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Irizarry RA, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 60.Howlett SE, Grandy SA, Ferrier GR. Calcium spark properties in ventricular myocytes are altered in aged mice. Am J Physiol Heart Circ Physiol. 2006;290(4):H1566–H1574. doi: 10.1152/ajpheart.00686.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.