

Abstract
Multidrug resistance is a serious barrier to successful treatment of many human diseases, including cancer, wherein chemotherapeutics are exported from target cells by membrane-embedded pumps. The most prevalent of these pumps, the ATP-Binding Cassette transporter P-glycoprotein (P-gp), consists of two homologous halves each comprising one nucleotide-binding domain and six transmembrane helices. The transmembrane region encapsulates a hydrophobic cavity, accessed by portals in the membrane, that binds cytotoxic compounds as well as lipids and peptides. Here we use mass spectrometry (MS) to probe the intact P-gp small molecule-bound complex in a detergent micelle. Activation in the gas phase leads to formation of ions, largely devoid of detergent, yet retaining drug molecules as well as charged or zwitterionic lipids. Measuring the rates of lipid binding and calculating apparent KD values shows that up to six negatively charged diacylglycerides bind more favorably than zwitterionic lipids. Similar experiments confirm binding of cardiolipins and show that prior binding of the immunosuppressant and antifungal antibiotic cyclosporin A enhances subsequent binding of cardiolipin. Ion mobility MS reveals that P-gp exists in an equilibrium between different states, readily interconverted by ligand binding. Overall these MS results show how concerted small molecule binding leads to synergistic effects on binding affinities and conformations of a multidrug efflux pump.
Keywords: mass spectrometry from native state, real time substrate monitoring
P-glycoprotein (P-gp) is an ATP-driven low-specificity efflux pump that plays an important role in the clearance of xenotoxins (1, 2). P-gp is also a member of the ATP-Binding Cassette (ABC) family of transporters and exports hydrophobic cytotoxic compounds as well as natural products, cyclic, and linear peptides (1, 3–5). Overexpression of P-gp in tumor cells impairs targeted drug delivery and is a major pitfall for chemotherapies. Small molecule substrates partition in the plasma membrane (6, 7), before binding in the internal hydrophobic cavity formed in the inward conformation of the pump (8). Export is then thought to proceed in an ATP-dependent way through conformational changes from the inward to the outward facing forms, evidenced by FRET spectroscopy (9). Recent high-resolution structures of eukaryotic P-gp from mouse (8) and Caenorhabditis elegans (10) were obtained in inward conformations. Two prokaryotic homologs of P-gp [Sav1866 (11) and MsbA (12)] were captured crystallographically in outward-facing conformers, reflecting ATP-bound states, as well as two different inward states for MsbA. From these X-ray structures it is possible to build up a picture of P-gp, alternating between inward- and outward-facing conformations.
Despite decades of careful biochemical studies (13), and recent insights from crystallography, many questions remain, however. Specifically it has not yet been possible to trap P-gp in an outward-facing state or to show how substrate binding activates ATPase activity. Furthermore since ATPase activity and drug efflux of P-gp are known to be influenced by detergent (14–16), the membrane bilayer (10, 17), and lipid binding (16, 18), monitoring drug binding against this background of lipid and detergent transport presents a major challenge.
Recently MS has been applied to the study of intact membrane protein complexes. By projecting these complexes into the gas phase, from detergent micelles formed in solution, intact membrane assemblies have been transmitted through a mass spectrometer revealing the subunit stoichiometry and presence of endogenous lipids or nucleotides (19–21). Conformational and compositional heterogeneity, captured in the gas phase, can be resolved using MS without resorting to additional separative methods. One of the remaining challenges for MS of membrane complexes, however, has been to study drug binding. Given the high concentration of the detergent that has to be removed, via collisional activation in the gas phase, together with the backdrop of lipid binding, preservation of drug binding is a significant challenge.
Here we use MS to study binding of lipids, nucleotides, and drugs to P-gp. Our results show that P-gp is able to bind two molecules of cyclosporin A (CsA), a cyclic peptide inhibitor used to restore the sensitivity of cancer cells to chemotherapeutic agents. Real-time monitoring enabled us to study binding of a variety of different lipids, revealing their stoichiometry, binding rates, and relative affinity. Preference was observed for negatively charged over zwitterionic double chain lipids. Binding was also monitored for bulkier four-chain cardiolipins (CLs) where negative charge promotes binding, with shorter hydrocarbon chains binding preferentially over their longer chain counterparts. Competition experiments show that P-gp can bind simultaneously to lipids and CsA. Based on these experiments we used molecular docking to assemble models of the inward conformation of P-gp bound to CsA and CLs. These models were stable in lipid bilayer molecular dynamics (MD) simulations, supporting our experimental data. Ion mobility (IM) coupled to MS revealed the surprising result that nucleotide and substrate binding are able to perturb the equilibrium between two states, increasing the population of a smaller conformation. Overall this study highlights the ability to probe ligand binding to membrane proteins in the gas phase and to observe directly allosteric effects.
Results
Mass Spectra of P-gp Reveal Detergent Binding.
The mass spectrum of mouse P-gp was obtained following nanoflow electrospray from a solution of n-dodecyl-β-d-maltoside (DDM), a detergent in which the protein remains fully active (8). Using a gas phase activation strategy described previously (19), we obtained a relatively broad distribution of charge states (22+ to 29+) (Fig. 1A). While the vast majority of P-gp molecules are in the apo form, less than 50% have one or two DDM attached while only 10% carry three or more DDM molecules. Of the ∼100 DDM molecules that constitute the micelle in solution (22), a very small subset are retained (Fig. 1A and Fig. S1). This is in contrast to observations for other membrane complexes in which all detergent molecules are lost during gas phase activation (20). We speculate that the low numbers of DDM attached are partially sequestered in the hydrophobic cavity, consistent with the ability of P-gp to transport detergent as substrates and with the X-ray structure of P-gp from C. elegans wherein two molecules of n-undecyl-β-maltoside (UDM) were located inside the cavity (10, 15).
Fig. 1.

Interaction of P-gp with detergent and phospholipids. Mass spectra of P-gp recorded following release in the gas phase from detergent micelles formed in solution from DDM at 2 × the critical micelle concentration (CMC) (A). Selected charge states for mass spectra recorded 15 min after incubation with two equivalents of zwitterionic lipids (POPE and POPC) (B) or anionic lipids (POPA and POPG) (C). Plot of the relative abundance of seven different species [(P-gP POPG)0.0.7] as a function of time (D). Apparent KD values calculated from time courses recorded for binding of the xth lipid where x ≤ 6 (E).
Binding of Lipids to P-gp Reveals a Preference for Negatively Charged Head Groups.
P-gp transports lipids as well as detergents (23–25); we therefore incubated solutions of P-gp in DDM micelles with seven different phosphoglyceride lipids. Monitoring in real-time the peak widths and mass differences over a period of 20 min reveals that lipids bind much more favorably than detergents, displacing DDM with up to four zwitterionic lipids or six anionic lipids in complex with P-gp (Fig. 1 B and C, Fig. S1B, and Table S1). Following the time course for formation of the various lipid-bound states, we see that after 5 min apo forms of P-gp are depleted by ∼35% and ∼50% for zwitterionic and anionic lipids, respectively (Fig. 1D and Fig. S2). Using identical activation conditions, apparent KD values were calculated. These values imply favorable binding for negatively charged 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (POPA) with a µM KD value, similar to anionic lipids with bulkier headgroups (Fig. 1E, Fig. S3, and Table S2). Lower occupancy is observed for zwitterionic lipids with the same hydrophobic chain as the negatively charged lipids. Changes in saturation of the hydrocarbon chains produced much smaller effects than changes in charge of the headgroup.
Reports of P-gp in human mitochondria (26) as well as the many reports of P-gp acting as a flippase (24, 27) prompted us to study binding of mitochondrial CLs (28), with four hydrophobic chains and a net charge (–2). Monitoring the real-time binding of CL14:1 (3)–15:1 (CL14) or CL24:1 (3)–14:1 (CL24) revealed that up to three or one lipid, respectively, could be detected in complex with P-gp after a 20 min incubation period (Fig. 2 A and B). Plotting the evolution of the different lipid-bound states as a function of time allows us to extract apparent KD values (Fig. 2). These KD values are most favorable for negatively charged lipids and CLs bind more favorably than zwitterionic lipids (Fig. 1E and Fig. S4).
Fig. 2.
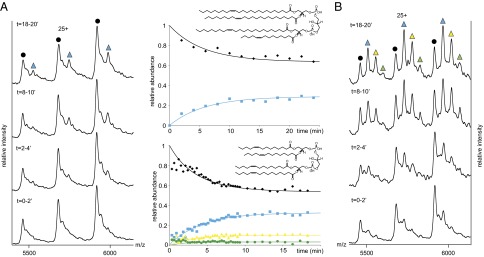
CL binding to P-gp in real time. Two equivalents of (A) CL24 and (B) CL14 were added to P-gp in DDM micelles. Peaks are assigned as apo P-gp (black) and incorporation of the first, second, and third CL, labeled cyan, yellow, and green, respectively. Plots of the relative intensity of the peaks assigned to CL24 (Upper) and CL14, against time (Lower).
To determine how size affects lipid binding, we compared lipids of the same net charge (–2, –1, or 0) on a plot of KD versus the experimental collision cross-section (CCS). We deduced CCS via IM–MS (29–31), a method that reports on the CCS of ions based on their ability to traverse a gas-filled mobility cell under a weak electric field. Results show that the smaller the lipid, the more favorable the interactions with P-gp, suggesting that steric effects are important (Fig. S3A). Lipid binding to P-gp, whether within the cavity or at the membrane interface, is therefore dictated both by steric effects and net charge.
Substrate Binding to P-gp.
A key question concerns the location of the lipids, particularly given recent X-ray structures of ABCB10 in which lipids and detergents were found both within transmembrane helices and outside the cavity [Protein Data Bank (PDB) ID codes 4AYT and 4AYX] and of P-gp wherein two molecules of detergent were observed within the cavity (10). To explore whether lipids could theoretically be accommodated within the cavity, we calculated a maximum molecular volume for all molecules detected in complex with P-gp and multiplied these values by the maximum stoichiometry detected, albeit at very low intensity. The following values were obtained: DDM (∼10*459 Å3 = 4,590 Å3), double-chain phospholipids (∼6*700 Å3 = 4,200 Å3), and CLs 14:1 (3)-15:1 (∼3*1,357 Å3 = 4,071 Å3). Interestingly the close similarity of these values implies that a maximum of six phospholipids, 10 detergents, or three CL14 could be accommodated within the inner cavity (∼6,000 Å3). However, we anticipate that steric repulsion likely leads to substrate binding both inside and outside the pocket.
Since the ligand-binding cavity is formed when P-gp is in an open inward-facing state, we probed the conformations of P-gp in solution using IM–MS. IM data show that apo P-gp exhibits essentially one narrow arrival time distribution (ATD), consistent with predominantly one conformation. A shoulder, left of the major peak, is observed, consistent with a smaller conformation with higher mobility (Fig. 3A). Incubating one equivalent of CL24 for 30 min with P-gp showed no appreciable difference in the population of these two different conformers.
Fig. 3.

Apo P-gp exists predominantly in an open inward conformation. (A) Mass spectra and corresponding ATD for the 26+ charge state recorded after 30 min of incubation of P-gp with one equivalent of CL24. (B) Structures of the P-gp homolog MsbA obtained in the outward (PDB ID code: 3B5Z), inward open (PDB ID code 3B5W), and inward closed (PDB ID code 3B5X) conformations. The CCSs were calculated from the respective PDB files using Driftscope (Waters). (C) Docking of CL14 to P-gp showing the interaction with residues K822 and K230. Oxygen and phosphorous atoms are red and orange, respectively. A simulated spectrum for lipid binding is shown (red line).
To relate these conformations to structures of P-gp, we calculated the CCS difference between the inward- and outward-facing forms of the homolog MsbA (12). We used MsbA structures as MsbA is structurally and functionally very close to P-gp and because there are no published X-ray structures of full-length P-gp in additional conformations. The difference calculated between the outward and open inward conformers of MsbA is 13.5% (Fig. 3 B and C). This value corresponds closely to the experimental difference measured for P-gp (13%). Since the two ATD peaks are well-resolved, they suggest a well-defined difference between two discrete states rather than a continuum of interconverting populations. While we cannot rule out the existence of additional conformations of P-gp or the potential for perturbation of P-gp structure in the gas phase, the close similarity with the difference between the two MsbA structures is compelling.
Using a molecular docking approach (SI Methods), we identified three potential binding pockets for the smaller CL14 in the open inward conformation of P-gp. The first CL14 was located at the top of the cavity in 58% of the models (Fig. S5A). The second CL14 was located in two binding sites beneath the first one in 52% of the models (Fig. S5B). In most cases, hydrophobic interactions with nonpolar residues account for the binding of the small CL14 within the cavity. We also identified electrostatic interactions between phosphate groups and K230 or K822, which could explain the preference for negatively charged lipids (Fig. 3C). In theory the cavity could also accommodate two of the larger CL24 molecules (2*1,916 Å3 = 3,832 Å3). This was not observed in the mass spectra, however, consistent with molecular docking, wherein only one large CL24 can be accommodated within the cavity, in three different orientations (Fig. S5C).
Drug Binding to P-gp.
To determine if drug binding could be retained following release of the membrane protein from the detergent micelle in the gas phase, we added one, five, and 10 equivalents of CsA, an immunosuppressant known to inhibit P-gp ATPase activity (32). Well-resolved peaks were detected for one and two molecules of CsA binding to P-gp (Fig. 4). The broad baseline is attributed to the low-level background of detergent molecules, also present within the cavity. Similar experiments confirmed the binding of six other cyclic peptides (Fig S6A) in line with the polyspecificity of the pocket (8) and highlight the potential for MS to uncover drug binding to P-gp. Moreover, following incubation of drug or lipid with P-gp and another membrane protein in the same solution, we observe binding exclusively to P-gp. This allows us to conclude that the binding observed between P-gp and lipids and drugs is not simply a function of interaction with any hydrophobic membrane protein surface (Fig. S7).
Fig. 4.

CsA binding to P-gp. Nano-electrospray mass spectra obtained before (Lower) and after (Upper) 5 min incubation with 1, 5, and 10 equivalents of CsA. Peaks assigned to apo P-gp and DDM are labeled (black and red, respectively). Molecular docking reveals the likely location of the first (cyan) and second (yellow) CsA molecule to P-gp. The presence of a low level of DDM molecules in addition to CsA is implied through the high mass tail on all peaks.
In support of this experimental data, molecular docking located the first CsA in 98% of all models at the top of the cavity (Fig. S5D). For the second CsA, 85% have CsA molecules located in the same binding site, beneath the first one. Two other clusters were found with the second CsA binding either inside or at the entrance of the cavity (Fig. S5E). The third CsA was located inside the cavity or at the entrance portal (Fig. S5F). MD simulations of P-gp, bound to different numbers of CsA and CL14, were performed after molecular docking and energy refinement. Within the timescale of the simulations (∼100 ns), we observed greater rmsd of both CsA and CL14 atoms (i.e., reduced stability) for systems with more than two ligand molecules docked in the binding site (Fig. S8 A–D). This is in accord with the low-intensity MS peak corresponding to binding of the third CsA/CL14. MD simulations, following molecular docking, also revealed that CsA and CLs molecules were more stable at the apex and bottom of the cavity, respectively, than in alternative locations (Fig. S8 E–G). Irrespective of the binding site, however, CL14 was found to be more stable in the cavity than CsA, providing a rationale for the ready dissociation of CsA observed in MS (Figs. S6B and S8G). Interestingly, for the two different CLs and CsA, ∼10% of molecules found in the TM region, but outside the cavity, were located at an entrance portal (Fig. S5, pink clusters). The position of these molecules, at the same location as the hydrophobic tail of UDM in the recent X-ray structure (10), suggests a favored access route into the cavity. Given the close agreement between binding stoichiometry, relative size of ligands, and dimensions of the cleft, it is tempting to speculate that the ligands bind inside the cavity. In a physiological context, steric repulsion between hydrophobic molecules would likely prevent full occupancy of the cavity and alternative modes of interactions, along the favored access route, are highly probable.
Concomitant Binding of Lipids, Nucleotides, and Drugs.
To explore simultaneous binding of lipids, nucleotides, and drugs to P-gp, we first incubated apo P-gp in a solution containing DDM micelles separately with each component to establish the extent of binding. Addition of a 20-fold excess of ATP to apo P-gp revealed two further well-resolved peaks, consistent with binding of one and two nucleotides. Similar results were obtained for a nonhydrolysable analog ATPγS. Under identical MS conditions, but with a fivefold excess of CsA, two further well-resolved peaks are attributed to CsA-bound forms.
Since lipids could affect entry of drugs (8) or adapt the cavity for drug recognition (33), we added lipids before binding of CsA and recorded the mass spectrum. Comparison (Fig. 5B, i, red line) with the summed mass spectral peaks for lipid and drug binding alone (red lines Figs. 3C and 5A, i) reveals no appreciable difference in intensity implying that lipid binding does not affect subsequent drug binding. Interestingly, however, if P-gp is preincubated with CsA, an unexpected ∼twofold increase in binding is observed for CL24 or CL14 (Fig. 5B, i, red line and Fig. S9). These results imply, therefore, that CsA is depleted when binding of CL takes place, possibly through conformational changes or adaptation of the cavity that favors CL binding at the expense of CsA.
Fig. 5.

Binding of nucleotides lipids, and drugs. Spectra recorded after 30 min of incubation of P-gp with 5, 20, or one equivalent of CsA, ATP, or CL24 individually (A, i) or concomitantly as indicated (B, i). Binding of the first and second ATP, CsA, and CL (24)3–14 are labeled cyan and yellow, respectively. Simulated spectra for lipid and drug binding alone and summed for concomitant binding are shown (red lines). Corresponding ATDs for the 26+ charge state of P-gp and ligands as indicated (A, ii and B, ii). Different conformations are represented schematically with predominant species in darker green.
Conformational changes of P-gp associated with concomitant ligand binding were investigated using IM–MS. First the effects of individual ligand binding were established and similar to binding of CL14, CsA, ATP, or nonhydrolysable nucleotides with Mg2+ showed no significant shift in the equilibrium (Fig. 5A, ii and Fig. S9 D and E). Surprisingly, however, when P-gp is incubated concomitantly with nucleotide and CsA, a significant increase in population of the smaller conformer occurs (Fig. 5B, ii and Fig. S9D). Similarly, when CL and CsA are present simultaneously, population of the two conformers is approximately equal. We conclude, therefore, that concomitant binding of nucleotides, lipids, or drugs induces a greater population of the smaller conformer than would be anticipated for independent binding of small molecules.
Discussion
We have shown that we can detect independent and simultaneous binding of lipids, nucleotides, and drugs to a membrane protein complex using MS. Monitoring lipid binding in real-time demonstrated a preference for lipids with an overall negative charge and showed that bulky lipids, such as CLs, were also able to access the hydrophobic pocket. Relative KD values were obtained from time course reactions and found to be in the μM range. These values correlate well with values obtained for other small molecules binding to P-gp, demonstrating the potential for MS to deliver relative affinity measurements (34).
Binding of CsA, or six further cyclic peptides, to P-gp was also studied using MS. The observation of drug binding is a significant advance given the difficulties of introducing these complexes in detergent micelles and the background of detergent and lipid molecules transported by this indiscriminate pump. Our results are consistent with CsA adapting the cavity for subsequent lipid binding. It is interesting to speculate that CsA binding occurs at the apex of the hydrophobic pocket, the favorable binding sites defined by molecular docking and identified crystallographically (8). This CsA binding event holds P-gp in an open conformation for subsequent enhanced CL binding, which then promotes expulsion of CsA. Given that CL also likely binds at the apex of the binding pocket (Fig. S5) but that subsequent CsA binding was not enhanced, we propose that the long hydrocarbon chains of the CLs stabilize the open conformation of P-gp but do not enhance CsA binding, possibly due to steric clashes. Consistent with this proposal, subsequent CsA binding is less favorable with prior binding of CL24 than with CL14 (Fig. S9C). Overall, therefore, while both CL and CsA could stabilize open conformations of P-gp for subsequent binding, the effect is only observed with CsA as there are fewer potential steric clashes for the incoming CL.
The two conformations identified by IM–MS were unanticipated. The close correlation between values for the differences calculated for open and closed MsbA structures and the experimental difference measured here imply that P-gp exists predominantly in a larger conformation, most likely the inward-facing conformer defined crystallographically (8). Independent binding of nucleotides, lipids, or CsAs is not sufficient to induce a significant shift in the equilibrium. Given the preponderance of X-ray structures for related ABC transporters, with nucleotide-binding driving formation of an outward state (12, 20, 35, 36), we might have expected an increased population of the smaller conformer. Recently, however, an ABC transporter (ABCB10) was crystallized, with nucleotides present, in an inward conformation (PDB ID codes 4AYT and 4AYX). While apo forms of P-gp from mouse and C. elegans were crystallized in the open inward conformation, as was the GltPh aspartate transporter, the latter was shown by EPR spectroscopy to be equally distributed between outward- and inward-facing states (37, 38). This result, together with the absence of X-ray structures for P-gp with nucleotides or CsA bound, in an outward-facing form (39), imply that nucleotide or drug binding alone is not sufficient to stabilize an outward-facing conformer (9, 37, 38). Concomitant binding of nucleotides, lipids, and drugs did, however, show an increase in the population of a smaller conformer. Our results, together with those from FRET, which monitored the distances between nucleotide-binding domain during drug and/or nucleotide binding and found them to be independent and additive events (9), allow us to speculate that the smaller conformer arises from an outward-facing form. Consequently, P-gp exists in a delicately balanced equilibrium, predominately in the open inward form.
Synergistic binding of lipids, nucleotides, and drugs appears sufficient to perturb the equilibrium between states, populating more of the outward-facing state, poised for efflux. We cannot rule out the possibility that lipids might affect conformational dynamics through binding to the surface of the protein. The stoichiometry obtained for the CL binding, however, is in good agreement with docking in the cavity, indicating that lipids might modify the conformational state of P-gp either through binding at the membrane interface or from within the cavity. Since it is established that drugs can affect the distribution of lipids within membranes (40), and given that lipids and drugs are transported indiscriminately through P-gp (41), distinguishing the effects of small molecule binding in this way is an exciting advance. Conformational states of membrane transporters are currently studied mainly by FRET (9) or EPR spectroscopy (37, 38) where labeled residues are inserted in the protein of interest, providing distance measurement between residues. However, such methods cannot distinguish directly the effects of unlabeled lipids and drugs when added simultaneously. As such, the method exemplified here, for probing the effects of drugs and lipids on the equilibrium in P-gp, is likely to have important consequences for studying drug binding to a broad range of membrane protein complexes.
Methods
Materials.
Lipids were purchased from Avanti Polar Lipids. CsA, ammonium acetate, and nucleotides were purchased from Sigma-Aldrich.
Protein Purification.
His-tagged P-gp was expressed in Pichia pastoris and purified as described previously (8). Before analysis P-gp was buffer exchanged into 200 mM ammonium acetate pH 7 supplemented with 0.02% DDM, using micro Bio-Spin-6 devices (Bio-Rad Laboratories).
Ligand Binding.
The protein concentration was determined with the bicinchoninic acid protein assay kit (Pierce) using BSA as a reference. Lipids were first prepared at 2 mM in chloroform and then diluted to 20 μM in 200 mM ammonium acetate pH 7 supplemented with 0.02% DDM. Cyclosporin was first dissolved at 8 mM in methanol and then diluted to 5–100 μM in 200 mM ammonium acetate pH 7, supplemented with 0.02% DDM.
Mass Spectrometry.
Samples were introduced into the ion source via gold-coated nanoflow capillaries prepared in house (42). Calibration was performed using a 10 mg/mL solution of cesium iodide. MS and IM–MS spectra were acquired on a Q-TOF2 (43) and a Synapt2 (44) (Micromass) modified for the transmission and detection of high-mass complexes. Parameters were set as described in SI Methods. Theoretical CCSs were calculated using the projection approximation method implemented in MOBCAL and scaled using a scaling factor of 1.14, as described previously (45).
Simulation of Spectra and Kinetic Fits.
Data analysis was performed using our in-house software Massign described recently (46) (SI Methods).
Modeling and Hierarchical Clustering.
The docking of CsA and CL was performed using Patchdock and refined with FireDock. The best scored models were clustered using in-house scripts as described in SI Methods.
MD Simulations.
All MD simulations were performed at constant temperature (300K) using GROMACS v4.5.4 at 300K (47) (SI Methods).
Supplementary Material
Acknowledgments
We thank Elizabeth Carpenter for helpful discussion and Martin Caffrey for providing diacylglycerol kinase alpha. We also acknowledge funding from European Research Council Integral Membrane Proteins Resolution of Stoichiometry and Structure, the Royal Society, and The Wellcome Trust.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303888110/-/DCSupplemental.
References
- 1.Darby RA, Callaghan R, McMahon RM. P-glycoprotein inhibition: The past, the present and the future. Curr Drug Metab. 2011;12(8):722–731. doi: 10.2174/138920011798357006. [DOI] [PubMed] [Google Scholar]
- 2.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 3.Sharom FJ, Lu P, Liu R, Yu X. Linear and cyclic peptides as substrates and modulators of P-glycoprotein: Peptide binding and effects on drug transport and accumulation. Biochem J. 1998;333(Pt 3):621–630. doi: 10.1042/bj3330621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharom FJ. Shedding light on drug transport: Structure and function of the P-glycoprotein multidrug transporter (ABCB1) Biochem Cell Biol. 2006;84(6):979–992. doi: 10.1139/o06-199. [DOI] [PubMed] [Google Scholar]
- 5.Sauna ZE, Ambudkar SV. About a switch: How P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol Cancer Ther. 2007;6(1):13–23. doi: 10.1158/1535-7163.MCT-06-0155. [DOI] [PubMed] [Google Scholar]
- 6.Dey S, Ramachandra M, Pastan I, Gottesman MM, Ambudkar SV. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc Natl Acad Sci USA. 1997;94(20):10594–10599. doi: 10.1073/pnas.94.20.10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu P, Liu R, Sharom FJ. Drug transport by reconstituted P-glycoprotein in proteoliposomes. Effect of substrates and modulators, and dependence on bilayer phase state. Eur J Biochem. 2001;268(6):1687–1697. [PubMed] [Google Scholar]
- 8.Aller SG, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323(5922):1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verhalen B, Ernst S, Börsch M, Wilkens S. Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. J Biol Chem. 2012;287(2):1112–1127. doi: 10.1074/jbc.M111.301192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin MS, Oldham ML, Zhang Q, Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature. 2012;490(7421):566–569. doi: 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawson RJP, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443(7108):180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 12.Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci USA. 2007;104(48):19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharom FJ. The P-glycoprotein multidrug transporter. Essays Biochem. 2011;50(1):161–178. doi: 10.1042/bse0500161. [DOI] [PubMed] [Google Scholar]
- 14.Regev R, Katzir H, Yeheskely-Hayon D, Eytan GD. Modulation of P-glycoprotein-mediated multidrug resistance by acceleration of passive drug permeation across the plasma membrane. FEBS J. 2007;274(23):6204–6214. doi: 10.1111/j.1742-4658.2007.06140.x. [DOI] [PubMed] [Google Scholar]
- 15.Li-Blatter X, Nervi P, Seelig A. Detergents as intrinsic P-glycoprotein substrates and inhibitors. Biochim Biophys Acta. 2009;1788(10):2335–2344. doi: 10.1016/j.bbamem.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 16.Doige CA, Yu X, Sharom FJ. The effects of lipids and detergents on ATPase-active P-glycoprotein. Biochim Biophys Acta. 1993;1146(1):65–72. doi: 10.1016/0005-2736(93)90339-2. [DOI] [PubMed] [Google Scholar]
- 17.Clay AT, Sharom FJ. Lipid bilayer properties control membrane partitioning, binding, and transport of p-glycoprotein substrates. Biochemistry. 2013;52(2):343–354. doi: 10.1021/bi301532c. [DOI] [PubMed] [Google Scholar]
- 18.Sharom FJ, Yu X, Chu JW, Doige CA. Characterization of the ATPase activity of P-glycoprotein from multidrug-resistant Chinese hamster ovary cells. Biochem J. 1995;308(Pt 2):381–390. doi: 10.1042/bj3080381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrera NP, Di Bartolo N, Booth PJ, Robinson CV. Micelles protect membrane complexes from solution to vacuum. Science. 2008;321(5886):243–246. doi: 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- 20.Velamakanni S, et al. A multidrug ABC transporter with a taste for salt. PLoS ONE. 2009;4(7):e6137. doi: 10.1371/journal.pone.0006137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou M, et al. Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science. 2011;334(6054):380–385. doi: 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.le Maire M, Champeil P, Moller JV. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim Biophys Acta. 2000;1508(1-2):86–111. doi: 10.1016/s0304-4157(00)00010-1. [DOI] [PubMed] [Google Scholar]
- 23.Romsicki Y, Sharom FJ. The membrane lipid environment modulates drug interactions with the P-glycoprotein multidrug transporter. Biochemistry. 1999;38(21):6887–6896. doi: 10.1021/bi990064q. [DOI] [PubMed] [Google Scholar]
- 24.Romsicki Y, Sharom FJ. Phospholipid flippase activity of the reconstituted P-glycoprotein multidrug transporter. Biochemistry. 2001;40(23):6937–6947. doi: 10.1021/bi0024456. [DOI] [PubMed] [Google Scholar]
- 25.Eckford PD, Sharom FJ. The reconstituted P-glycoprotein multidrug transporter is a flippase for glucosylceramide and other simple glycosphingolipids. Biochem J. 2005;389(Pt 2):517–526. doi: 10.1042/BJ20050047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roundhill EA, Burchill SA. Detection and characterisation of multi-drug resistance protein 1 (MRP-1) in human mitochondria. Br J Cancer. 2012;106(6):1224–1233. doi: 10.1038/bjc.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharom FJ. Flipping and flopping—Lipids on the move. IUBMB Life. 2011;63(9):736–746. doi: 10.1002/iub.515. [DOI] [PubMed] [Google Scholar]
- 28.Houtkooper RH, Vaz FM. Cardiolipin, the heart of mitochondrial metabolism. Cell Mol Life Sci. 2008;65(16):2493–2506. doi: 10.1007/s00018-008-8030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clemmer DE, Hudgins RR, Jarrold MF. Naked protein conformations: Cytochrome C in the gas phase. J Am Chem Soc. 1995;117(40):10141–10142. [Google Scholar]
- 30.Gill AC, Jennings KR, Wyttenbach T, Bowers MT. Conformations of biopolymers in the gas phase: A new mass spectrometric method. Int J Mass Spectrom. 2000;195(SI):685–697. [Google Scholar]
- 31.Ruotolo BT, et al. Evidence for macromolecular protein rings in the absence of bulk water. Science. 2005;310(5754):1658–1661. doi: 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- 32.Loor F, Tiberghien F, Wenandy T, Didier A, Traber R. Cyclosporins: Structure-activity relationships for the inhibition of the human MDR1 P-glycoprotein ABC transporter. J Med Chem. 2002;45(21):4598–4612. doi: 10.1021/jm0109863. [DOI] [PubMed] [Google Scholar]
- 33.Kimura Y, Kioka N, Kato H, Matsuo M, Ueda K. Modulation of drug-stimulated ATPase activity of human MDR1/P-glycoprotein by cholesterol. Biochem J. 2007;401(2):597–605. doi: 10.1042/BJ20060632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melchior DL, et al. Determining P-glycoprotein-drug interactions: Evaluation of reconstituted P-glycoprotein in a liposomal system and LLC-MDR1 polarized cell monolayers. J Pharmacol Toxicol Methods. 2012;65(2):64–74. doi: 10.1016/j.vascn.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hollenstein K, Dawson RJ, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007;17(4):412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Seeger MA, van Veen HW. Molecular basis of multidrug transport by ABC transporters. Biochim Biophys Acta. 2009;1794(5):725–737. doi: 10.1016/j.bbapap.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 37.Hänelt I, Wunnicke D, Bordignon E, Steinhoff HJ, Slotboom DJ. Conformational heterogeneity of the aspartate transporter Glt(Ph) Nat Struct Mol Biol. 2013;20(2):210–214. doi: 10.1038/nsmb.2471. [DOI] [PubMed] [Google Scholar]
- 38.Georgieva ER, Borbat PP, Ginter C, Freed JH, Boudker O. Conformational ensemble of the sodium-coupled aspartate transporter. Nat Struct Mol Biol. 2013;20(2):215–221. doi: 10.1038/nsmb.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tao H, et al. Design and synthesis of Selenazole-containing peptides for cocrystallization with P-glycoprotein. ChemBioChem. 2011;12(6):868–873. doi: 10.1002/cbic.201100048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eckford PD, Sharom FJ. Interaction of the P-glycoprotein multidrug efflux pump with cholesterol: effects on ATPase activity, drug binding and transport. Biochemistry. 2008;47(51):13686–13698. doi: 10.1021/bi801409r. [DOI] [PubMed] [Google Scholar]
- 41.Eckford PD, Sharom FJ. P-glycoprotein (ABCB1) interacts directly with lipid-based anti-cancer drugs and platelet-activating factors. Biochem Cell Biol. 2006;84(6):1022–1033. doi: 10.1139/o06-196. [DOI] [PubMed] [Google Scholar]
- 42.Hernández H, Robinson CV. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat Protoc. 2007;2(3):715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- 43.Sobott F, Hernández H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal Chem. 2002;74(6):1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 44.Bush MF, et al. Collision cross sections of proteins and their complexes: A calibration framework and database for gas-phase structural biology. Anal Chem. 2010;82(22):9557–9565. doi: 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- 45.Hall Z, Politis A, Bush MF, Smith LJ, Robinson CV. Charge-state dependent compaction and dissociation of protein complexes: Insights from ion mobility and molecular dynamics. J Am Chem Soc. 2012;134(7):3429–3438. doi: 10.1021/ja2096859. [DOI] [PubMed] [Google Scholar]
- 46.Morgner N, Robinson CV. Massign: An assignment strategy for maximizing information from the mass spectra of heterogeneous protein assemblies. Anal Chem. 2012;84(6):2939–2948. doi: 10.1021/ac300056a. [DOI] [PubMed] [Google Scholar]
- 47.Van Der Spoel D, et al. GROMACS: Fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.