

Abstract
The regulation of gonadotropin synthesis by GnRH plays an essential role in the neuroendocrine control of reproduction. The known signaling mechanisms involved in gonadotropin synthesis have been expanding. For example, involvement of β-catenin in LHβ induction by GnRH has been discovered. We examined the role of β-catenin in FSHβ gene expression in LβT2 gonadotrope cells. GnRH caused a sustained increase in nuclear β-catenin levels, which was significantly reduced by c-Jun N-terminal kinase (JNK) inhibition. Small interfering RNA-mediated knockdown of β-catenin mRNA demonstrated that induction of FSHβ mRNA by GnRH depended on β-catenin and that regulation of FSHβ by β-catenin occurred independently of the JNK-c-jun pathway. β-Catenin depletion had no impact on FSHβ mRNA stability. In LβT2 cells transfected with FSHβ promoter luciferase fusion constructs, GnRH responsiveness was conferred by the proximal promoter (−944/−1) and was markedly decreased by β-catenin knockdown. However, none of the T-cell factor/lymphoid enhancer factor binding sites in that region were required for promoter activation by GnRH. Chromatin immunoprecipitation further corroborated the absence of direct interaction between β-catenin and the 1.8-kb FSHβ promoter. To elucidate the mechanism for the β-catenin effect, we analyzed approximately 1 billion reads of next-generation RNA sequencing β-catenin knockdown assays and selected the nuclear cofactor breast cancer metastasis-suppressor 1-like (Brms1L) as one candidate for further study. Subsequent experiments confirmed that Brms1L mRNA expression was decreased by β-catenin knockdown as well as by JNK inhibition. Furthermore, knockdown of Brms1L significantly attenuated GnRH-induced FSHβ expression. Thus, our findings indicate that the expression of Brms1L depends on β-catenin activity and contributes to FSHβ induction by GnRH.
GnRH plays a key role in the neuroendocrine control of reproduction through the stimulation of biosynthesis and secretion of the gonadotropins (LH and FSH) by pituitary gonadotropes. LH and FSH are heterodimeric glycoproteins composed of a common α-subunit (CGA) and a specific β-subunit (LHβ and FSHβ, respectively) (1). The pulsatile release of GnRH is crucial for normal gonadal development and function, because it determines the pattern of gonadotropin differential expression: whereas LHβ gene expression increases much more than FSHβ at higher GnRH pulse frequencies, FSHβ gene expression is favored at lower GnRH pulse frequencies (2). The establishment of the immortalized gonadotrope cell lines αT3–1 and LβT2 has broadened our understanding of GnRH signaling (3–7). GnRH receptor (GnRHR) coupling to Gαq/11 leads to activation of the protein kinase C (PKC)-dependent pathway, followed by several MAPK cascades including ERK, c-Jun N-terminal kinase (JNK), and p38MAPK, and subsequent regulation of gonadotropin subunit gene expression (for review, see Ref. 8). GnRHR (GnRH receptor) is also coupled to Gαs leading to cAMP accumulation in the LβT2 cells (9), which may enhance LHβ gene transcription (10, 11).
Additional signaling components or pathways/mechanisms have been reported to contribute to the gonadotrope response to GnRH. Steroid hormone feedback is implicated in the regulation of gonadotropin gene expression; for instance, estrogen augments GnRH-induced transcription of LHβ and CGA genes through regulation of stimulatory and suppressive transcription factors Egr-1 and Zeb1, respectively (12). A member of the TGFβ superfamily, activin is a potent inducer of the FSHβ gene, because it stimulates endogenous FSHβ mRNA expression and acts via the Smad signaling pathway to regulate the FSHβ promoter (4, 13). Through induction of cyclooxygenase-2, GnRH stimulates the synthesis of prostaglandin F2α, which subsequently reduces GnRH-stimulated LHβ gene transcription, highlighting a cross talk between GnRH and prostanoid receptors (14). Phosphatidylinositol 3-kinase may be involved in GnRH-induced CGA and FSHβ gene expression in LβT2 cells, because treatment with a phosphatidylinositol 3-kinase inhibitor results in an increase in GnRH-induced gonadotropin promoter activity (15). GnRH induces an increase in phosphorylated PEA-15 through PKC activation, leading to ERK nuclear translocation; the scaffold protein also has a modulatory effect on LHβ and CGA gene expression (16). GnRH also induces the synthesis and secretion of inhibin, which regulates FSHβ in an autocrine manner (11). Overall, understanding the variety of mechanisms that are engaged in the regulation of gonadotropin expression in response to GnRH may not only shed light on gonadotrope physiology, but also have potential therapeutic applications in the treatment of infertility and hormone-dependent cancers (17, 18).
A key mediator of the Wnt developmental signaling pathway, β-catenin accumulates in the cytoplasm and then translocates to the nucleus where it acts as a cofactor to T-cell factor (TCF)/lymphoid enhancer factor (LEF) transcription factors to promote the transcription of Wnt target genes, such as oncogenes cyclin D1 and c-Myc (for review, see Ref. 19). β-Catenin also plays a structural role in cell-cell adhesion by linking cadherins to the actin cytoskeleton (20). Interestingly, the Wnt/β-catenin pathway is thought to play a critical role in pituitary tumorigenesis (21–23). In fact, it was reported to induce Pitx2 (24), a transcription factor the overexpression of which in nonfunctioning pituitary adenoma tumorigenesis may have an antiapoptotic effect (22) and which also plays an important role in gonadotrope cell lineage specification during pituitary development (25). β-Catenin itself has been linked to GnRH-regulated gene expression (for review, see Ref. 26). A previous study demonstrated that GnRH induces nuclear accumulation of β-catenin and up-regulation of c-jun in LβT2 cells, suggesting a potential for cross talk between GnRH and Wnt signaling (27). Additionally, β-catenin was shown to be involved in GnRH-stimulated LHβ transcription through coactivation of SF-1 and Egr-1 (28). The same group proposed that GnRH-mediated induction of the immediate early gene c-jun requires β-catenin as a coactivator, thereby affecting the transcription of CGA, one of Jun target genes (29). Collectively, there is emerging evidence of an important role played by β-catenin in GnRH signaling in the gonadotropes.
In the present study, we sought to 1) decipher how GnRH signals to β-catenin; 2) determine whether β-catenin contributes to basal and/or GnRH-mediated FSHβ gene expression; and 3) understand the molecular basis of this gene regulation in the LβT2 gonadotrope cell line. We show that GnRH induces a sustained accumulation of β-catenin in the nucleus via the JNK pathway and report a new role for β-catenin in the regulation of FSHβ gene expression in response to GnRH treatment. Furthermore, we identify breast cancer metastasis-suppressor 1-like (Brms1L) as a mediator of the β-catenin effect on GnRH-induced FSHβ gene expression.
Materials and Methods
Materials
The following chemicals and reagents were obtained from the indicated sources: SP600125 (Calbiochem, EMD Biosciences Inc, La Jolla, CA); dimethylsulfoxide (Sigma, St. Louis, MO); GnRH (Bachem Biosciences, Torrance, CA). JNK small interfering RNA (siRNA) (ON-TARGET plus SMARTpool L-040128–00-0005 for JNK1 and L-040134–00-0005 for JNK2), β-catenin siRNA (ON-TARGET plus SMARTpool L-040628–00-0005), Brms1L siRNA (siGENOME SMARTpool M-055459–01-005) and scrambled siRNA (D-001600–01-20) were purchased from Dharmacon (Lafayette, CO). Mouse monoclonal anti-phospho-JNK, rabbit polyclonal anti-JNK, rabbit polyclonal anti-c-jun and anti-phospho-c-jun, rabbit polyclonal anti-lysine (K)-specific demethylase 1 (LSD1), and rabbit polyclonal anti-phospho-glycogen synthase kinase (GSK)-3α/β (pSer21/9) antibodies were from Cell Signaling Technology (Beverly, MA). Mouse monoclonal anti-β-catenin and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies were from Santa Cruz Biotechnology, Inc. (Santa-Cruz, CA). Horseradish peroxidase-coupled secondary antibodies were from Santa Cruz Biotechnology. Fluorophore-conjugated secondary antibodies were from Li-Cor Biosciences (Lincoln, NE). Ortho-nitrophenyl-beta-D-galactopyranoside was ordered from Thermo Fisher Scientific (Rockford, IL). Restriction enzymes were purchased from New England Biolabs (Ipswich, MA). Formaldehyde and Actinomycin D were purchased from Sigma (St. Louis, MO).
Western blot analysis
Western blot analysis was performed as previously described (16). Nuclear fractions were extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific), according to manufacturer's recommendations. Protein concentrations were measured using the Bio-Rad protein assay (Bio-Rad Laboratories, Inc., Hercules, CA). Briefly, 50 μg proteins were loaded onto a 4–20% precasted SDS-PAGE gel (Bio-Rad). After transfer to a nitrocellulose membrane (Li-Cor Biosciences), blocking for 1 h at room temperature using blocking buffer (Li-Cor Biosciences, Lincoln, NE) was followed by overnight incubation with primary antibody at 4 C. Anti-JNK1/2 and anti-β-catenin primary antibodies were used at a 1:1000 dilution. Anti-GAPDH and anti-LSD1 antibodies were used at a 1:5000 dilution. Horseradish peroxidase-coupled secondary antibodies were diluted at 1:5000, and fluorophore-conjugated secondary antibodies were diluted at 1:10,000. Incubation with the secondary antibody was performed at room temperature for 2 h. Odyssey infrared fluorescent Western blot system (Li-Cor Biosciences) was used to detect and quantify protein bands of interest.
Cell culture and transient transfection
LβT2 cells obtained from Pr. Pamela Mellon (University of California, San Diego, CA) were maintained at 37 C in a humidified air atmosphere of 5% CO2 in DMEM (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (Gemini, Calabasas, CA) and l-glutamine (GIBCO Invitrogen, Carlsbad, CA). Cells were transfected in electroporation cuvettes using the Amaxa Cell Line Nucleofector Kit L, following the manufacturer's instructions (Lonza Walkersville, Inc., Walkersville, MD). Briefly, 1 million LβT2 cells were transfected with 600 ng of either plasmid construct or siRNA (2 μl of a 20 μm stock in a 20-μl reaction) using Nucleofector program DS-137. Immediately after transfection, cells were plated and incubated in fresh medium for 48 h. Cells were then serum starved and ultimately subjected to hormonal treatment.
Reporter plasmid construction
The −1830/−1 FSHβ promoter region was cloned from mouse genomic DNA by nested PCR. An approximately 2-kb fragment was initially cloned using the following pair of primers: sense, 5′-GCC ACC CCT GTC CTT CTA CA-3′; antisense, 5′-TGT CTC TTC TGG GGA AAG CTG-3′. Then, a 1.8-kb PCR product was generated using a pair of nested primers designed to place NheI and HindIII restriction sites at the 5′- and 3′-ends, respectively: sense, 5′-TTG CTA GCA CAT CCC ATC TCC ATG TGA GCA-3′; antisense, 5′-TTA AGC TTA CTG AGT CAA GTT ACA CCT CAT CTT TT-3′. The PCR program was as follows: 94 C for 2 min; 40 cycles of 94 C for 45 sec, 54 C for 45 sec, 72 C for 4 min; 72 C for 15 min. The 1.8-kb PCR product was inserted into a pBluescript vector (Stratagene, Santa Clara, CA). The pBluescript vector containing the 1.8-kb fragment and a pGL3 plasmid (kindly provided by Dr. Natarajan Balasubramanian) were digested with NheI and HindIII. The 1.8-kb mouse FSHβ fragment was then ligated into pGL3 using DNA ligase.
The −944/−1 FSHβ promoter region was subcloned from the −1830/−1 FSHβ-luciferase pGL3construct using the following PCR primers: sense, 5′-CTT AAA ATC CTA CAG TGG ACC AA-3′; antisense, 5′-CCA GCG GTT CCA TCT TCC AG-3′. The PCR program was as follows: 94 C for 3 min; 40 cycles of 94 C for 30 sec, 50 C for 30 sec, 72 C for 2.5 min; 72 C for 10 min. The PCR product was digested with NcoI at 37 C for 4 h. The pGL3 vector was first digested with SmaI at 25 C for 1.5 h, and then with NcoI for 1.5 h at 37 C. The 944-bp fragment was ligated into pGL3 using DNA ligase.
The −1000/−1 FSHβ-luciferase pGL3construct was kindly provided by the Mellon laboratory (56). Using the Multisite-directed mutagenesis kit (Stratagene), PstI restriction sites (5′-CTGCAG-3′) were introduced in the −944/−1 FSHβ-luciferase pGL3construct to replace the three TCF/LEF putative binding sites (5′-NCAAAG-3′). The following three mutagenesis PCR primers (100 ng of each) were used: 5′ CTG TCT TTT CTT TCC ACT GCA GGT CTT GAA ACT TTA CCA CAC AGA GG-3′, 5′-CCC CCT TTT TCT GAT ACT GCA GTA AGA GCA TCA GAA ATT GTA ATT CCC-3′, 5′-GAA TCC TTT CTG AAC ATT ACT GCA GAT CCA TTG CGA AGG TAA ACC AAA-3′. The PCR program was as follows: 95 C for 1 min; 30 cycles of 95 C for 1 min, 50 C for 1 min, 65 C for 13 min; 65 C for 10 min. The resulting PCR product was digested by DpnI for 3 h at 37 C. A 2-μl volume of DpnI-digested PCR product was used for bacterial transformation. XL-blue competent cells (Stratagene) were used for cloning selection. All plasmids were verified by sequencing using the following primer: 5′-CTT TAT GTT TTT GGC GTC TTC CA-3′.
Luciferase and β-galactosidase assays
Transfected cells were seeded into 24-well plates at 0.3 million/well. Upon hormonal treatment, cells were lysed in 200 μl reporter lysis buffer, and the cell lysate was incubated at room temperature for 30 min under gentle agitation. The Luciferase assay kit was purchased from Promega Corp. (Madison, WI). The cell lysate (5 μl) was mixed with luciferase substrate (25 μl), and luciferase activity was measured using Sirius single tube luminometer (Berthold Detection Systems, Huntsville, AL). For β-galactosidase activity, 20 μl cell lysate were mixed with 50 μl Buffer A [100 mm NaH2PO4-Na2HPO4, (pH 7.5); 10 mm KCl, 1 mm MgSO4, 50 mm β-mercaptoethanol) at room temperature for 5 min, and 17 μl ortho-nitrophenyl-beta-D-galactopyranoside (at 4 mg/ml) was added to the mixture and incubated at 37 C until appearance of a yellow color. The reaction was terminated by addition of 30 μl of 1 m Na2CO3. Absorbance at 420 nm was measured in a spectrometer (Certified GeneTool, Inc. Pleasanton, CA). Samples were measured in triplicate in both assays. Luciferase activity was normalized to β-galactosidase activity.
Pulse administration
Cells were seeded into 12-well plates at 1 million/well and were exposed to 5-min pulses of 5 nm GnRH either every 30 min (fast pulses) or every 2 h (slow pulses) for a total duration of 6 h. Cells were washed 30 min after the last pulse and lysed for RNA extraction. Using two distinct frequencies of media pulses, we verified that our pulse administration itself had no significant effect on FSHβ gene expression as compared with the no-pulse conditions (data not shown).
Quantitative real-time PCR
For quantitative real-time PCR experiments, total RNA was isolated using Stratagene Absolutely 96 RNA microprep kit (Stratagene, La Jolla, CA). RNA quantity and quality were assessed with NanoDrop (NanoDrop Technologies, Inc., Wilmington, DE). After reverse transcription of 1 μg of RNA, cDNA samples were diluted to 1:50 in dH2O. SYBR green quantitative PCR was performed according to the manufacturer's recommendations, using 5 μl of cDNA template and 5 μl of master mix containing the specific primers for the targeted gene. The PCR program was as follows: 95 C for 2 min, followed by 40 cycles of 95 C for 15 sec, 55 C for 15 sec, 72 C for 30 sec. Samples were measured in triplicate using ABI Prism 7900HT detection system and software version 2.2.2 (Applied Biosystems, Foster City, CA). Results were exported as Ct values for subsequent analysis. Data were normalized to either RPS11 or GAPDH housekeeping gene. Primer sequences were as follows: FSHβ/sense, 5′-TGG AGA CTC TGG CAT GAT TG-3′; FSHβ/antisense, 5′-GAG TTG AGC AGC CTA ACC TT-3′; β-catenin/Sense, 5′-CAT TAC TAA CTG GGA GCG TG-3′; β-catenin/antisense, 5′-GAC CCC GTG AGT CTT TAC AG-3′; rps11/sense, 5′-CGT GAC GAA GAT GAA GAT GC-3′; rps11/antisense, 5′-GCA CAT TGA ATC GCA CAG TC-3′; GAPDH/sense, 5′-TGC GAC TTC AAC AGC AAC TC-3′; GAPDH/antisense, 5′-CTT GCT CAG TGT CCT TGC TG-3′; c-jun/sense, 5′-TGA AAG CTG TGT CCC CTG TC-3′; c-jun/antisense, 5′-ATC ACA GCA CAT GCC ACT TC-3′; Brms1L/sense, 5′-TGA AAT CCA GGC TTC TCG ACA GCA-3′; Brms1L/antisense, 5′-TAA TGT CGA TGC TGT GCC TGT CCT-3′.
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation was performed using the chromatin immunoprecipitation kit EZ-CHIP (Millipore, Billerica, MA), in accordance with the manufacturer's protocol. In brief, cells were serum starved overnight and stimulated with either vehicle or 10 nm GnRH for either 1 h or 4 h. Formaldehyde was then added to cross-link the proteins to the DNA. Genomic DNA-protein complexes were sheared into 500- to 1000-bp fragments using a Bioruptor UCD-200 sonicator at high power (30 sec “ON” and 30 sec “OFF”). Specific antibody or control IgG (3 μg) was added to the clarified sample and incubated at 4 C overnight. DNA was then purified and quantitative real-time PCR was carried out as described above using the following primers: GAPDH/sense, 5′-CCT CGT CCC GTA GAC AAA ATG-3′; GAPDH/antisense, 5′-AAA GGC GGA GTT ACC AGA GG-3′; FSHβ-1/sense, 5′-GAA AGA ATA GTC TAG ACT CTA GAG TCA CA-3′; FSHβ-1/antisense, 5′-ACT GAG TCA AGT TAC ACC TCA TCT TTT A-3′; FSHβ-2/sense, 5′-GGT CAT AAA GAA AGA CAC AGC CCA-3′; FSHβ-2/antisense, 5′-ACC CAC TCC CTC ACC TTG TAA-3′; FSHβ-3/sense, 5′-GTG AAG CAC CTC TGT GTG GTA A-3′; FSHβ-3/antisense, 5′-GGA AGT ACT TCT GCA TCT TGT TCC TA-3′; FSHβ-4/sense, 5′-TAC ATT TCT GAC ATG CTC CCC TAA-3′; FSHβ-4/antisense, 5′-AGT TCT TGA AAC TTT ACC ACA CAG A-3′; FSHβ-5/sense, 5′-CAT GCC TCA CCT GTG TTAC TAC AA-3′; FSHβ-5/antisense, 5′-GGA TCA ATA GTG TAA AAA TGT TAG GGG A-3′; FSHβ-6/sense, 5′-GAC ACA TCA CAG TCA ACA TAT CTA TAC AGA-3′; FSHβ-6/antisense, 5′-GAG AAA CAC GGT AGA AGC AGG AA-3′; FSHβ-7/sense, 5′-GAC ACT TTC TCA ACT TCA TTT GGA TAG AA-3′; FSHβ-7/antisense, 5′-GAT GGA ATG GTT AAG GAT GTG CA-3′; FSHβ-8/sense, 5′-TCC CCT GAC ATG TGA GCC AA-3′; FSHβ-8/antisense, 5′-GAT ACA ATG AAT CTT CTT CCT CAT CCT-3′; LHβ primer sequences were as follows: LHβ/sense, 5′-CTG CAG TGG CCT CCC CTT TA-3′; LHβ/antisense: 5′-TCT TGA TAC CTT CCC TAC CTT GG-3′ (28).
RNA-Seq assays
LβT2 cells were transfected with either scrambled or β-catenin siRNA for 48 h, serum starved overnight, and stimulated with 5 nm GnRH or vehicle for either 45 min or 2 h. There was a total of six different groups or experimental conditions, and each group was comprised of four replicates. Total RNA (2.5 μg) from each replicate was sequenced at the Mount Sinai Genomics Core Facility using an Illumina platform (Illumina, Inc., San Diego, CA) and a HiSeq 2000 sequencing system (100-nucleotide length, single read type, multiplexing three samples per lane, eight lanes in total). For data analysis, the RNA-Seq reads were processed using RNA-Seq unified mapper (30). The reads per kilobase of transcript per million mapped reads (RPKM) values for RefSeq transcripts generated by the RNA-Seq unified mapper pipeline were used to assess differential expression. The smallest non-zero value was added to all RMPK values (to allow taking logs), and the data were log2 transformed and quantile normalized. We computed mean expression for each transcript across all experiments, and the bottom 30% was excluded from differential analysis. Differential expression analysis was performed using the Limma R package (31).
Statistical analysis
Statistical analysis in β-catenin nuclear accumulation, endogenous FSHβ, and β-catenin gene expression as well as reporter assays were done by GraphPad Prism version 5.04. One-way and two-way ANOVAs were applied for overall effect, and specific comparison was examined with Bonferroni corrections. Statistical significance was set as indicated in each figure legend with at least a P < 0.05.
Results
GnRH induces a sustained increase in nuclear β-catenin levels in LβT2 cells
β-Catenin nuclear accumulation in response to GnRH was previously demonstrated in HEK293 cells expressing the GnRHR (27). To assess the pattern of β-catenin nuclear accumulation in response to GnRH in LβT2 gonadotrope cells, we measured the effect of GnRH on β-catenin nuclear levels over time. Protein levels of β-catenin in nuclear extracts were assayed by Western blotting. We observed nuclear accumulation of β-catenin after a 15-min GnRH stimulation. This effect was sustained for hours (Supplemental Fig. 1, A and B, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org).
The increase in nuclear β-catenin induced by GnRH is JNK-dependent
To identify the signaling pathway(s) involved in GnRH-induced nuclear accumulation of β-catenin, GnRH-stimulated cells were pretreated with pharmacological inhibitors of various kinases, namely protein kinase A, ERK, JNK, p38, steroid receptor coactivator, and GSK. SP600125, a well-characterized JNK inhibitor in gonadotropes (32, 33), blocked the accumulation of β-catenin (Fig. 1, A and B) in a dose-dependent manner (Supplemental Fig. 2, C and D), whereas the other inhibitors tested had no significant effects (data not shown). To verify the dependency of GnRH-induced nuclear accumulation of β-catenin on JNK, we employed JNK inhibitor III, a JNK inhibitor that has a different mode of action than SP600125. Treatment of LβT2 cells with JNK inhibitor III impaired GnRH-mediated β-catenin nuclear accumulation (Supplemental Fig. 2, A and B), thus confirming that GnRH-induced β-catenin nuclear accumulation is significantly dependent on the JNK pathway.
Fig. 1.

Attenuation of GnRH-induced β-catenin nuclear accumulation and FSHβ mRNA expression by JNK inhibition. A, Effect of SP600125 on GnRH-induced nuclear accumulation of β-catenin in LβT2 cells. Cells were serum starved overnight, pretreated with 40 μm SP600125 or vehicle for 30 min, and stimulated with 10 nm GnRH or vehicle for 15 min. Nuclear extracts were subjected to a quantitative Western blot analysis using a β-catenin-specific antibody. LSD1 (in red) was used as a loading control. B, Quantification of Western blot densitometry from three independent experiments, plotted as mean ± sem. One-way ANOVA (n = 3; *, P < 0.05). C, Effect of SP600125 on GnRH-induced FSHβ mRNA levels. Cells were serum starved overnight, pretreated with 40 μm SP600125 or vehicle for 30 min, and stimulated with 5 nm GnRH or vehicle for 6 h. Relative mRNA copy numbers of FSHβ were determined by quantitative real time-PCR. Two-way ANOVA (n = 4; **, P < 0.01).
In the absence of Wnt ligand, β-catenin is held in a destruction complex that includes GSK3. GSK3 is a kinase composed of two distinct isoforms, GSK3α and GSK3β, which are inhibited by phosphorylation at Ser21 and Ser9, respectively (34, 35). Upon Wnt binding, GSK activity is phospho inhibited, allowing β-catenin to translocate to the nucleus (19). Hence, we wondered whether GnRH and/or JNK could augment GSK3 phosphorylation, thereby promoting β-catenin nuclear translocation. We observed an increase, rather than a decrease, in the levels of both Ser21 phospho-GSK3α and Ser9 phospho-GSK3β in the presence of SP600125. Furthermore, these effects were independent of GnRH stimulation (Supplemental Fig. 3). Collectively, these data suggest that JNK-dependent nuclear accumulation of β-catenin in GnRH-stimulated cells is not mediated by GSK3 phosphorylation.
GnRH-induced FSHβ mRNA level is dependent on both β-catenin and JNK expression
We investigated whether JNK and β-catenin were involved in the transcriptional response of FSHβ to GnRH. Inhibition of JNK using SP600125 decreased the GnRH-induced increase in FSHβ mRNA by 80% (Fig. 1C). Next, LβT2 cells were transfected via electroporation with siRNA targeting either β-catenin or JNK1/2 and stimulated with GnRH. The extent of gene knockdown was assessed by quantitative Western blot analysis (Fig. 2A). β-Catenin protein levels were reduced by about 90% compared with control cells, providing evidence for a specific and significant down-regulation of β-catenin protein expression by β-catenin siRNA. Likewise, JNK1 and JNK2 protein levels were reduced by about 70%. β-Catenin siRNA knockdown resulted in a reduction in GnRH-induced FSHβ gene expression by 90% (Fig. 2B). Additionally, β-catenin knockdown led to a decrease in basal FSHβ expression, yet it was not significant. Consistent with the inhibitory effect of SP600125 on GnRH-stimulated FSHβ expression, JNK1/2 gene knockdown resulted in an 80% decrease in GnRH-induced FSHβ expression. Altogether, these data suggest that GnRH-induced FSHβ gene expression is dependent on both JNK and β-catenin.
Fig. 2.

Effect of siRNA-mediated silencing of β-catenin and JNK on GnRH-induced FSHβ mRNA levels. A, LβT2 cells were transfected with either scrambled, β-catenin, JNK1, or JNK2 siRNA for 72 h using the nucleofection method (as described in Materials and Methods). Whole-cell lysates were subjected to a Western blot analysis using JNK- and β-catenin-specific antibodies. GAPDH was used as a loading control. B, Effect of β-catenin or JNK1/2 knockdown on FSHβ gene expression. LβT2 cells were transfected with either scrambled, β-catenin, or JNK1/2 siRNA for 48 h, serum starved overnight, and stimulated with 5 nm GnRH or vehicle for 8 h. RNA copy numbers of FSHβ were determined by quantitative real time-PCR. Two-way ANOVA (n = 4; **, P < 0.01). C, Effect of β-catenin or JNK1/2 knockdown on FSHβ gene expression under low-frequency GnRH pulses. LβT2 cells were transfected with either scrambled, β-catenin, or JNK1/2 siRNA for 48 h, serum-starved overnight, and stimulated with slow pulses of 5 nm GnRH or vehicle for 6 h. Each pulse lasted 5 min. The time intervals between GnRH pulses (media replacement) were 2 h. Samples were collected 30 min after the last pulse. Relative mRNA copy numbers of FSHβ were determined by quantitative real time-PCR. Two-way ANOVA (n = 4; **, P < 0.01).
Low GnRH pulse frequency-induced FSHβ transcript level involves β-catenin and JNK
Gonadotropes can distinguish between high-frequency vs. low-frequency GnRH pulses, decode this information and convert, it into differential expression of the gonadotropins LH and FSH. We investigated the impact of β-catenin on GnRH pulse frequency-dependent regulation of FSHβ gene expression. Cells were transfected with siRNA targeting β-catenin and stimulated with either high- or low-frequency GnRH pulses (i.e. every 30 min or every 2 h for 6 h). As expected, low-frequency GnRH pulses induced a marked (4-fold) increase in FSHβ mRNA levels as compared with the control (Fig. 2C). In contrast, high-frequency GnRH pulses had little effect on FSHβ gene expression (data not shown). Knockdown of β-catenin resulted in a significant attenuation of low pulse frequency-induced FSHβ expression. Similarly, knockdown of JNK and JNK inhibition by JNK inhibitor III caused a significant decrease in low pulse frequency-induced FSHβ expression (Fig. 2C and Supplemental Fig. 2E, respectively). Thus, our findings support the conclusion that induction of FSHβ gene expression by slow pulses of GnRH is significantly mediated by JNK and β-catenin.
β-Catenin regulation of FSHβ gene expression is independent of the JNK-c-jun pathway
Because AP-1 (Fos/Jun) was previously shown to be involved in GnRH response of the FSHβ promoter in LβT2 cells (36, 37), we wondered whether regulation of FSHβ expression by β-catenin was linked to activation of the JNK-c-jun pathway. To test this hypothesis, we performed siRNA-mediated knockdown of β-catenin and examined its effect on the JNK-c-jun signaling pathway in GnRH-stimulated cells. As expected, phospho-JNK and phospho-c-jun protein levels were increased under GnRH stimulation, whereas total JNK protein level was unchanged (Fig. 3, A and B). Consistent with GnRH activation of immediate early gene expression in the gonadotropes (38–40), GnRH induced an increase in both c-jun mRNA and protein levels (Fig. 3, A–C). β-Catenin gene silencing had no effect on the protein and phosphorylation levels of either JNK or c-jun (Fig. 3, A and B). Finally, whereas GnRH-induced c-jun transcripts were reduced by 50% by JNK knockdown, they were unaffected by β-catenin knockdown (Fig. 3C). Although GnRH-induced β-catenin nuclear accumulation depends on the JNK pathway, these results suggest that β-catenin regulation of FSHβ gene expression is independent of JNK-c-jun signaling.
Fig. 3.

Effect of β-catenin knockdown on GnRH-induced JNK-c-jun activation. A, LβT2 cells were transfected with either scrambled or β-catenin siRNA for 48 h, serum-starved overnight, and stimulated with 10 nm GnRH or vehicle for 30 min (for JNK) and 1 h (for c-jun). Whole-cell lysates were subjected to a Western blot analysis using β-catenin-, JNK-, phospho-JNK-, c-jun-, and phospho-c-jun-specific antibodies 72 h after transfection. LSD1 was used as a loading control. B, Quantification of Western blot densitometry from three independent experiments, plotted as mean ± sem. Two-way ANOVA. C, Effect of β-catenin or JNK1/2 knockdown on c-jun gene expression. LβT2 cells were transfected with either scrambled, β-catenin, or JNK1/2 siRNA for 48 h, serum starved overnight, and stimulated with 10 nm GnRH or vehicle for 1 h. RNA copy numbers of c-jun were determined by quantitative real time-PCR. Two-way ANOVA with post Bonferroni corrections (n = 3; *, P < 0.05).
β-Catenin does not regulate FSHβ mRNA stability
To assess the potential effect of β-catenin on the stability of FSHβ transcripts, we inhibited new mRNA transcription with actinomycin D in cells transfected with either β-catenin or scrambled siRNA and stimulated with GnRH. After the addition of actinomycin D, the decay of FSHβ mRNA over time was measured by quantitative real-time PCR. As shown in Fig. 4, the half-life of FSHβ mRNA (2.68 ± 0.20 h) was not significantly altered by β-catenin siRNA knockdown (2.61 ± 0.10 h). The estimated half-life of FSHβ mRNA is compatible with a previous study showing that FSHβ mRNA has a rapid turnover (41). Thus, β-catenin did not influence the stability of FSHβ mRNA.
Fig. 4.
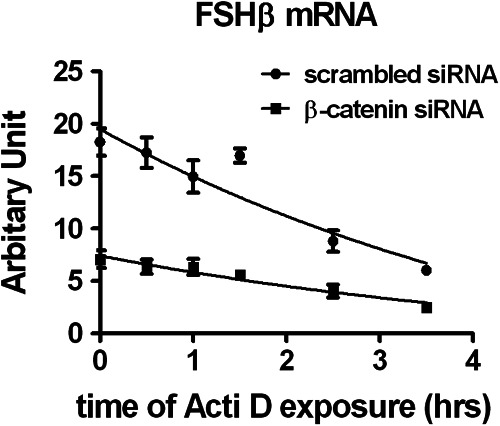
Effect of β-catenin knockdown on FSHβ mRNA stability. LβT2 cells were transfected with either scrambled or β-catenin for 48 h, serum starved overnight, and stimulated with 5 nm GnRH for 2 h. Medium was replaced with fresh culture medium for another 4 h. Cells were then exposed to 2 μg/ml actinomycin D (Acti D, 1.6 μm) and harvested at the indicated time points. RNA copy numbers of FSHβ were determined by quantitative real time-PCR. Decay curves were fitted to the data using GraphPad Prism (n = 4); Student's t test.
GnRH responsiveness of the FSHβ promoter is dependent on β-catenin expression
To explore the regulation of FSHβ expression by β-catenin at the transcriptional level, we subcloned 1.8 kb (−1830/−1) of the murine FSHβ promoter region upstream of the luciferase reporter gene. In transfected LβT2 cells, β-catenin knockdown reduced GnRH-induced FSHβ promoter activity by about 60% (Fig. 5B), a result that was in accordance with β-catenin knockdown effect on endogenous FSHβ mRNA levels (see Fig. 2B). Thus, our data were consistent with β-catenin having a significant role in GnRH-induced FSHβ gene transcription.
Fig. 5.

Effect of β-catenin gene silencing on GnRH-stimulated FSHβ gene promoter activity. A, The 1.8-kb (−1830/−1) FSHβ promoter region was cloned from mouse genomic DNA upstream of a luciferase reporter gene. Using the TRANSFAC database, analysis against the T-cell factor/lymphoid enhancer factor (TCF/LEF) family core binding signature revealed six putative TCF/LEF binding sites. Sequences of the consensus and the six putative TCF/LEF sites are shown. “Mutation” designates the PstI restriction site that was used in block replacement mutagenesis in D. B (top panel), Structure of the 1.8-kb FSHβ promoter luciferase (Luc) construct. The TCF/LEF sites are numbered 1–6, as in panel A. B (bottom panel), LβT2 cells were cotransfected with the 1.8-kb FSHβ promoter luciferase construct, a control β-galactosidase expression vector (internal standard for transfection efficiency), and either scrambled or β-catenin siRNA. Forty eight hours after transfection, cells were serum starved for 4 h and stimulated with 10 nm GnRH or vehicle for 16 h. Luciferase activity was measured and normalized to β-galactosidase. Two-way ANOVA (n = 3; **, P < 0.01). C (top panel), Structure of the −1000/−1 FSHβ promoter luciferase construct. C (bottom panel), Cells were cotransfected with a −1000/−1 FSHβ promoter luciferase construct, a control β-galactosidase expression vector, and either scrambled or β-catenin siRNA. Cells were then treated as described in panel B. Two-way ANOVA (n =3; **, P < 0.01). D (top panel), Structure of the −944/−1 FSHβ promoter luciferase construct and its mutant counterpart. Block replacement mutations symbolized by black triangles were introduced at the three most proximal TCF/LEF sites in the context of the −944/−1 construct. D (bottom panel), Either the wild-type or the mutated −944/−1 promoter construct was transfected into LβT2 cells, as described in panel B. Two-way ANOVA (n = 3; *, P < 0.05; **, P < 0.01). The data shown are mean ± sem of triplicate samples from one experiment and are representative of three independent experiments.
Indirect mechanism of β-catenin regulation of FSHβ transcription
We hypothesized that β-catenin might act as a cofactor to TCF/LEF transcription factors to promote FSHβ gene transcription. Accordingly, we analyzed the FSHβ promoter sequence for the presence of TCF/LEF binding sites using the TRANSFAC database (42), a broad compilation of experimentally verified binding sites summarized as position weight matrices. The region 2 kb upstream of the FSHβ transcription start site was analyzed by the TRANSFAC MATCH (43) algorithm with a cutoff, as defined within the database, chosen to minimize the sum of false-positive and false-negative binding site predictions. The analysis was performed for all binding signatures annotated to the mouse TCF/LEF proteins within the 2010.3 release of TRANSFAC. The analysis identified six putative TCF/LEF sites located at −1422/−1414, −994/−986, −974/−966, −634/−626, −498/−490, and −324/−316 (Fig. 5A). To determine which putative TCF/LEF binding site(s) were involved in the promoter responsiveness to β-catenin, we employed three 5′-deletion mutants with 5′-termini located at −1000 (five TCF/LEF sites), −944 (three TCF/LEF sites), and −565 (two TCF/LEF sites), respectively. Cells were cotransfected with each of these various constructs and β-catenin siRNA and then exposed to GnRH. Both the −1000/−1 and the −944/−1 promoter constructs remained responsive to GnRH and sensitive to β-catenin knockdown (Fig. 5, C and D) as compared with the −1.8-kb construct (Fig. 5B). The −565/−1 construct was responsive neither to GnRH, nor to β-catenin knockdown (data not shown). Therefore, the −944/−1 promoter construct was selected for further scrutiny.
We used block replacement mutagenesis to mutate simultaneously the three proximal TCF/LEF sites present in the −944/−1 promoter construct (Fig. 5D). Block replacement mutations consisted of substituting the wild-type sequences with PstI restriction sites, as signified in Fig. 5A and detailed in Materials and Methods. Notably, the mutated FSHβ promoter construct was responsive to GnRH, and the induction remained sensitive to β-catenin knockdown. Hence, these results suggested that the regulation of FSHβ expression by β-catenin was independent of putative TCF/LEF binding sites.
To identify any physical interaction between β-catenin and the endogenous FSHβ promoter under GnRH stimulation, we performed ChIP assays on genomic DNA fragments from LβT2 cells treated with GnRH. Chromatin was initially precipitated with an antibody to dimethyl-histone H3-Lys4 as a positive control (Fig. 6A). As expected, the constitutively expressed GAPDH gene promoter displayed clear Lys4 methylation of Histone H3, with a nearly 500-fold enrichment as compared with the mouse IgG (negative control). We then verified that the anti-β-catenin antibody was effective in our hands for ChIP in LβT2 cells by analyzing the immunoprecipitation of a known target gene, LHβ (28). The LHβ proximal promoter exhibited a 5-fold enrichment subsequent to β-catenin immunoprecipitation, as compared with the IgG control. Similar to the results obtained by Nilson's group (28), we observed a significant increase (2.11 ± 0.36-fold) in the amount of β-catenin associated with the LHβ proximal promoter after a 1-h treatment with GnRH, as compared with the vehicle-treated control (Fig. 6B). We then used a series of PCR primers spanning the entire 1.8-kb FSHβ promoter region that showed β-catenin responsiveness when contained in the reporter construct. These assays showed no enrichment of β-catenin association with this promoter region (Fig. 6C). The demonstration of β-catenin-sensitive regulation of the 1.8-kb promoter region, together with the absence of β-catenin association with the region and the absence of any effect of disruption of the TCF/LEF sites, suggested that the regulation of the 1.8-kb segment of the FSHβ promoter by β-catenin most likely occurs indirectly.
Fig. 6.
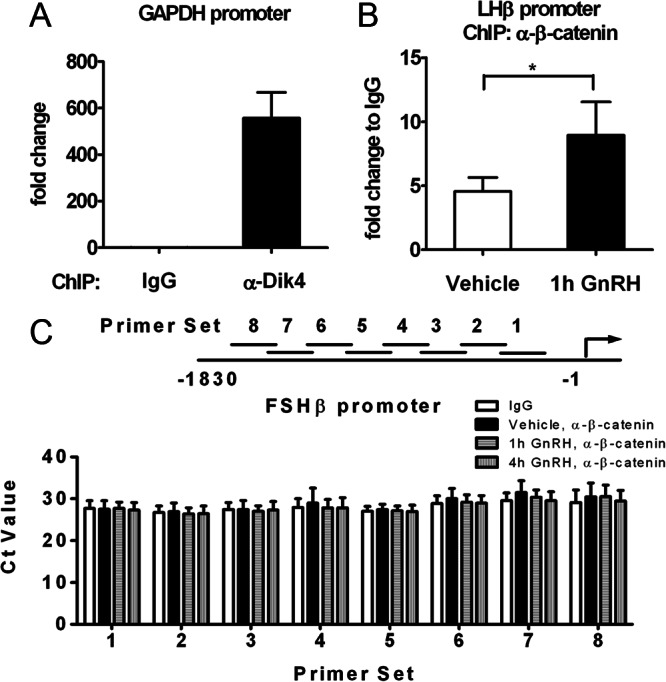
Chromatin immunoprecipitation of the β-catenin protein to the FSHβ promoter. Quantification of the ChIP assays performed on LβT2 cells is shown. A, The presence of dimethyl-histone H3-Lys4 binding to the GAPDH promoter was assessed with an antidimethylhistone H3-Lys4 antibody (α-Dik4, as indicated) and served as a positive control for the ChIP assay. A nonspecific antibody, normal mouse IgG was used as a negative control. Data were normalized to the IgG control and represent the mean ± sem of four experiments. B, The binding of β-catenin to the LHβ promoter in the presence of GnRH was assessed with an anti-β-catenin antibody (α-β-catenin, as indicated) and served as a positive control for β-catenin immunoprecipitation. Cells were serum starved overnight and stimulated with GnRH (10 nm) or vehicle for 1 h. Chromatin immunoprecipitation was performed on genomic DNA fragments from those cell nuclei. Data were normalized to the IgG control and represent the mean ± sem of four experiments. Paired t test (n = 4; *, P < 0.05). C, Cells were serum-starved overnight and stimulated with either vehicle or GnRH (10 nm) for 1 h or 4 h. The presence of β-catenin binding to the FSHβ promoter under GnRH stimulation was assessed with the same antibody as in panel B, through the use of eight primer sets (1–8, as indicated) encompassing the six putative TCF/LEF sites within the 1.8-kb promoter region. Results are expressed as Ct values and represent the mean ± sem of three independent experiments.
β-Catenin regulates FSHβ transcription through Brms1L
Analysis of previous extensive microarray experiments performed in LβT2 cells identified no plausible candidates for the indirect regulation of FSHβ by β-catenin (Ref. 39 and Ebersole, B.J., T. Yuen, E. Wurmbach, and S.C. Sealfon, unpublished data). Therefore, we used a more sensitive next-generation RNA-Seq approach to attempt to find the mechanism responsible for the effect of β-catenin. To increase the sensitivity of this screen, we analyzed two different time points (45 min and 2 h) for the GnRH stimulation conditions, assayed four samples per experimental condition, and sequenced only three samples per lane (high-depth sequencing). The entire experiment contained more than 970 million reads. All but two samples had at least 30,000,000 reads. Of 29,942 RefSeq transcripts at time zero (no GnRH), 24,998 transcripts had at least one read, and 17,658 transcripts had log2(RPKM) > −2. Based on previous analyses, features with log2(RPKM) > −2 are extremely unlikely to be a result of genomic contamination (44). Using a cutoff of Benjamini-Hochberg false discovery rate of 0.05 at the 45-min GnRH time point, we found that 1292 transcripts were differentially expressed relative to time zero, and 917 transcripts were differentially expressed under β-catenin knockdown. The RNA-Seq data were deposited in Genome Expression Omnibus (GSE42120).
Among the transcripts identified by RNA-Seq, we focused on candidates that were novel, regulated, and had homology to known factors involved in gene regulation. This selection process yielded two novel factors that were selected for further study: KDM3A and Brms1L (breast cancer metastasis-suppressor 1-like). In contrast to studies of Brms1L, KDM3A knockdown had no effect on FSHβ expression (data not shown) and was eliminated from consideration. As shown in Fig. 7A, knockdown efficiency of Brms1L siRNA was high, because basal Brms1L mRNA levels were reduced by 60% compared with scrambled siRNA-transfected cells. Reducing Brms1L led to a major attenuation of GnRH-induced FSHβ expression (50%; Fig. 7B). To confirm that Brms1L was regulated by β-catenin, we studied the effect of β-catenin knockdown on Brms1L mRNA expression in either the presence or absence of GnRH stimulus. β-Catenin knockdown caused a significant decrease in Brms1L mRNA levels in either GnRH-treated or untreated cells (30%–50%; Fig. 7C). Given the dependency of β-catenin nuclear accumulation on JNK, we wondered whether the regulation of Brms1L expression was also JNK-dependent. Hence, we examined the impact of JNK inhibition on Brms1L mRNA levels. As illustrated in Fig. 7D, inhibition of JNK by SP600125 led to a significant reduction in Brms1L expression (∼ 50%). The intensity of this inhibitory effect was comparable to that of β-catenin knockdown on Brms1L expression (Fig. 7C). We also observed a decrease in Brms1L mRNA expression in the presence of GnRH (30%–40%; Fig. 7, C and D), that might contribute to negative feedback mechanisms (see Discussion). Taken together, these data indicate that expression of Brms1L is dependent on β-catenin and JNK, and Brms1L is implicated in the induction of FSHβ expression by GnRH. Thus, these data suggest that the mechanism for the dependence of FSHβ induction on β-catenin is due to the involvement of JNK and β-catenin in Brms1L expression.
Fig. 7.
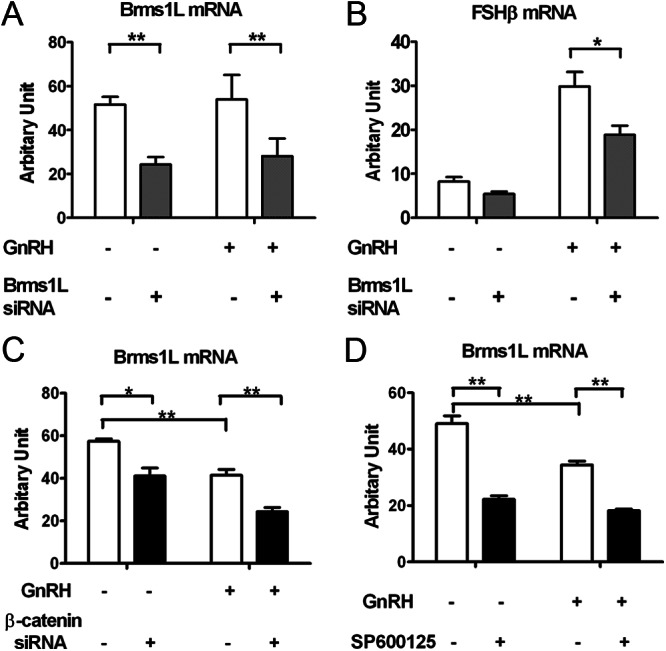
Role of Brms1L in β-catenin regulation of GnRH-induced FSHβ expression. A, Knockdown efficiency of Brms1L siRNA in LβT2 cells. LβT2 cells were transfected with either scrambled or Brms1L siRNA for 48 h, serum starved overnight, and stimulated with 5 nm GnRH or vehicle for 2 h. Medium was replaced with fresh culture medium for another 4 h. B, Effect of siRNA-mediated silencing of Brms1L on GnRH-induced FSHβ mRNA levels. Cells were treated as described in panel B. C, Effect of siRNA-mediated silencing of β-catenin on Brms1L mRNA levels. Cells were transfected with either scrambled or β-catenin siRNA for 48 h, serum starved overnight, and stimulated with 5 nm GnRH or vehicle for 2 h. D, Effect of JNK inhibition on Brms1L mRNA levels. Cells were serum starved overnight, pretreated with 40 μm SP600125 or vehicle for 30 min, and stimulated with 5 nm GnRH or vehicle for 6 h. Relative mRNA copy numbers of Brms1L and FSHβ were determined by quantitative real time-PCR. Two-way ANOVA (n = 4; **, P < 0.05). Results represent the mean ± sem of three independent experiments.
Discussion
We demonstrate that GnRH-induced FSHβ expression in LβT2 cells is dependent on β-catenin. These results provide insight into the role played by β-catenin in gonadotropin regulation and in gonadotrope physiology. The mechanism by which β-catenin is required for full GnRH induction of FSHβ transcription involves Brms1L expression.
We provide evidence that in gonadotropes, β-catenin nuclear activation depends on GnRH-induced activation of the JNK cascade. JNK had previously been implicated in mediating β-catenin activation in bone marrow-derived stromal cells. Wu and colleagues showed that Rac1 activates JNK2, which in turn phosphorylates β-catenin on critical serine residues (Ser191 and Ser605) and controls its nuclear translocation in response to Wnt signals. Mutation of these serine residues significantly affects β-catenin nuclear accumulation (45). Another report using keratinocytes demonstrated that JNK phosphorylates β-catenin at Ser (37) and Thr41, both of which are also phosphorylated by GSK-3β; inhibition of JNK activity reduced phosphorylation of β-catenin and promoted its localization to adherens junctions (46). Because it was previously reported by Wu et al. that mutation at Ser191 prevents β-catenin from translocating to the nucleus, we speculate that GnRH may promote β-catenin nuclear translocation in the gonadotropes via phosphorylation of this residue by JNK.
In the present study, we demonstrate that GnRH induces β-catenin accumulation in the nucleus in a sustained manner. Our data are in agreement with a previous report in LβT2 cells showing β-catenin nuclear accumulation in response to a 30-min stimulation with GnRH (27). Likewise, we find no association between β-catenin nuclear accumulation and GSK3β inactivation in LβT2 gonadotropes. Our data further suggest that JNK-dependent nuclear accumulation of β-catenin is not mediated by GSK3 phospho-inhibition (either at Ser21 in GSK3α or at Ser9 in GSK3β). Importantly, Gardner et al. (27) showed that GnRH stimulation of TCF/β-catenin nuclear transcriptional activity can be blocked by a Gαq/11 inhibitor, suggesting that GnRH may regulate β-catenin via PKC. Whether PKC is an upstream activator of JNK in the context of GnRH-induced β-catenin nuclear accumulation remains to be established.
Based on both pharmacological inhibition and siRNA-mediated knockdown of JNK, we demonstrate that the induction of FSHβ transcripts by GnRH relies on JNK. Additionally, we detect a large induction of c-jun mRNA by GnRH, as well as a GnRH-induced increase in c-jun protein levels and phospho-c-jun protein levels. Consistent with these findings, GnRH was previously shown to stimulate the ovine FSHβ promoter via the JNK pathway in LβT2 cells (47), and GnRH-induced JNK stimulated c-jun protein expression in αT3–1 gonadotrope cells (33). The Mellon group (36) also demonstrated that AP-1 (Fos/Jun) contributed to GnRH response of the FSHβ promoter in LβT2 cells. Further, using β-catenin knockdown, we show that β-catenin regulation of FSHβ expression is distinct from the JNK-c-jun pathway. Therefore, it appears that the dependence of FSHβ mRNA on JNK occurs partly through c-jun activation, and partly through the β-catenin-dependent Brms1L mechanism we have identified.
Our findings are also consonant with previous work showing that β-catenin inactivation in Amhr2cre mice in the pituitary leads to down-regulation of pituitary FSHβ mRNA (48). Furthermore, conditional deletion of β-catenin in those Amhr2cre mice causes female infertility due to impaired development of the oviduct and uterus. Thus, these data underline the physiological implication of β-catenin in the pituitary-gonadal axis, in particular in the gonadotropes (27–29).
The inhibitory effect of β-catenin gene knockdown on GnRH-induced activity of FSHβ promoter luciferase constructs concurs with its deleterious impact on GnRH-stimulated expression of the endogenous FSHβ gene. Such concordance further supports the role of β-catenin in the regulation of GnRH-activated FSHβ expression. Interestingly, we found that the −944-bp proximal FSHβ promoter was responsive to GnRH, whereas the −565-bp promoter construct was not. This result is in contrast with the Mellon group's previous report (36), showing that the proximal 398 bp of the mouse FSHβ promoter was sufficient for GnRH response in LβT2 cells. Because our experiments involve siRNA transfection requiring a longer transfection period, and different methods of transfection than the studies previously reported, it is possible that the different time courses of the experiments account for these differences.
The data obtained from the luciferase reporter gene mutagenesis studies imply that regulation of FSHβ gene transcription by β-catenin is independent of activity at the putative TCF/LEF sites. Additionally, ChIP analysis showed no evidence for β-catenin recruitment to the endogenous proximal 1.8-kb FSHβ promoter. Although we cannot exclude a role for β-catenin binding to a region located upstream of the −1830/−1 FSHβ promoter, our data strongly suggest that the regulation of FSHβ expression by β-catenin occurs via an indirect mechanism. We hypothesized that this may occur through activation of an intermediate gene encoding a transcription factor, which in turn would contribute to FSHβ transcription. β-Catenin regulation of c-jun has been implicated in contributing to the modulation of the common gonadotropin subunit (CGA) gene expression (29). Other genes that have been reported to be transcriptionally regulated by β-catenin and implicated in FSHβ expression include Nur77 (49) and Pitx2 (50). However, we detected no alterations in either their basal or GnRH-induced mRNA expression levels (Supplemental Fig. 4).
We carried out RNA-Seq experiments and identified Brms1L as a potential target gene of β-catenin. Our subsequent studies indicated that Brms1L contributes to the induction of FSHβ resulting from β-catenin regulation. Brms1L knockdown reduces induction of FSHβ expression by GnRH. β-Catenin knockdown enhances the suppressive effect of GnRH on Brms1L expression. Furthermore, GnRH-stimulated β-catenin signaling opposes the overall suppressive effects of GnRH signaling on Brms1L, i.e. the reduction of Brms1L caused by GnRH is much greater in the absence of catenin signaling. Overall, these results establish the role of Brms1L and β-catenin in FSHβ regulation by GnRH.
Little is known about Brms1L and further study will be needed to establish its mechanism of action. Brms1L is highly homologous to breast cancer metastasis-suppressor 1 (Brms1), which is thought to be involved in growth control (51, 52). Indeed, Brms1 was previously shown to restore gap junctional intercellular communication and alter connexin expression in tumor cells, implying its role in metastasis inhibition (53). Similar to Brms1, overexpression of Brms1L inhibited cell growth in human cells. Additionally, Brms1L was identified as a component of a mammalian histone deacetylase complex, which acts as a transcription repressor (54). JNK inhibition reduces basal expression of Brms1L in the absence of GnRH, indicating a GnRH-independent role of JNK in Brms1L regulation. Brms1L may play a direct stimulatory role at the FSHβ gene. Although Brms1 typically has suppressive activity, it has also been reported to stimulate expression (55). Chromatin-immunoprecipitation studies will be useful when a suitable antibody is developed. The overall effect of GnRH stimulation is to reduce Brms1L expression, although β-catenin signaling opposes this suppression. We speculate that the modest reduction in Brms1L by GnRH may serve an overall negative feedback regulatory function. The underlying molecular circuitry is complex and may require frequency response studies, time course studies, and mathematical modeling to clarify its function. Nonetheless, the identification of Brms1L as a novel FSHβ-regulatory factor that contributes to the β-catenin effects is established by the data reported.
Our study reinforces the importance of β-catenin for gonadotropin regulation and underscores a new role for JNK in GnRH signaling, in addition to its role in regulation of c-jun. We demonstrate that the regulation of the FSHβ gene by GnRH requires β-catenin. Further, our data show that β-catenin deficiency attenuates FSHβ sensitivity to slow GnRH pulses. The mechanism through which β-catenin regulates FSHβ gene expression is indirect and depends on the expression of Brms1L. Several lines of evidence imply that the Wnt/β-catenin pathway is important in pituitary tumorigenesis, and up-regulation of Wnt pathway target genes Pitx2 and cyclin D1 has been found in nonfunctioning pituitary adenoma (21, 23). The present work implicates β-catenin in FSH physiology and increases its significance as a potential target for treatment of reproductive and neoplastic disorders.
Supplementary Material
Acknowledgments
We thank Professor Pamela L. Mellon (University of California, San Diego, La Jolla, California) for providing the LβT2 cells and the −1 kb FSHβ-luciferase pGL3construct; Dr. Natarajan Balasubramanian (University of Colorado School of Medicine, Aurora, Colorado) for giving us the pGL3 plasmid; Dr. Venugopalan Nair (Mount Sinai School of Medicine, New York, New York) for providing the ChIP-grade antibody against dimethyl-histone H3-Lys4 and PCR primers for the GAPDH promoter; and Dr. John H. Nilson (Washington State University, Pullman, Washington) for providing LHβ primer sequences.
This work was supported by the National Institute of Health Grant DK46943.
Disclosure Summary: Q.W., M.C., E.Z., and H.P. have nothing to declare. S.C.S. is an inventor on U.S. Patents 5,985,583 and 5,750,366 and has received royalties for these patents.
Footnotes
- Brms1L
- Breast cancer metastasis-suppressor 1-like
- CGA
- common α-glycoprotein subunit
- ChIP
- chromatin immunoprecipitation;
- GnRHR
- GnRH receptor
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- GSK
- glycogen synthase kinase
- JNK
- c-Jun N-terminal kinase
- LSD1
- lysine (K)-specific demethylase 1
- PKC
- protein kinase C
- RPKM
- reads per kilobase of transcript per million mapped reads
- siRNA
- small interfering RNA
- TCF/LEF
- T-cell factor/lymphoid enhancer factor.
References
- 1. Pierce JG, Parsons TF. 1981. Glycoprotein hormones: structure and function. Annu Rev Biochem 50:465–495 [DOI] [PubMed] [Google Scholar]
- 2. Burger LL, Haisenleder DJ, Dalkin AC, Marshall JC. 2004. Regulation of gonadotropin subunit gene transcription. J Mol Endocrinol 33:559–584 [DOI] [PubMed] [Google Scholar]
- 3. Alarid ET, Windle JJ, Whyte DB, Mellon PL. 1996. Immortalization of pituitary cells at discrete stages of development by directed oncogenesis in transgenic mice. Development 122:3319–3329 [DOI] [PubMed] [Google Scholar]
- 4. Pernasetti F, Vasilyev VV, Rosenberg SB, Bailey JS, Huang HJ, Miller WL, Mellon PL. 2001. Cell-specific transcriptional regulation of follicle-stimulating hormone-β by activin and gonadotropin-releasing hormone in the LβT2 pituitary gonadotrope cell model. Endocrinology 142:2284–2295 [DOI] [PubMed] [Google Scholar]
- 5. Thomas P, Mellon PL, Turgeon J, Waring DW. 1996. The L β T2 clonal gonadotrope: a model for single cell studies of endocrine cell secretion. Endocrinology 137:2979–2989 [DOI] [PubMed] [Google Scholar]
- 6. Turgeon JL, Kimura Y, Waring DW, Mellon PL. 1996. Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol Endocrinol 10:439–450 [DOI] [PubMed] [Google Scholar]
- 7. Windle JJ, Weiner RI, Mellon PL. 1990. Cell lines of the pituitary gonadotrope lineage derived by targeted oncogenesis in transgenic mice. Mol Endocrinol 4:597–603 [DOI] [PubMed] [Google Scholar]
- 8. Naor Z. 2009. Signaling by G-protein-coupled receptor (GPCR): studies on the GnRH receptor. Front Neuroendocrinol 30:10–29 [DOI] [PubMed] [Google Scholar]
- 9. Liu F, Usui I, Evans LG, Austin DA, Mellon PL, Olefsky JM, Webster NJ. 2002. Involvement of both G(q/11) and G (s) proteins in gonadotropin-releasing hormone receptor-mediated signaling in L β T2 cells. J Biol Chem 277:32099–32108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferris HA, Walsh HE, Stevens J, Fallest PC, Shupnik MA. 2007. Luteinizing hormone β promoter stimulation by adenylyl cyclase and cooperation with gonadotropin-releasing hormone 1 in transgenic mice and LβT2 Cells. Biol Reprod 77:1073–1080 [DOI] [PubMed] [Google Scholar]
- 11. Choi SG, Jia J, Pfeffer RL, Sealfon SC. 2012. G proteins and autocrine signaling differentially regulate gonadotropin subunit expression in pituitary gonadotrope. J Biol Chem 287:21550–21560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kowase T, Walsh HE, Darling DS, Shupnik MA. 2007. Estrogen enhances gonadotropin-releasing hormone-stimulated transcription of the luteinizing hormone subunit promoters via altered expression of stimulatory and suppressive transcription factors. Endocrinology 148:6083–6091 [DOI] [PubMed] [Google Scholar]
- 13. Bernard DJ. 2004. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone β subunit in mouse gonadotrope cells. Mol Endocrinol 18:606–623 [DOI] [PubMed] [Google Scholar]
- 14. Naor Z, Jabbour HN, Naidich M, Pawson AJ, Morgan K, Battersby S, Millar MR, Brown P, Millar RP. 2007. Reciprocal cross talk between gonadotropin-releasing hormone (GnRH) and prostaglandin receptors regulates GnRH receptor expression and differential gonadotropin secretion. Mol Endocrinol 21:524–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mutiara S, Kanasaki H, Harada T, Oride A, Miyazaki K. 2008. The involvement of phosphatidylinositol 3-kinase in gonadotropin-releasing hormone-induced gonadotropin α- and FSHβ-subunit genes expression in clonal gonadotroph LβT2 cells. Mol Cell Endocrinol 283:1–11 [DOI] [PubMed] [Google Scholar]
- 16. Choi SG, Ruf-Zamojski F, Pincas H, Roysam B, Sealfon SC. 2011. Characterization of a MAPK scaffolding protein logic gate in gonadotropes. Mol Endocrinol 25:1027–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Del Mastro L, Catzeddu T, Venturini M. 2006. Infertility and pregnancy after breast cancer: current knowledge and future perspectives. Cancer Treat Rev 32:417–422 [DOI] [PubMed] [Google Scholar]
- 18. Kim DK, Yang JS, Maiti K, Hwang JI, Kim K, Seen D, Ahn Y, Lee C, Kang BC, Kwon HB, Cheon J, Seong JY. 2009. A gonadotropin-releasing hormone-II antagonist induces autophagy of prostate cancer cells. Cancer Res 69:923–931 [DOI] [PubMed] [Google Scholar]
- 19. Moon RT, Kohn AD, De Ferrari GV, Kaykas A. 2004. WNT and β-catenin signalling: diseases and therapies. Nat Rev Genet 5:689–699 [DOI] [PubMed] [Google Scholar]
- 20. Nelson WJ, Nusse R. 2004. Convergence of Wnt, β-catenin, and cadherin pathways. Science 303:1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moreno CS, Evans CO, Zhan X, Okor M, Desiderio DM, Oyesiku NM. 2005. Novel molecular signaling and classification of human clinically nonfunctional pituitary adenomas identified by gene expression profiling and proteomic analyses. Cancer Res 65:10214–10222 [DOI] [PubMed] [Google Scholar]
- 22. Acunzo J, Roche C, Defilles C, Thirion S, Quentien MH, Figarella-Branger D, Graillon T, Dufour H, Brue T, Pellegrini I, Enjalbert A, Barlier A. 2011. Inactivation of PITX2 transcription factor induced apoptosis of gonadotroph tumoral cells. Endocrinology 152:3884–3892 [DOI] [PubMed] [Google Scholar]
- 23. Elston MS, Gill AJ, Conaglen JV, Clarkson A, Shaw JM, Law AJ, Cook RJ, Little NS, Clifton-Bligh RJ, Robinson BG, McDonald KL. 2008. Wnt pathway inhibitors are strongly down-regulated in pituitary tumors. Endocrinology 149:1235–1242 [DOI] [PubMed] [Google Scholar]
- 24. Briata P, Ilengo C, Corte G, Moroni C, Rosenfeld MG, Chen CY, Gherzi R. 2003. The Wnt/β-catenin–>Pitx2 pathway controls the turnover of Pitx2 and other unstable mRNAs. Mol Cell 12:1201–1211 [DOI] [PubMed] [Google Scholar]
- 25. Suh H, Gage PJ, Drouin J, Camper SA. 2002. Pitx2 is required at multiple stages of pituitary organogenesis: pituitary primordium formation and cell specification. Development 129:329–337 [DOI] [PubMed] [Google Scholar]
- 26. Salisbury TB, Binder AK, Nilson JH. 2008. Welcoming β-catenin to the gonadotropin-releasing hormone transcriptional network in gonadotropes. Mol Endocrinol 22:1295–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gardner S, Maudsley S, Millar RP, Pawson AJ. 2007. Nuclear stabilization of β-catenin and inactivation of glycogen synthase kinase-3 β by gonadotropin releasing hormone: targeting Wnt signaling in the pituitary gonadotrope. Mol Endocrinol 21:3028–3038 [DOI] [PubMed] [Google Scholar]
- 28. Salisbury TB, Binder AK, Grammer JC, Nilson JH. 2007. Maximal activity of the luteinizing hormone β-subunit gene requires β-catenin. Mol Endocrinol 21:963–971 [DOI] [PubMed] [Google Scholar]
- 29. Salisbury TB, Binder AK, Grammer JC, Nilson JH. 2009. GnRH-regulated expression of Jun and JUN target genes in gonadotropes requires a functional interaction between TCF/LEF family members and β-catenin. Mol Endocrinol 23:402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grant GR, Farkas MH, Pizarro AD, Lahens NF, Schug J, Brunk BP, Stoeckert CJ, Hogenesch JB, Pierce EA. 2011. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM). Bioinformatics 27:2518–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3:Article 3 [DOI] [PubMed] [Google Scholar]
- 32. Roberson MS, Zhang T, Li HL, Mulvaney JM. 1999. Activation of the p38 mitogen-activated protein kinase pathway by gonadotropin-releasing hormone. Endocrinology 140:1310–1318 [DOI] [PubMed] [Google Scholar]
- 33. Xie J, Bliss SP, Nett TM, Ebersole BJ, Sealfon SC, Roberson MS. 2005. Transcript profiling of immediate early genes reveals a unique role for activating transcription factor 3 in mediating activation of the glycoprotein hormone α-subunit promoter by gonadotropin-releasing hormone. Mol Endocrinol 19:2624–2638 [DOI] [PubMed] [Google Scholar]
- 34. Cohen P, Frame S. 2001. The renaissance of GSK3. Nat Rev Mol Cell Biol 2:769–776 [DOI] [PubMed] [Google Scholar]
- 35. Woodgett JR. 1990. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J 9:2431–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coss D, Jacobs SB, Bender CE, Mellon PL. 2004. A novel AP-1 site is critical for maximal induction of the follicle-stimulating hormone β gene by gonadotropin-releasing hormone. J Biol Chem 279:152–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mistry DS, Tsutsumi R, Fernandez M, Sharma S, Cardenas SA, Lawson MA, Webster NJ. 2011. Gonadotropin-releasing hormone pulse sensitivity of follicle-stimulating hormone-β gene is mediated by differential expression of positive regulatory activator protein 1 factors and corepressors SKIL and TGIF1. Mol Endocrinol 25:1387–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ruf F, Fink MY, Sealfon SC. 2003. Structure of the GnRH receptor-stimulated signaling network: insights from genomics. Front Neuroendocrinol 24:181–199 [DOI] [PubMed] [Google Scholar]
- 39. Wurmbach E, Yuen T, Ebersole BJ, Sealfon SC. 2001. Gonadotropin-releasing hormone receptor-coupled gene network organization. J Biol Chem 276:47195–47201 [DOI] [PubMed] [Google Scholar]
- 40. Yuen T, Wurmbach E, Ebersole BJ, Ruf F, Pfeffer RL, Sealfon SC. 2002. Coupling of GnRH concentration and the GnRH receptor-activated gene program. Mol Endocrinol 16:1145–1153 [DOI] [PubMed] [Google Scholar]
- 41. Attardi B, Winters SJ. 1993. Decay of follicle-stimulating hormone-β messenger RNA in the presence of transcriptional inhibitors and/or inhibin, activin, or follistatin. Mol Endocrinol 7:668–680 [DOI] [PubMed] [Google Scholar]
- 42. Matys V, Fricke E, Geffers R, Gössling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, Kloos DU, Land S, Lewicki-Potapov B, Michael H, Münch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E. 2003. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31:374–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kel AE, Gössling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. 2003. MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res 31:3576–3579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hebenstreit D, Fang M, Gu M, Charoensawan V, van Oudenaarden A, Teichmann SA. 2011. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol Syst Biol 7:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. 2008. Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell 133:340–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee MH, Koria P, Qu J, Andreadis ST. 2009. JNK phosphorylates β-catenin and regulates adherens junctions. FASEB J 23:3874–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bonfil D, Chuderland D, Kraus S, Shahbazian D, Friedberg I, Seger R, Naor Z. 2004. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone β-subunit promoter. Endocrinology 145:2228–2244 [DOI] [PubMed] [Google Scholar]
- 48. Hernandez Gifford JA, Hunzicker-Dunn ME, Nilson JH. 2009. Conditional deletion of β-catenin mediated by Amhr2cre in mice causes female infertility. Biol Reprod 80:1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu H, Lin Y, Li W, Sun Z, Gao W, Zhang H, Xie L, Jiang F, Qin B, Yan T, Chen L, Zhao Y, Cao X, Wu Y, Lin B, Zhou H, Wong AS, Zhang XK, Zeng JZ. 2011. Regulation of Nur77 expression by β-catenin and its mitogenic effect in colon cancer cells. FASEB J 25:192–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kioussi C, Briata P, Baek SH, Rose DW, Hamblet NS, Herman T, Ohgi KA, Lin C, Gleiberman A, Wang J, Brault V, Ruiz-Lozano P, Nguyen HD, Kemler R, Glass CK, Wynshaw-Boris A, Rosenfeld MG. 2002. Identification of a Wnt/Dvl/β-Catenin → Pitx2 pathway mediating cell-type-specific proliferation during development. Cell 111:673–685 [DOI] [PubMed] [Google Scholar]
- 51. Seraj MJ, Samant RS, Verderame MF, Welch DR. 2000. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res 60:2764–2769 [PubMed] [Google Scholar]
- 52. Shevde LA, Samant RS, Goldberg SF, Sikaneta T, Alessandrini A, Donahue HJ, Mauger DT, Welch DR. 2002. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp Cell Res 273:229–239 [DOI] [PubMed] [Google Scholar]
- 53. Saunders MM, Seraj MJ, Li Z, Zhou Z, Winter CR, Welch DR, Donahue HJ. 2001. Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res 61:1765–1767 [PubMed] [Google Scholar]
- 54. Nikolaev AY, Papanikolaou NA, Li M, Qin J, Gu W. 2004. Identification of a novel BRMS1-homologue protein p40 as a component of the mSin3A/p33ING1b/HDAC1 deacetylase complex. Biochem Biophys Res Commun 323:1216–1222 [DOI] [PubMed] [Google Scholar]
- 55. Hurst DR, Edmonds MD, Scott GK, Benz CC, Vaidya KS, Welch DR. 2009. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer Res 69:1279–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Thackray VG, McGillivray SM, Mellon PL. 2006. Androgens, progestins, and glucocorticoids induce follicle-stimulating hormone beta-subunit gene expression at the level of the gonadotrope. Mol Endocrinol 20:2062–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.