

Abstract

The protein kinase ERK5 (MAPK7) is an emerging drug target for a variety of indications, in particular for cancer where it plays a key role mediating cell proliferation, survival, epithelial–mesenchymal transition, and angiogenesis. To date, no three-dimensional structure has been published that would allow rational design of inhibitors. To address this, we determined the X-ray crystal structure of the human ERK5 kinase domain in complex with a highly specific benzo[e]pyrimido[5,4-b]diazepine-6(11H)-one inhibitor. The structure reveals that specific residue differences in the ATP-binding site, compared to the related ERKs p38s and JNKs, allow for the development of ERK5-specific inhibitors. The selectivity of previously observed ERK5 inhibitors can also be rationalized using this structure, which provides a template for future development of inhibitors with potential for treatment of disease.
Introduction
ERK5, also known as MAPK7 and BMK1 (big map kinase), was first identified in 1995.1,2 It is the terminal kinase in the ERK5 signaling pathway that involves MEKK2/MEKK3 and MEK5, a pathway required for angiogenesis and placental development3−5 and for neural differentiation in Xenopus embryos,6 although experiments in mice show that ERK5 knockout does not block neural development but does affect the number of interneurons.7 Among other stimuli, ERK5 is activated by epidermal growth factor (EGF).8 It is expressed in many cell types1,2,9 and phosphorylates various substrates including c-Fos and Fra-1,10 Sap1A,11 myocyte enhancer factor 2 (MEF2),12 MEF2C,13 and c-Myc.14 Structurally, ERK5 differs from other MAPK family members in that it has an extended C-terminal region (hence, the name big map kinase), which may have an autoinhibitory role.9 The C-terminus also contains a transcriptional activation domain that interacts with MEF2D15 and that enhances the transactivation activity of activator protein 1 (AP-1), after it has itself been autophosphorylated by the activated ERK5 kinase domain.16 The region N-terminal to the kinase domain contains sequences for targeting to the cytoplasm, while in the C-terminal region there is a nuclear localization sequence (residues 505–539).17 ERK5 is found in both cytoplasmic and nuclear locations.9
The kinase domain itself has closest similarity to the kinase domains of MAPK3 (ERK1, 51%), MAPK1 (ERK2, 51%), MAPK11 (p38β, 47%), MAPK14 (p38α, 46%), MAPK13 (p38δ, 43%), NLK (nemo-like kinase, 43%), and MAPK12 (p38γ, 38%). Crystal structures have so far been determined for all human p38 and JNK MAPKs. Of the ERK family, there are structures for ERK1,18 ERK2,19 and ERK3 (PDB code 2I6L, unpublished). The only MAP kinase structures currently remaining unsolved are ERK5, ERK7, and the atypical MAP kinase ERK4. (Atypical MAP kinases have an alternative activation loop phosphorylation motif SEG, which has only one phosphorylation site compared to the TXY motif of typical MAPKs. A recent paper has shown that atypical MAPKs are phosphorylated on their activation loop by group 1 p21-activated kinases (PAKs), which leads to their activation.20) ERK5 is activated by phosphorylation on Thr219 and Tyr221 by MEK5 after which ERK5 autophosphorylates its C-terminal region,21 including a nuclear localization signal motif that allows ERK5 to translocate to the nucleus.
ERK5 is a potential drug target for a number of indications including cancers.22,23 For instance, ERK5 hyperactivation and overexpression have been observed in particular in a large fraction of prostate and breast cancer,24 and high ERK5 expression levels have been associated with poor prognosis25 as well as bone and lymph node metastasis.26,27 In addition, the ERK5 locus is amplified in about 50% of all primary HCC (hepatocellular carcinoma).28 ERK5 is also a key regulator of tumor angiogenesis which has been demonstrated by the phenotype of ERK5 knockout mice which display multiple vascular defects3−5 and by targeted deletion in endothelial cells resulting in reduced mass and vascular density in xenograft models.29,30
To establish a structural model for the rational design of potent and selective inhibitors, we determined the X-ray crystal structure of the ERK5 kinase domain. In addition, we characterized the molecular mechanisms determining the specificity of selective benzo[e]pyrimido[5,4-b]diazepine-6(11H)-one inhibitors (25, 26), a privileged scaffold for the development of selective ATP competitive inhibitors.31,32
Results
Structure Determination
ERK5 protein was expressed and purified from baculovirus-infected insect cells. Multiple constructs comprising different regions of ERK5 could be used to produce soluble protein, each with an N-terminal hexahistidine purification tag. Crystals were obtained from a construct expressing residues 1–397 of human ERK5, which enabled the ERK5 structure to be determined to 2.8 Å resolution (Table 1). The electron density for the bound inhibitor 25 was of good quality and together with the three-dimensional shape of 25 allowed the inhibitor to be modeled with confidence. ERK5 residues 47–393 were resolved in the electron density with the exception of the activation loop residues 208–214, which were disordered (Figure 1), and residues 288–292, which form a short loop in the CMGC-specific insert. In other MAP kinase structures such as ERK219 phosphorylation of the Thr and Tyr of the activation loop TXY motif causes the activation loop to fold into an ordered structure. In the ERK5 structure the activation loop residues Thr219 and Tyr221, which would be phosphorylated by MEK5 during activation, were not phosphorylated according to electrospray ionization mass spectrometry, and no sign of phosphorylation was seen in the electron density. This segment of the activation loop was instead involved in crystal packing interactions, including Tyr221 which packs against the inhibitor 25 that is bound to another ERK5 molecule in the crystal (Supporting Information Figure 1). Therefore, it is unlikely that phosphorylated, activated ERK5 would crystallize in the same crystal form as the structure presented here. It is also possible that variation of the parts of the inhibitor that sit external to the ATP-binding site would prevent crystallization in this crystal form. Indeed, the proximity of the symmetry-related Tyr221 to the methoxy moiety of 25 is most likely the reason that we obtained crystals with 25 but not with 26 which has instead an ethoxy moiety.
Table 1. Data Collection and Refinement Statistics for Cocrystal Structure of ERK5 with 25.
| PDB code | 4B99 |
| space group | P41212 |
| no. of molecules in the asymmetric unit | 1 |
| unit cell dimensions | |
| a (Å) | 95 |
| b (Å) | 95 |
| c (Å) | 119 |
| α (deg) | 90 |
| β (deg) | 90 |
| γ (deg) | 90 |
| Data Collection | |
| beamline | Diamond I24 |
| resolution range (Å)a | 74.42–2.80 (2.99–2.80) |
| unique observationsa | 13868 (2466) |
| average multiplicitya | 4.1 (4.1) |
| completeness (%)a | 98.9 (99.2) |
| Rmerge a | 0.094 (0.711) |
| mean I/σ(I) a | 10.5 (2.4) |
| Refinement | |
| resolution range (Å) | 74.42–2.80 |
| R, Rfree | 22.5, 28.7 |
| rms deviation from ideal bond length (Å) | 0.006 |
| rms deviation from ideal bond angle (deg) | 1.2 |
Values within parentheses refer to those of the highest resolution shell.
Figure 1.

(A) Structure of ERK5 bound to 25. The N-terminal lobe of the kinase is shown in blue and the C-terminal lobe in green. The activation loop and αC helix are shown in red, and the CMGC kinase family specific insert in the C-terminal lobe is shown in pink. The inhibitor ERK5-IN-1 (25) is shown in yellow. Dashed lines represent residues that were not observed in the structure because of disorder. (B) Chemical structures of inhibitors.
Structural Similarity to Other CMGC Kinases
The structure shows the features typical of an ERK family kinase including the CMGC-specific insert in the C-terminal lobe of the kinase domain between αG and αH, and the additional C-terminal helix that packs above and against the mechanistically important helix αC (Figure 1). In emphasis of the structural similarity, despite only 51% sequence identity, the ERK5 structure nevertheless superimposes with the inhibitor-bound structures of ERK2 (PDB code 2Z7L) and p38β (PDB code 3GP0) with root-mean-square deviations of 1.14 Å over 283 Cα atoms and 1.11 Å over 245 Cα atoms, respectively.
Comparison of the surface charges of ERK5 and ERK2 (Supporting Information Figure 2) reveals that despite the surfaces containing a large proportion of the nonconserved residues, there is a relatively similar pattern of surface charge. ERK5 contains a mostly positively charged substrate binding region, although there would be modification of parts of this surface upon phosphorylation and ordering of the activation loop. The MAPK docking peptide region is also quite well conserved as shown by comparison with the structure of the complex between ERK2 and a DUSP6 (MKP3) kinase interaction motif (KIM) docking peptide33 (Supporting Information Figure 2). The N-terminus of the KIM motif (R/K)2–3-X1–6-Φ-X-Φ (Φ = hydrophobic residue) binds to a negatively charged region on the back of the kinase domain, and the hydrophobic residues bind in a groove adjacent to the kinase hinge region. Both of these surfaces are present in the ERK5 structure, although the negatively charged region is less pronounced.
Inhibitor Binding to ERK5
Initial potent and selective ERK5 inhibitors have been previously identified by a scaffold expansion of the well-known 2-aminopyrido[2,3-d]pyrimidine kinase inhibitors leading to the discovery of benzo[e]pyrimido[5,4-b]diazepine-6(11H)-ones.34 This privileged inhibitor class led to the development of a number of highly selective kinase inhibitors targeting LRRK2, TTK, and Aurora.31,32 Further optimization of this scaffold for LRRK2 and ERK5 is discussed in a subsequent manuscript46 and led to the discovery of ERK5-IN-1 (26, Figure 1), a highly selective ERK5 inhibitor. Since 25 differs from 26 only in a solvent-exposed methyl ether, the following discussion of the ERK5/25 structure applies also to inhibitor 26.
While the inhibitor 25 binds to ERK5 in the same general binding mode as previously observed with this inhibitor scaffold (Figure 2), the core forms a much more three-dimensional shape, with the N-cyclopentyl substituent pointing upward toward the glycine-rich loop and binding against Ile61, and the 6,7,6 ring system curved around Leu189 on β7 in the base of the ATP-binding site. The inhibitor forms two hydrogen bonds with the backbone of the kinase hinge region at Met140 and a hydrogen bond via a water molecule to the backbone nitrogen of the DFG motif Asp200 and Glu102 from αC (Figure 2A,C). There are also possibilities for hydrogen bond formation via the inhibitor’s methoxy group, or amide oxygen, but at the resolution of the structure the assignment of these cannot be made unequivocally. The methoxyphenyl moiety binds parallel to the hinge region residue Ser142 and its peptide bonds to Glu141 and Asp143 (Figure 2C). Inhibitor 25 has excellent shape complementarity to the binding site, filling the majority of the available space (Figure 2B). The piperidine–piperazine moiety points out of the ATP binding site and lies adjacent to the glycine rich loop. However, the positions of the two rings may be more determined by the crystal packing (Supporting Information Figure 1), and in solution it is likely that they would be flexible.
Figure 2.

Active site of ERK5 with inhibitor 25 bound. (A) The N-terminal lobe is colored blue. The C-terminal lobe is colored green. The activation loop and αC helix are colored red, and compound 25 is colored yellow. (B) As in (A) showing the surface of the ERK5 ATP-binding site around the inhibitor. (C) View from an alternative angle to (A), with the glycine-rich loop (strands β1 and β2) removed for clarity. (D) Experimental 2mFo – DFc electron density contoured at 1.0σ around the inhibitor from two different viewing angles.
The structure explains why any substituent on the aromatic ring of the anthranilic acid, in particular at the 3 or 4 position, significantly decreased binding to ERK5. This aromatic ring binds against the backbone atoms of Gly199 and Asn187 (Figure 2A), and any larger substituent would clash; because of the N-cyclopentyl substituent, there is insufficient space above the inhibitor to allow the core 6,7,6 ring system to move upward to accommodate any additional substituent. The structure also explains why addition of an N-methyl group increased binding affinity, as this methyl group forms a favorable hydrophobic packing with Ile115 (Figure 2C).
Cellular Activity of 26
We used Western blot analysis to detect ERK5 autophosphorylation in HeLa cells. After activation by MEK5 phosphorylation on the ERK5 activation loop, ERK5 autophosphorylates at many other locations,21 and these many additional phosphorylations cause a mobility shift of autophosphoryated ERK5 on polyacrylamide gel electrophoresis.21 Dose response experiments demonstrated low nanomolar cellular activity of 26 as judged by the significant dose-dependent reduction of mobility shifted phosphorylated ERK5 bands from sorbitol stimulated cells. As expected, total and activated levels of ERK1 and ERK2 remained unaffected indicating selective inhibition of the MEK5/ERK5 but not the MEK/ERK1/2 pathway (Figure 3A).
Figure 3.

Inhibition of ERK5 autophosphorylation in HeLa cells and effect on AP-1 activity. (A) Shown are Western blot analysis of cellular extracts stimulated by sorbitol using total ERK5 antibodies (top panel), phospho ERK1/2 specific antibodies (middle panel), and an antibody detecting total ERK1/2 (lower panel). Phosphorylation of ERK5 is evident by the mobility shift of the ERK5 band. Increasing concentration of 26 leads to significant reduction of ERK5 autophosphorylation levels. (B) Compound 26 inhibits ERK5-mediated AP1 transcriptional activity. The pAP1-luciferase reporter and pRL-CMV-Renilla plasmids were co-transfected with plasmids encoding for ERK5 and constitutively active MEK5 (MEK5DD) in HEK293 cells. Cells were left alone (control) or treated with the indicated amounts of compound 26 for 24 h, and lysates were subjected to the dual-luciferase assay. Each value is expressed as % of controls (no inhibitor), as the mean ± SD of three different determinations and normalized using the Renilla values. From the three different experiments the EC50 for compound 26 against ERK5 was 4.2 ± 0.69 μM.
Once activated, ERK5 translocates to the nucleus where it enhances the transcriptional activity of several transcription factors, including the AP1 transcriptional complex.10,16 Therefore, we tested if 26 would inhibit ERK5-mediated AP1 transcriptional activity, to monitor the activity of 26 over one of ERK5’s cellular functions. An AP1-dependent luciferase gene reporter assay in HEK293 cells overexpressing ERK5 and a constitutively active MEK5 (MEK5-DD, S311D/T315D) was used (Supporting Information Figure 3). Compound 26 completely inhibited the ERK5-mediated AP1 transcriptional activity at 30 μM and had an EC50 of 4.2 ± 0.7 μM (Figure 3B). By contrast 24 had an EC50 of >30 μM (Supporting Information Figure 4).
Comparison of Inhibitor Binding to Mps1-IN-2 Bound to TTK
The TTK inhibitor Mps1-IN-2 (30, Figure 1B) from this series adopts a similar general binding arrangement, as seen in the TTK:30 cocrystal structure (PDB code 3H9F).32 However, the additional aromatic ring that converts the 6,7 ring system into a 6,7,6 system with a terminal anthranilic acid moiety in our new inhibitor series over 30 causes the inhibitor to adopt a much less planar shape (Figure 4). The additional aromatic ring imposes additional conformational restrictions on the central seven-membered ring such that, because of the N-cyclopentyl substituent, the observed conformation with the additional aromatic ring pointing down toward the DFG motif is the only possible arrangement.
Figure 4.

Comparison of binding of 25 to ERK5 with that of Mps1-IN-2 to TTK (PDB code 3H9F(32)). (A) The TTK protein is colored cyan with the activation loop, and αC helix is colored red. Inhibitor Mps1-IN-2 bound to TTK is colored green. The additional atoms present in 25 that are not present in Mps1-IN-2 are colored magenta, with the remainder of 25 colored yellow. (B) Surface of the TTK ATP-binding site around the inhibitor Mps1-IN-2.
Comparison of the ERK5:25 structure with the TTK:30 structure (Figure 4) shows that the additional aromatic ring would clash with Ile663 of TTK, explaining why the new series has lost TTK-binding ability (KINOMEscan data in ref (46)). This comparison shows clearly that these inhibitors require a small residue before the DFG motif at Gly199. Furthermore, the N-cyclopentyl substituent on the seven-membered ring forces the compound into a conformation with the 6–7–6 ring system curved around Leu189, making that residue essential too. In the absence of the N-cyclopentyl group the seven-membered ring could adopt an alternative conformation that would result in a flatter compound structure or even one in which the terminal aromatic ring pointed upward toward the top of the binding site and away from Gly199. KINOMEscan profiling of 25 and 26 compared to compounds without the N-cyclopentyl group shows that 25 and 26 have a strong binding preference for kinases that have a small residue before the DFG motif (ERK5:Gly199), and a Leu on β7 in the base of the ATP-binding site (Leu189), whereas those without the N-cyclopentyl are more relaxed in their requirements.46
Reasons for the Specificity of Inhibitors 25 and 26
Profiling of binding data shows that these inhibitors have an unusual distribution of protein targets, binding strongly only to a few isolated kinases from across the phylogenetic tree.46 The N-cyclopentyl-substituted compounds 25 and 26 in particular select isolated kinases from a subfamily of closely related members. The non-N-cyclopentyl-substituted compounds, including LRRK2-IN-1 (15), also partly show this unusual pattern except with binding to the families of RSK C-terminal kinase domains, PLKs, and TNK1/TNK2.
As a comparison with the binding of Mps1-IN-2 (30) shows, a small residue preceding the DFG motif (Gly199 in ERK5) and Leu in the bottom of the ATP-binding site (Leu189) are important for binding of the compounds that contain the N-cyclopentyl substituent on the seven-membered ring. A Gly and Leu in these positions are commonly found in human protein kinases; however, among ERKs, JNKs, and p38s, which are otherwise quite highly related, only ERK5 has this combination (Figure 5A). For example, ERK3 and ERK4 do have a small residue (Gly) before the DFG motif but they both have a Phe in place of Leu189. LRRK2 has an Ala and a Leu in these positions, fulfilling the favorable criteria for binding. The compounds without the N-cyclopentyl substituent can bind to certain kinases that do not fulfill these criteria, such as the C-terminal kinase domains of RSKs/MSKs, MYLK, or MKNK1/MKNK2 (Supporting Information Figure 5), which explains the lesser selectivity of 15.
Figure 5.

Important residues for inhibitor selectivity. (A) Sequence alignment of ERK, JNK, and p38 kinases covering ERK5 Leu189 and Gly199. Only this combination of Leu and Gly is favorable for binding of the N-cyclopentyl-substituted inhibitors. (B) Sequence alignment of DCAMKLs covering the loop between αC and β4. An Ile at this position is most favored. (C) Interactions of Ile115 in the ERK5 crystal structure. (D) Sequence alignment of PLKs as in (A).
Further illustrating the effect of the N-cyclopentyl substituent in making Leu189 essential, the binding data against the PLKs clearly show that the compounds with an N-cyclopentyl substituent bind only to PLK4, the only isoform with Leu; the other isoforms that have Phe in this position bind only the non N-cyclopentyl-substituted compounds (Figure 5D).
The Ile at the back of the pocket (Ile115) is important. This residue forms a hydrophobic interaction with the N-methyl group of the inhibitor (Figure 5C). The cross-reacting kinases DCAMKL1, DCAMKL2, and DCAMKL3 all have favorable equivalents to Gly199 and Leu189, but only DCAMKL2 has an Ile equivalent to Ile115; DCAMKL1 and DCAMKL3 have Val (Figure 5B), and the binding data show that the inhibitors bind to DCAMKL2 with significantly greater affinity. This residue is strongly conserved as Ile, Val, Leu, or Phe in the kinome; however, the structure suggests that any hydrophobic residue other than Ile would not form as good a binding interaction.
The Asp at the end of the hinge region (Asp143) appears to be important. Across the kinome, a Gly at this position is the most common residue. However, all kinases bound by this inhibitor series have a larger residue such as Asp (ERK5), Ser (LRRK2), Leu (PLK1, PLK3, DCAMKLs, RIPK5), Met (PLK4), or Val (AURKA). This residue position is presumably important to sandwich the 2-methoxy-4-carboxamidyl-anilino moiety against the hinge region to which it forms hydrogen bonds (Figure 2C).
Overall, these inhibitors appear to achieve their surprising ERK5 specificity by relying on steric constraints against specific active site residues to prevent inhibitor binding to kinases otherwise closely related to ERK5; with more distantly related kinases that could bind the inhibitors without steric hindrance, the affinities are presumably lower because of each kinase not having the other residues important for binding affinity. It is also possible that some kinases are unable to position their glycine-rich loop to allow enough space for the inhibitor’s N-cyclopentyl group. Although rotation of a kinase N-lobe relative to the C-lobe is commonly seen when binding to diverse inhibitors, the rigid three-dimensional core of this inhibitor series couples the rotation of the N-lobe to the hydrogen bonding on the hinge region, while the inhibitor’s orientation relative to the C-lobe is fixed by the interactions with Leu189 (Figure 6); it may be that some kinases cannot appropriately position their glycine-rich loops while maintaining the hinge hydrogen bonds.
Figure 6.
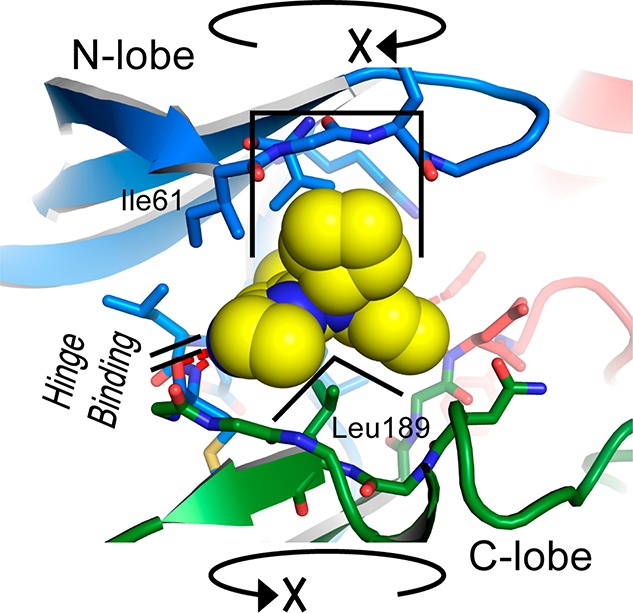
Three-dimensional core of the inhibitor selects a specific conformation of the kinase with respect to rotation of the N-lobe against the C-lobe.
Among the weaker bound kinases there are nevertheless some outliers that are not so easily explained, such as TNK1/TNK2 which have favorable equivalents to Gly199, Leu189, and Ile115, except TNK1 which has a Val instead of Ile115; but it is TNK1 that has the slightly higher binding affinity compared to TNK2 (data in ref (46)). However, overall there are no strongly bound intrasubfamily selectivities that are not explained by the criteria above.
A fuller sequence alignment of kinases relevant to the discussion here can be found in the Supporting Information.
Previously the kinase inhibitors BIX02188 and BIX02189 have been reported by Boehringer Ingelheim as MEK5 inhibitors but then also shown to have strong binding to ERK5, as well as good selectivity for MEK5 and ERK5.35 These inhibitors are based on a different scaffold to 25/26 with an indolin-2-one core. BIX02188 and BIX02189 differ by the addition of two methyl groups to the 6-carboxamide moiety in BIX02189. BIX02188 and BIX02189 have IC50 values against ERK5 of 810 and 59 nM, respectively.35 However, no experimental binding mode of these inhibitors has been reported.
Although BIX02188 and BIX02189 have good selectivity for MEK5 and ERK5 among other MAPK family members, they also potently inhibit various tyrosine kinases (TKs).35 This is to be expected, since the 3-substituted indolin-2-ones have been seen in a variety of TK-targeting inhibitors such as sunitinib (SU11248, Pfizer Inc.). Compared to 25/26, BIX02188 and BIX02189 have greater conformational flexibility, and it may be that a more rigid derivative of the BIX02189 scaffold could be a starting point for more specific inhibitors against various kinases. Both possible modes of binding to ERK5 suggest that joining the phenyl and 3-((dimethylamino)methyl)aniline moieties to form a macrocycle might be the basis of a useful chemical series.
Conclusion
The structure of ERK5 helps to explain the observed structure–activity relationships of known ERK5 inhibitors, identifies active site features specific to ERK5, and provides a template to aid in the design of additional inhibitor series.
Materials and Methods
Cloning
DNA for MAPK7 isoform 1 residues 1–397 (gi|20986497) was PCR amplified and subcloned into an in-house pFASTBAC-based vector pFB-LIC-Bse36 from DNA in the Mammalian Gene Collection (IMAGE consortium clone identifier 4111084), using ligation-independent cloning. The resulting plasmid was used to generate a recombinant baculovirus by the Bac-to-Bac method (Invitrogen). The resulting baculovirus expressed the kinase domain of ERK5, with an N-terminal hexahistidine tag and TEV (tobacco etch virus) protease tag cleavage site (extension MGHHHHHHSSGVDLGTENLYFQ*S-).
Expression and Purification
The baculovirus was used to infect 3 L of Spodoptera frugiperda cells (Sf9) in suspension culture at a density of 2 × 106 cells/mL in Insect-XPRESS medium (Lonza). The flasks were shaken at 27 °C for 48 h. The cells were harvested by centrifugation at 1000g, resuspended in 45 mL of lysis buffer (50 mM Tris, pH 7.8, 200 mM NaCl, 20 mM imidazole, 0.5 mM TCEP, 1:2000 dilution of Sigma protease inhibitor cocktail), and frozen at −20 °C until further use. The frozen cell pellet was thawed, and the cells were lysed by sonication. Polyethyleneimine (PEI) was added to a concentration of 0.1%, and the insoluble debris was removed by centrifugation.
The supernatant was bound to 6 mL of Ni Sepharose resin (GE Healthcare) and washed with 50 mL of lysis buffer, 50 mL of lysis buffer containing 40 mM imidazole and 1 M NaCl, and 50 mL of lysis buffer containing 60 mM imidazole. Finally the resin was eluted with 36 mL of lysis buffer containing 250 mM imidazole. The 60 and 250 mM imidazole elution fractions were combined, and tobacco etch virus (TEV) protease was added. After overnight incubation at 4 °C the protein was concentrated by ultrafiltration. The concentrated protein was injected onto an S200 16/60 gel filtration column (GE Healthcare) in 20 mM Tris, pH 7.8, 200 mM NaCl, 0.5 mM TCEP (GF buffer). Fractions containing ERK5 were pooled and passed through 1 mL of Ni Sepharose resin. The resin was eluted with 5 mL of GF buffer, then 5 mL of GF buffer containing 10 mM and then 20 mM imidazole. The flow-through and elution fractions were combined, concentrated by ultrafiltration, and injected a second time onto an S200 16/60 gel filtration column in GF buffer. Fractions containing ERK5 were pooled.
Protein identity was confirmed by mass spectrometry under denaturing conditions (expected 44 942.7 Da, observed 44 943.7 Da).
Crystallization and Data Collection
Inhibitor 25 was added to the protein such that the final inhibitor concentration after subsequent concentration of the sample would be 1 mM. The sample was concentrated by ultrafiltration to a protein concentration of 12.6 mg/mL (measured by absorbance at 280 nm).
Crystals were obtained using the sitting drop vapor diffusion method at 4 °C. Crystals grew from a mixture of 75 nL of protein and 75 nL of a well solution containing 0.05 M CaCl2, 0.1 M MES, pH 6.0, 20% PEG 6000, 10% ethylene glycol. Crystals were cryoprotected using the well solution supplemented to 25% ethylene glycol and flash-frozen in liquid nitrogen.
X-ray diffraction data was collected at the Diamond synchrotron, beamline I24. Data collection statistics can be found in Table 1.
Structure Determination
The diffraction data were indexed and integrated using MOSFLM37 and scaled using AIMLESS.38 The structure was solved by molecular replacement using Phaser39 and the structure of p38β (PDB code 3GC9)40 as a search model. There was one molecule in the asymmetric unit. The model was built using Coot41 and refined with REFMAC5.42 Rebuilding and refinement (including refinement of TLS parameters and addition of atoms for the inhibitor) resulted in the final model. Atomic restraints for the inhibitor were generated using PRODRG,43 and the model was validated using MolProbity.44
Western Blot Analysis
HeLa cells were maintained in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 50 U/mL penicillin G, and 50 μg/mL streptomycin. Before use HeLa cells were serum starved for 16 h in DMEM (Dulbecco’s modified Eagle medium) supplemented with 2 mM l-glutamine, 50 U/mL penicillin G, and 50 μg/mL streptomycin. HeLa cells were then incubated with ERK5-IN-1 at the indicated concentrations for 1 h prior to stimulation with 0.5 M sorbitol for 30 min. Cells were lysed in Triton lysis buffer (50 mM Tris-HCl, pH 7.5, 1 mM EGTA, 1 mM EDTA, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 1 mM sodium pyrophosphate, 0.27 M sucrose, 1 μM microcystin-LR, 1% (v/v) Triton X-100, 0.1% (v/v) 2-mercaptoethanol) and 20 μg of protein loaded per well. Samples were run on 8% polyacrylamide gels using standard methods. Proteins were transferred onto nitrocellulose membranes and specific proteins detected by immunoblotting. Total ERK5 antibody was obtained from Sigma, and total ERK1/2 and phospho ERK1/2 were purchased from Cell Signaling Technology. HRP-conjugated secondary antibodies were purchased from Pierce (Cheshire, U.K.), and detection was performed using the ECL reagent from Amersham Biosciences.
AP1-Dependent Luciferase Gene Reporter Assay
DNA Constructs. We used the pEBG-2T vector encoding for GST-tagged full-length human ERK5 and a pCMV plasmid encoding HA-tagged human MEK5-DD.45 AP1-luciferase vector was purchased from Stratagene, and pRL-CMV-Renilla was purchased from Promega.
Reporter Luciferase Assay. HEK293 cells were cultured at 37 °C under humidified air (5% CO2), using DMEM supplemented with 10% FBS (fetal bovine serum) and penicillin/streptomycin antibiotics. Cells were transfected using PEI (Warrington, U.S.). HEK293 cells cultured in 12-well plates were transfected with 500 ng of DNA, which contained plasmids encoding for AP-1-driven luciferase reporter (150 ng), Renilla (50 ng), ERK5 (100 ng), and MEK5-DD (200 ng). Three hours after transfection, the medium was changed and inhibitor compounds (dissolved in DMSO) were added at the indicated final concentrations. The concentration of DMSO in the culture medium did not exceed 0.3%. At 36 h later, luciferase activity assay was performed using the dual-luciferase reporter assay kit (Promega) in a Clarity luminescence microplate reader (BioTek Instruments). Results are presented as AP1-luciferase values normalized against Renilla luciferase activity. Data were obtained from triplicate determinations from three different experiments and analyzed by nonlinear regression using GraphPad software (GraphPad Software Inc.).
Acknowledgments
J.M.E., J.W., and S.K. are grateful for financial support by the SGC, a registered charity (No. 1097737) that receives funds from the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada, GlaxoSmithKline, Pfizer, Eli Lilly, Takeda, AbbVie, the Novartis Research Foundation, the Ontario Ministry of Research and Innovation, and the Wellcome Trust (No. 092809/Z/10/Z).
Supporting Information Available
Figures showing inhibitor interaction, charged surfaces, activation of ERK5, EC50 data, sequence alignment of N-terminal regions, and protein sequences. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhou G.; Bao Z. Q.; Dixon J. E. Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 1995, 270, 12665–12669. [DOI] [PubMed] [Google Scholar]
- Lee J. D.; Ulevitch R. J.; Han J. Primary structure of BMK1: a new mammalian map kinase. Biochem. Biophys. Res. Commun. 1995, 213, 715–724. [DOI] [PubMed] [Google Scholar]
- Sohn S. J.; Sarvis B. K.; Cado D.; Winoto A. ERK5MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J. Biol. Chem. 2002, 277, 43344–43351. [DOI] [PubMed] [Google Scholar]
- Regan C. P.; Li W.; Boucher D. M.; Spatz S.; Su M. S.; Kuida K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 9248–9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L.; Carr J.; Ashby P. R.; Murry-Tait V.; Thompson C.; Arthur J. S. C. Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev. Biol. 2003, 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto S.; Kusakabe M.; Nishida E. Requirement of the MEK5-ERK5 pathway for neural differentiation in Xenopus embryonic development. EMBO Rep. 2005, 6, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J.; Pan Y.-W.; Wang Z.; Chang S.-Y.; Wang W.; Wang X.; Tournier C.; Storm D. R.; Xia Z. Targeted deletion of ERK5MAP kinase in the developing nervous system impairs development of GABAergic interneurons in the main olfactory bulb and behavioral discrimination between structurally similar odorants. J. Neurosci. 2012, 32, 4118–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y.; Tapping R. I.; Huang S.; Watson M. H.; Ulevitch R. J.; Lee J. D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998, 395, 713–716. [DOI] [PubMed] [Google Scholar]
- Buschbeck M.; Ullrich A. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J. Biol. Chem. 2005, 280, 2659–2067. [DOI] [PubMed] [Google Scholar]
- Terasawa K.; Okazaki K.; Nishida E. Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes Cells 2003, 8, 263–273. [DOI] [PubMed] [Google Scholar]
- Kamakura S.; Moriguchi T.; Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J. Biol. Chem. 1999, 274, 26563–26571. [DOI] [PubMed] [Google Scholar]
- Yang C. C.; Ornatsky O. I.; McDermott J. C.; Cruz T. F.; Prody C. A. Interaction of myocyte enhancer factor 2 (MEF2) with a mitogen-activated protein kinase, ERK5/BMK1. Nucleic Acids. Res. 1998, 26, 4771–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y.; Kravchenko V. V; Tapping R. I.; Han J.; Ulevitch R. J.; Lee J. D. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997, 16, 7054–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English J. M.; Pearson G.; Baer R.; Cobb M. H. Identification of substrates and regulators of the mitogen-activated protein kinase ERK5 using chimeric protein kinases. J. Biol. Chem. 1998, 273, 3854–3860. [DOI] [PubMed] [Google Scholar]
- Kasler H. G.; Victoria J.; Duramad O.; Winoto A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 2000, 20, 8382–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto H.; Kondoh K.; Nishimoto S.; Terasawa K.; Nishida E. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J. Biol. Chem. 2007, 282, 35449–35456. [DOI] [PubMed] [Google Scholar]
- Yan C.; Luo H.; Lee J. D.; Abe J.; Berk B. C. Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J. Biol. Chem. 2001, 276, 10870–10878. [DOI] [PubMed] [Google Scholar]
- Kinoshita T.; Yoshida I.; Nakae S.; Okita K.; Gouda M.; Matsubara M.; Yokota K.; Ishiguro H.; Tada T. Crystal structure of human mono-phosphorylated ERK1 at Tyr204. Biochem. Biophys. Res. Commun. 2008, 377, 1123–1127. [DOI] [PubMed] [Google Scholar]
- Canagarajah B. J.; Khokhlatchev A.; Cobb M. H.; Goldsmith E. J. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell 1997, 90, 859–869. [DOI] [PubMed] [Google Scholar]
- Déléris P.; Trost M.; Topisirovic I.; Tanguay P.-L.; Borden K. L. B.; Thibault P.; Meloche S. Activation loop phosphorylation of ERK3/ERK4 by group I p21-activated kinases (PAKs) defines a novel PAK-ERK3/4-MAPK-activated protein kinase 5 signaling pathway. J. Biol. Chem. 2011, 286, 6470–6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody N.; Campbell D. G.; Morrice N.; Peggie M.; Cohen P. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem. J. 2003, 372, 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken S. R. C.; Ramsay A.; Heer R.; Mathers M. E.; Jenkins B. L.; Edwards J.; Robson C. N.; Marquez R.; Cohen P.; Leung H. Y. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene 2008, 27, 2978–2988. [DOI] [PubMed] [Google Scholar]
- Lochhead P. A.; Gilley R.; Cook S. J. ERK5 and its role in tumour development. Biochem. Soc. Trans. 2012, 40, 251–256. [DOI] [PubMed] [Google Scholar]
- Hsieh F. C.; Cheng G.; Lin J. Evaluation of potential Stat3-regulated genes in human breast cancer. Biochem. Biophys. Res. Commun. 2005, 335, 292–299. [DOI] [PubMed] [Google Scholar]
- Montero J. C.; Ocana A.; Abad M.; Ortiz-Ruiz M. J.; Pandiella A.; Esparis-Ogando A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS One 2009, 4, e5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P. B.; Jenkins B. L.; McCarthy L.; Thilak L.; Robson C. N.; Neal D. E.; Leung H. Y. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene 2003, 22, 1381–1389. [DOI] [PubMed] [Google Scholar]
- Sticht C.; Freier K.; Knopfle K.; Flechtenmacher C.; Pungs S.; Hofele C.; Hahn M.; Joos S.; Lichter P. Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC). Neoplasia 2008, 10, 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zen K.; Yasui K.; Nakajima T.; Zen Y.; Zen K.; Gen Y.; Mitsuyoshi H.; Minami M.; Mitsufuji S.; Tanaka S.; Itoh Y.; Nakanuma Y.; Taniwaki M.; Arii S.; Okanoue T.; Yoshikawa T. ERK5 is a target for gene amplification at 17p11 and promotes cell growth in hepatocellular carcinoma by regulating mitotic entry. Genes, Chromosomes Cancer 2009, 48, 109–120. [DOI] [PubMed] [Google Scholar]
- Hayashi M.; Kim S. W.; Imanaka-Yoshida K.; Yoshida T.; Abel E. D.; Eliceiri B.; Yang Y.; Ulevitch R. J.; Lee J. D. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J. Clin. Invest. 2004, 113, 1138–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M.; Lee J. D. Role of the BMK1/ERK5 signaling pathway: lessons from knockout mice. J. Mol. Med. (Berlin) 2004, 82, 800–808. [DOI] [PubMed] [Google Scholar]
- Deng X.; Dzamko N.; Prescott A.; Davies P.; Liu Q.; Yang Q.; Lee J. D.; Patricelli M. P.; Nomanbhoy T. K.; Alessi D. R.; Gray N. S. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N.; Jelluma N.; Filippakopoulos P.; Soundararajan M.; Manak M. S.; Kwon M.; Choi H. G.; Sim T.; Deveraux Q. L.; Rottmann S.; Pellman D.; Shah J. V; Kops G. J. P. L.; Knapp S.; Gray N. S. Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nat. Chem. Biol. 2010, 6, 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Sun J.-P.; Zhou B.; Zhang Z.-Y. Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 5326–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Yang Q.; Kwiatkowski N.; Sim T.; McDermott U.; Settleman J. E.; Lee J. D.; Gray N. S. Discovery of a benzo[e]pyrimido-[5,4-b][1,4]diazepin-6(11H)-one as a potent and selective inhibitor of big MAP kinase 1. ACS Med. Chem. Lett. 2011, 2, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatake R. J.; O’Neill M. M.; Kennedy C. A.; Wayne A. L.; Jakes S.; Wu D.; Kugler S. Z.; Kashem M. A.; Kaplita P.; Snow R. J. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem. Biophys. Res. Commun. 2008, 377, 120–125. [DOI] [PubMed] [Google Scholar]
- Savitsky P.; Bray J.; Cooper C. D. O.; Marsden B. D.; Mahajan P.; Burgess-Brown N. A.; Gileadi O. High-throughput production of human proteins for crystallization: the SGC experience. J. Struct. Biol. 2010, 172, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A. G. W.; Powell H. R. Processing diffraction data with Mosflm. Evol. Methods Macromol. Crystallogr. 2007, 245, 41–51. [Google Scholar]
- Evans P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. 2011, D67, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S. B.; Cameron P. M.; O’Keefe S. J.; Frantz-Wattley B.; Thompson J.; O’Neill E. A.; Tennis T.; Liu L.; Becker J. W.; Scapin G. The three-dimensional structure of MAP kinase p38beta: different features of the ATP-binding site in p38beta compared with p38alpha. Acta Crystallogr. 2009, D65, 777–785. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr. 2010, D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. 2011, D67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüttelkopf A. W.; Van Aalten D. M. F. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. 2004, D60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. 2010, D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iñesta-Vaquera F. A.; Campbell D. G.; Tournier C.; Gómez N.; Lizcano J. M.; Cuenda A. Alternative ERK5 regulation by phosphorylation during the cell cycle. Cell. Signalling 2010, 22, 1829–1837. [DOI] [PubMed] [Google Scholar]
- Deng X.; Elkins J.; Zhang J.; Yang Q.; Choi H. G.; Dzamko N.; Lee J.-D.; Sim T.; Kim N.; Knapp S.; Alessi D. R.; Gray N. S.. Structure determinants for big MAP kinase 1 and leucine rich repeat kinase 2 activities of benzo[e]pyrimido[5,4-b]diazepine-6(11H)-ones. J. Med. Chem., manuscript submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.