

Microsomal epoxide hydrolase (mEH, EPHX1) is a critical xenobiotic-metabolizing enzyme, catalyzing both detoxification and bioactivation reactions, including the carcinogenic metabolites of several polycyclic aromatic hydrocarbons. Here, another human EPHX1 transcript, E1-b′, is identified. It possesses unusual functional features in its 5′-untranslated region, including a GC-rich leader sequence and two upstream AUGs that encode for short peptides of 26 and 17 amino acids in length, respectively. The E1-b′ upstream open reading frames (uORFs) are functional, with their presence markedly inhibiting the translation of EPHX1 protein, both in cis and in trans configurations.
Keywords: microsomal epoxide hydrolase, EPHX1, upstream open reading frame (uORF), translational regulation, 5′-untranslated region (5′ UTR), gene expression
Abstract
Microsomal epoxide hydrolase (mEH, EPHX1) is a critical xenobiotic-metabolizing enzyme, catalyzing both detoxification and bioactivation reactions that direct the disposition of chemical epoxides, including the carcinogenic metabolites of several polycyclic aromatic hydrocarbons. Recently, we discovered that a previously unrecognized and primate-specific EPHX1 transcript, termed E1-b, was actually the predominant driver of EPHX1 expression in all human tissues. In this study, we identify another human EPHX1 transcript, designated as E1-b′. Unusually, both the E1-b and E1-b′ mRNA transcripts are generated from the use of a far upstream gene promoter, localized ∼18.5 kb 5′-upstream of the EPHX1 protein-coding region. Although expressed at comparatively lower levels than E1-b, the novel E1-b′ transcript is readily detected in all tissues examined, with highest levels maintained in human ovary. The E1-b′ mRNA possesses unusual functional features in its 5′-untranslated region, including a GC-rich leader sequence and two upstream AUGs that encode for short peptides of 26 and 17 amino acids in length, respectively. Results from in vitro transcription/translation assays and direct transfection in mammalian cells of either the E1-b′ transcript or the encoded peptides demonstrated that the E1-b′ upstream open reading frames (uORFs) are functional, with their presence markedly inhibiting the translation of EPHX1 protein, both in cis and in trans configurations. These unique uORF peptides exhibit no homology to any other known uORF sequences but likely function to mediate post-transcription regulation of EPHX1 and perhaps more broadly as translational regulators in human cells.
INTRODUCTION
Microsomal epoxide hydrolase (mEH, EPHX1) catalyzes the hydration of numerous xenobiotic epoxides; in particular, those derived by cytochrome P-450 (CYPs) mediated oxidation (Rappaport et al. 1996; Abdel-Rahman et al. 2005). In these respects, EPHX1 contributes important detoxification function, for example, in the metabolism of the occupationally important neurotoxins and potential carcinogens, styrene oxide and 1,2-epoxy-3-butene, to less reactive dihydrodiol derivatives (Fretland and Omiecinski 2000). However, EPHX1 also contributes critically to the bioactivation of carcinogenic polycyclic aromatic hydrocarbons, commonly encountered in smoke as combustion by-products. In concert with the CYPs, EPHX1 functions to convert these compounds into highly reactive electrophilic metabolites capable of forming covalent DNA adducts and thereby initiating mutational events (Buterin et al. 2000; Lloyd and Hanawalt 2000). The importance of the EPHX1 bioactivation function has been demonstrated directly in knockout mouse studies in which EPHX1-deficient mice are markedly resistant to 7,12-dimethylbenz[a]anthracene-induced tumorigenesis compared with their wild-type counterparts (Miyata et al. 1999). Although more poorly delineated, several lines of evidence also indicate endogenous roles for this enzyme, including that of steroid metabolism, bile acid transport, and in the vitamin K reductase complex (Guenthner et al. 1998).
In humans, EPHX1 activity levels appear quite variable among individuals (Hassett et al. 1998; Newman et al. 2005). While a variety of parameters may contribute to interindividual differences in expression, EPHX1 genetic polymorphism is one potential determinant. EPHX1 genetic variation described includes the existence of nonsynonymous polymorphisms in the gene’s coding region, as well as functional polymorphisms in the respective upstream promoter regions (Raaka et al. 1998; Yang et al. 2009). In these respects, molecular epidemiology reports have associated altered risk of lung and upper aerodigestive tract cancers with genetic variation in EPHX1 coding structure (Li et al. 2011). Variation within the EPHX1 5′-proximal gene promoter region was also suggested to contribute to differences in transcriptional regulation together with differential sensitivity of certain individuals to genotoxic effects resulting from 1,3-butadiene exposures (Raaka et al. 1998; Abdel-Rahman et al. 2005).
Although EPHX1 expression is detected in nearly all tissues, levels of expression are strikingly tissue selective, with high levels of enzymatic activity detected particularly in liver hepatocytes, as well as ovary and testes (Coller et al. 2001; Liang et al. 2005). Interestingly, the use of alternative promoters has been identified as an important regulatory feature driving tissue-specific expression of human EPHX1 transcripts containing differing first exons (Liang et al. 2005). One transcript, designated E1, initiates from the 5′-proximal promoter that directly flanks the structural coding region of the gene. However, the E1 transcript is expressed selectively in the liver (Liang et al. 2005). More recently, a far upstream promoter, designated E1-b, was identified that is localized ∼18.5 kb 5′ of the E1 exon, yet serves as the predominant driver of EPHX1 in all tissues examined, including liver (Liang et al. 2005). Because the human EPHX1 coding region extends from exon 2 to exon 9, the E1 and E1-b transcripts include distinct noncoding exon 1’s but otherwise are expected to generate the same EPHX1 protein. Importantly, the E1-b promoter region is conserved in nonhuman primates but has not been identified in rodents, indicating the evolution of differing transcriptional regulatory mechanisms across mammalian species.
Potentially important features of post-transcriptional regulatory control are often imbedded in the 5′-untranslated region of mRNA transcripts. In addition to the adoption of secondary structures that may characterize certain 5′ UTRs, short upstream open reading frames (ORFs) occur frequently in eukaryotic genes (Crowe et al. 2006). Estimates indicate that at up to 40% of mammalian mRNA sequences possess AUG trinucleotides upstream of the main coding sequence, with a substantial number of these motifs influencing translational efficiencies of the associated full-length transcripts (Morris and Geballe 2000; Churbanov et al. 2005; Crowe et al. 2006). The presence of 5′ uORFs may affect translation through several mechanisms, including as a barrier preventing the efficient scanning of the ribosomal complex through to the main downstream ORF, by regulating rates of ribosomal reinitiation, or through the generation of short peptides functionally encoded in the uORFs (Sachs and Geballe 2006).
In this study, we identified the existence of a novel human EPHX1 transcript, designated E1-b′. Like the previously characterized E1-b transcript, E1-b′ is also generated from the use of a far upstream promoter, in close genomic proximity to the E1-b start site. The E1-b′ transcript was detected in all human tissues examined, but its expression pattern was uniquely tissue-selectively regulated. Characterization of the 5′ UTR of the E1-b′ transcript revealed the presence of two uORFs, predicted to encode short peptides of 26 and 17 amino acids in length, respectively. We demonstrate that these imbedded uORFs impart a marked inhibitory effect on EPHX1 translation. These findings further demonstrate that expression of human EPHX1 is subjected to complex regulatory control, involving both transcriptional and post-transcriptional mechanisms.
RESULTS
Identification of the E1-b′ transcript by RCA-RACE
Using 5′-RACE methodology, two human tissue EPHX1 transcripts with unique first exons were previously identified (Liang et al. 2005). Significantly, these studies revealed that EPHX1 transcription is predominantly driven by a far upstream promoter, designated as E1-b. In the present study, we used a rolling circle amplification technique, i.e., Rapid Amplification of cDNA Ends (RCA-RACE) (Polidoros et al. 2006) to extend these analyses. This methodology uses gene-specific primers to eliminate the high background typically encountered with conventional RACE procedures, coupled with the use of a universal primer corresponding to the anchor sequence, to better enable the isolation of even low-level transcripts within a pool of amplified circular cDNA templates. The RACE results obtained using total RNA isolates from human A549 and HepG2 cells both revealed the presence of a unique EPHX1 transcript with a novel exon 1 sequence composition that we designate here as E1-b′. The presence of the E1-b′ transcript was further verified as present in a variety of human tissues, and its exon 1 sequence was localized by genomic mapping to an adjacent upstream promoter region to that previously identified as generating the E1-b transcript. A schematic of the overall EPHX1 gene structure from which the various transcripts are derived is presented in Figure 1A.
FIGURE 1.

Diverse 5′-leader sequence and translation efficiency of the EPHX1 variants. (A) Structural map of three human EPHX1 alternative exon 1s. Both E1-b and E1-b′ are localized ∼18.5 kb upstream of exon 2. Translation of the EPHX1 protein initiates in exon 2; therefore, each of the three exon 1 transcript variants encode the identical protein. (B) Exon 1 sequences of the E1-b and E1-b′ EPHX1 transcripts. E1-b′ was newly identified in this study. E1-b and E1-b′ are separated by 238-bp GC-rich genomic sequence. Two E1-b′ uAUGs are indicated. The main AUG within exon 2 is indicated in the lower portion of the figure. The open reading frames encoded by the first and second uAUGs are underlined with solid and dashed underlines, respectively. (C) Full-length E1, E1-b, and E1-b′ cDNAs were subcloned into pcDNA 3.1+ vectors along with a control template containing the main AUG initiating in exon 2. All constructs were expressed in an in vitro transcription/translation system. The resulting [35S]-methionine-labeled protein products were separated by SDS-PAGE, and the gels were dried and exposed to film. (D) Two micrograms of each of the constructs used in panel A was transfected into 293A cells. Twenty-four hours after transfection, equal amounts of total cell lysate were separated by SDS-PAGE, and EPHX1 protein levels were detected by Western blot using an anti-EPHX1 antibody. Computer densitometry was performed to assess relative quantification of EPHX1 protein expression, normalized to β-actin loading control, and the relative EPHX1 protein levels detected are indicated numerically under the respective lanes of the Western blot results.
Of interest, analysis of the NCBI AceView database (http://www.ncbi.nlm.nih.gov) that annotates cDNA-based transcript expression failed to identify the novel E-1b′ transcript. Nor was the E-1b′ transcript identified by review of the Genbank EST database (http://www.ncbi.nlm.nih.gov). Rather, these database sources indicated the presence of a yet independent EPHX1 transcript (e.g., HY168853, contributed by RIKEN), an apparent transcript comprising a combined [E1-b plus E1-b′] exon 1 structure. However, multiple RT-PCR-based experiments conducted with panels of human tissue mRNAs or with human cell lines were unsuccessful in corroborating the existence of the [E-1b plus E1-b′] EPHX1 exon 1 transcript (data not shown). Our results indicate that either the respective transcript has been mistakenly identified in the respective databases or that its level of expression is extremely low such that it may be of questionable biological significance.
The E1-b′ transcript identified in this study was characterized in detail and contains a 206-nt 5′ UTR that includes two upstream AUGs 5′ of the main ORF (uAUG1 and uAUG2) (Fig. 1B), with the main ORF residing in exon 2. The uAUG1 is in-frame with the EPHX1 coding region and predicted to encode a uORF composed of 26 amino acids that terminates 5′ of the full-length AUG start codon. Translation initiating at uAUG2 is predicted to encode an uORF generating a 17-amino-acid peptide. Unlike uORF1, the 17-amino-acid ORF is not in-frame with the main AUG of the full-length transcript and terminates at the +10 position, i.e., downstream from the main ORF translation initiation site. In its distal region, the E1-b′ 3′ UTR shares identical sequence with both the E1 and E1-b transcripts (Fig. 1B).
The E1-b′ 5′ UTR appears to possess a stable secondary structure, as modeled by Lasergene v.6 software (data not shown). The folding algorithms indicate the potential of this region to adopt a complex stem–loop configuration composed of nine stable individual hairpin structures with an overall Gibbs free energy value of ΔG = −65.58 kcal/mol. Secondary structures with a free energy value of less than −30 kcal/mol are considered stable and likely to impair translation (Kozak 1989). Together with the contributions of the two uORFs in this region, the features of the E1-b′ 5′ UTR are striking and may dictate regulatory control of E1-b′ translation.
In vitro translational efficiency of the 5′-UTR EPHX1 mRNAs
Expression vectors were constructed containing the full-length EPHX1 transcript coding region (the full-length ORF initiates translation in exon 2 and extends to its stop codon in exon 9) flanked by the various 5′-UTR sequences present in the endogenous E1, E1-b, or E1-b′ mRNAs. The constructs were tested using in vitro transcription/translation assays conducted in rabbit reticulocyte lysates. SDS-PAGE analysis of the derived 35S-methionine-labeled protein products demonstrated that the full-length E1 and E1-b products were translated at high efficiency; however, translation products of the E1-b′ 5′-UTR-containing sequence were barely detectable (Fig. 1C). Comparable results were obtained when the constructs were expressed in transiently transfected 293A cells; i.e., the translation mediated by the E1-b′ 5′-UTR transcript was ∼20% the level of the E1-b transcript (Fig. 1D). The results indicate that the presence of the E1-b′ 5′ UTR markedly impairs EPHX1 translation both in cell-free lysates and in transfected human cells.
Mutational and deletion effects on EPHX1 mRNAs translational efficiency
Previous reports have demonstrated that translational repression of a full-length mRNA can be assuaged when uAUG codons are mutated. As illustrated in Figure 2A, mutational analyses of the E1-b′ uAUGs were conducted, both individually and in combination. Mutation of the first uAUG (Mut1), which is in-frame with the full-length ORF and within a favorable Kozak translation initiation context, resulted in moderately increased translation efficiency (Fig. 2B). We anticipated that the uAUG2 (Mut2) mutation would affect translation to a lesser extent because this uAUG is localized within a poorer theoretical Kozak context for ribosome recognition. However, the Mut2 modification resulted in markedly enhanced EPHX1 protein translation, with levels generated comparable to the wild-type construct. The combined mutations, eliminating both uORFs, contributed the most dramatic effect on translation efficiency, increasing protein production fourfold to fivefold (Fig. 2B). These results suggest that both upstream AUGs within the E1-b′ 5′ UTR are efficiently recognized as initiation sites by the translational machinery and function to divert the ribosome’s efficacy of initiating translation at the downstream AUG that encodes the full-length cistron.
FIGURE 2.

(A) Schematic showing the E1-b′, Mut1, Mut2, and STOP1 constructs. In Mut1 and Mut2, the respective uAUGs were mutated individually to noninitiation codon GCAs. In STOP1, the stop codon of uORF1 was mutated to cysteine allowing the translation of the uORF1 to be extended through the EPHX1 coding region. (B) Full-length and mutant constructs were expressed using coupled in vitro transcription/translation reactions. In Mut1 + 2, both uAUGs were mutated together to noninitiation codon GCAs. The Del-50 and Del-110 lanes designate the use of templates where the 5′ leader sequence was deleted by either 50 or 110 nt from the 5-terminus, while preserving the normal uAUGs of the downstream uORFs. The STOP1 lane was obtained from a separate experiment indicating the existence of two translated products. One was a 50-kDa EPHX1 protein, as expected to be generated from the main AUG, while the other upper band is a higher-molecular-weight product resulting from the read-through translation of the mutated STOP1 uORF1, encoding an extended N terminus. (C) Computer densitometry was used to determine relative levels of quantification of EPHX1 protein expression from experiments conducted as described for panel A. Results from three separate experiments are indicated and compared with the control E1-b′ template versus the various mutated or deleted E1-b′ templates. (*) Significance at P < 0.05, as determined by ANOVA.
Functionality of the EPHX1 uAUG1
To demonstrate that the uORF1 is functional, the STOP1 mutation was created as illustrated in Figure 1A, altering the stop codon of uORF1 to a cysteine residue, thereby extending the uORF1 translation product in-frame through the main ORF to now encode a predicted ∼53-kDa protein. The E1-b′ and STOP1 mutation constructs were expressed in a cell-free transcription/translation system, and the products were visualized on an SDS-PAGE gel by 35S autoradiography. Translation of the E1-b′ construct yielded a single band, representing the full-length EPHX1 protein, whereas translation of the STOP1 mutated construct resulted in the production of two discrete protein bands, one derived from the full-length EPHX1 protein, and a second, larger protein product resulting from the uORF1 extension. These results, shown in Figure 2B, provide further evidence that ribosomal initiation can indeed occur at the uORF1, functionally interfering with the translation of the downstream main AUG.
Truncation of the 5′ UTR of E1-b′
The effects of sequential sequence deletions of E1-b′ 5′ UTR were analyzed using an in vitro transcription/translation system. Deletion of the initial 50 nt of the leader sequence resulted in an approximately twofold translational enhancement, whereas a 110-nt truncation, shortening the 5′ UTR to within 5 nt upstream of the first uAUG, resulted in an approximately threefold increase in translated product (Fig. 2C). Deletion of the 5′ UTR upstream of the uORFs would likely impact formation of secondary structure normally present within the 5′ region.
trans effects of the E1-b′ uORF1
To investigate potential trans effects of the E1-b′ uORFs, the natural EPHX1 E1 and E1-b constructs were translated simultaneously in the presence of the E1-b′ construct. E1 was used as a relative control because it does not contain any uAUGs within its 5′ UTR. Interestingly, the addition of the E1-b′ transcript to the reaction mix suppressed translation of both the E1 and E1-b transcripts (Fig. 3A). In a separate assay, increasing levels of the E1 transcript were added into the reactions and resulted in corresponding increases in the production of EPHX1 protein, demonstrating that the reaction mix components were not limiting in the translation system. These data suggest that the inhibitory effects of the E1-b′ transcript additions in translation reactions containing either E1 or E1-b transcripts result from trans-suppressive activities. To better assess the specificity of the trans effect, the uORF1-fs construct was created by frameshifting the uORF-1 peptide coding sequence such that the peptide product generated possesses a different amino acid sequence composite. The E1-b′ and uORF1-fs constructs were transfected into HepG2 human hepatoma cells, and their impact on the cellular levels of EPHX1 expression was analyzed by Western blot. Transfection of E1-b′ resulted in a dose-dependent reduction in the endogenous EPHX1 protein level (Fig. 3B), whereas transfection with the uORF1-fs construct was without effect.
FIGURE 3.
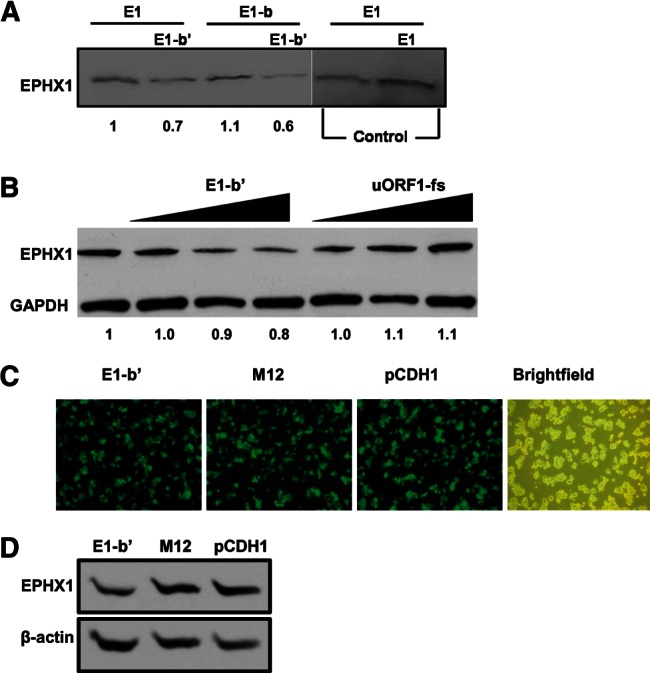
The effects of overexpression of E1-b′ on EPHX1 expression. (A) 0.5 µg of full-length EPHX1 E1 and E1-b plasmid constructs was expressed in the presence or absence of 0.5 µg of full-length EPHX1 E1-b′ or 0.5 µg of full-length EPHX1 E1 plasmid constructs. In vitro translated [35S]-methionine-labeled products were separated by SDS-PAGE, and gels were dried and exposed to film. The E1 transcript was used as a control to show that doubling the addition of this template to the reactions resulting in corresponding increased protein product formation, demonstrating that the translation resources were not limiting in these reactions. (B) Increasing amounts (2 µg, 3 µg, and 4 µg) of the full-length EPHX1 E1-b′ construct, or the mutated uORF1-fs construct, were transfected into HepG2 cells, and the effects on endogenous levels of EPHX1 protein expression were assessed. The uORF1-fs plasmid is a frameshifted mutation construct, in which uORF2 start codon (AUG) is mutated into noninitiating codon (GCA), and the entire uORF1 peptide coding sequence is frameshifted to allow for production of a new 18-amino-acid peptide with a completely different composition, and it was used as a control. Twenty-four hours after transfection, equal amounts of cell lysate were separated by SDS-PAGE, and EPHX1 proteins were detected in Western blots with an anti-EPHX1 antibody. Computer densitometry-based quantification of EPHX1 protein expression in each lane, normalized to loading control, is indicated under the Western blot results in both panels. (C) HepG2-C3A cells were transduced with pseudoviral supernatant carrying the full-length EPHX1 E1-b′ construct, M12 construct (in which both uAUGs are mutated into noninitiation codons GCAs) or control empty vector pCDH1. GFP expression in HepG2-C3A was driven by a promoter downstream from the E1-b′ and M12 construct cloning site. GFP images were captured with a fluorescent microscope at 200× magnification at day 3 post-lentiviral transduction, showing the transduction efficiency ∼80%–90%. (D) Transduced HepG2-C3A cells were subsequently harvested for Western blot analysis with an anti-EPHX1 antibody. The result shows that overexpression of the E1-b′ construct can decrease the total amount of EPHX1 protein detected, while overexpression of the M12 construct or empty vector control did not result in any significant changes.
Additionally, a lentivirus-based transduction system was used to express the E1-b′ transcript in HepG2 cells. A control in these studies included the use of the M12 vector, where the E1-b′ uAUGs were both mutated into noninitiation codons. High transduction efficiencies with these vectors, ∼80%–90%, were verified by expression of green fluorescent protein (GFP) driven by a separate downstream promoter present in the constructs (Fig. 3C). Lentivector-driven expression of the E1-b′ uORFs significantly reduced endogenous EPHX1 protein levels in the cells, compared with the similarly transduced M12 vector (Fig. 3D). These results corroborate those obtained with the HepG2 cell studies described above and support the concept that short peptides generated from the E1-b′ uORFs function to inhibit endogenous expression of cellular EPHX1 protein.
Inhibitory effect of synthetic E1-b′ uORF peptides on in vitro protein production
The 26- and 17-amino-acid peptides, predicted from the E1-b′ uORF1 and uORF2 sequences, respectively, were commercially synthesized and then tested directly for their effects on EPHX1 protein synthesis. The results obtained, using a mammalian cell-free translation system, demonstrated that addition of either or both peptides significantly inhibited the generation of EPHX1 protein in a sequence-specific and dose-dependent manner (Fig. 4B–E). Controls included synthetic scrambled peptides (see Fig. 4A). Although at high peptide concentration the scrambled uORF2 peptide also tended to inhibit EPHX1 protein synthesis, these effects were not statistically significant. Since the scrambled peptides share the same amino acid content as the uORF peptides, the inhibitory nature of uORF peptides may be contributed, at least in part, to overall charge dependency. To test this idea, a control peptide derived from soluble spoxide hydrolase (sEH; EPHX2) was substituted, representing a peptide of comparable length to the uORF2 EPHX1 peptide but possessing a completely different amino acid composition (the sEH peptide sequence is provided in Supplemental Table 2). Presence of the latter peptide did not inhibit EPHX1 protein synthesis, even at a relatively high concentration (100 µM) (Fig. 4F).
FIGURE 4.

E1-b′ uORF peptides can inhibit EPHX1 protein synthesis in mammalian cell lysate system. (A) Amino acid sequences of synthetic uORF1, uORF2 peptides, and corresponding scrambled peptide controls. (B,D) Rabbit reticulocyte lysates were programmed with full-length EPHX1 E1-b plasmid construct, and in vitro transcription/translation occurred in the presence of [35S]-methionine with increasing concentrations of uORF peptides and scrambled peptides. (C,E) Quantification of EPHX1 E1-b protein expression in the presence of varying peptide concentrations relative to control (no peptide). The data shown depict means and S.D. values from two separate experiments. (Varying peptide concentrations vs. no peptide; [*] P < 0.05, Student’s t-test.) (F) Rabbit reticulocyte lysates were programmed with EPHX1 E1-b transcript, and in vitro transcription/translation was performed as described previously in the presence of water solvent or 100 µM each peptide—uORF1 peptide, uORF2 peptide, or control sEH peptide.
E1-b′ uORF peptides do not contribute to nucleolytic or protein degradation pathways
When rabbit reticulocyte lysates were programmed with EPHX1 mRNA together with 40 µM each of the E1-b′ uORF peptides, the peptides were also inhibitory (data not shown), consistent with the data obtained using the in vitro transcription/translation system (Fig. 4). These results suggest that the E1-b′ uORF peptides inhibit EPHX1 protein production by interfering directly with translation. However, to test mechanistically whether potential nuclease or protease activities within the uORF peptide mix might be contributing to the reduced level of protein production observed, a kinetic analysis of EPHX1 E-1b mRNA stability was undertaken in the reticulocyte lysates in the absence or presence of 40 µM each uORF peptide or corresponding scrambled peptides. Fractionation of the [α-32P]ATP-labeled E1-b mRNAs was conducted in time-course studies using polyacrylamide gel electrophoresis. The RNA stability measures were unaffected by the presence of uORF peptides (Fig. 5A), indicating the absence of ribonucleolytic activity contributed by the peptides. The absence of proteolytic activity of the uORF peptide mix was similarly ascertained in reactions where EPHX1 mRNA was translated in the presence of [35S]-methionine for 45 min. Following additions of cycloheximide to stop the translations, the translation mixtures containing the nascent EPHX1 protein were incubated in the presence of 40 µM each peptide for 1 h and then analyzed by SDS-PAGE fractionation (Fig. 5B). The lack of detected degradation indicates that the uORF peptides are not contributing proteolytic activity and further support the idea that inhibitory effects contributed by the peptides involve direct interference with the translational process.
FIGURE 5.

(A) Kinetic analysis of RNA stability indicates uORF peptides do not contribute to nucleolytic activity. The 32P-labeled full-length EPHX1 E1-b mRNA was translated in the rabbit reticulocyte lysate system supplemented with 40 µM uORF peptides and equal amounts of corresponding scrambled peptides or just water solvent. An equal amount of lysate was removed from each individual reaction at different time points, and labeled RNA samples were extracted. Precipitated RNA samples were fractionated on a 4% polyacrylamide/8 M urea gel, dried, and exposed to film at −80°C with an intensifying screen. (B) uORF peptides do not induce nascent protein degradation. The 32P-labeled full-length EPHX1 E1-b mRNA was translated in the rabbit reticulocyte lysate system for 45 min at 30°C. Translation was stopped by the addition of cycloheximide at the final concentration of 500 ng/µL for 15 min at 30°C. Forty micromolar various peptides were then titrated into each reaction tube. After 1 h of incubation at room temperature, equal amounts of in vitro–translated [35S]-methionine-labeled EPHX1 E1-b were separated by an SDS-PAGE. Gels were dried and exposed to film overnight at −80°C. Each experiment was performed at least two times.
Translational inhibitory function of uORF peptides in mammalian cell lines
To test the functionality of the uORF peptides in intact mammalian cells, we used a peptide transfection reagent to deliver active uORF peptides into human lung carcinoma A549 cells. At 6 h or 24 h post-transfection; equal amounts of total protein were analyzed for EPHX1 protein expression level. The protein synthesis inhibitor cycloheximide was included as a positive control. Delivery of active uORF2 peptide (17 amino acids) into the mammalian cells resulted in significant inhibition of EPHX1 protein translational, as early as 6 h post-transfection, similar to the effects noted with cycloheximide. The uORF2 scrambled peptide (17 aaC) was without effect (Fig. 6A). At the 24 h time point, the effects of the uORF2 peptides were dissipated, likely due to their degradation over an extended period. The mock transfection group (R), where only the transfection reagent was added, and undergoing no transfection (N) (Fig. 6A), was also evaluated, but these treatments resulted in no detectable changes in the respective EPHX1 protein levels. Therefore, the uORF1 and uORF2 peptides exhibit their inhibitory function on EPHX1 translation within mammalian cells, effects are both potent and of rapid onset—clearly observable at 6 h post-transfection.
FIGURE 6.

(A) uORF peptides were transfected into A549 cells in a time-course experiment. At 6 h or 24 h post-transfection, cells were harvested for Western blot analysis. (17aa) Cells transfected with uORF2 peptide; (17aaC) cells transfected with uORF2 scrambled peptide; (CHX) cells treated with 10 μM cycloheximide; (R) mock transfected group; (N) normal cells. (B) uORF1 and uORF2 peptides were labeled with DyLight Fluor 633 and DyLight Fluor 488, respectively, at the N termini. Fluorescently labeled peptides were transfected into A549 cells in small amounts as tracers. Two hours later, the cells were harvested, fixed, stained with DAPI, and examined with confocal microscopy. (DIC) Differential interference contrast. (C) uORF peptides or scrambled peptides were transfected into A549 cells. Six hours later, cells were harvested and equal amounts of cell lysates were fractionated on an SDS-PAGE gel. Western blot analysis was performed to detect EPHX1 protein expression using a specific monoclonal antibody. β-Actin serves as loading control. Computer densitometry was performed to assess relative quantification of EPHX1 protein expression, normalized to β-actin loading control, and the relative EPHX1 protein levels detected are indicated numerically under the respective lanes of the Western blot results. Each experiment was performed at least two times.
To assess the delivery efficiencies of the uORF peptides in the transfected cells, the N termini of the uORF1 peptide and the uORF2 peptide were labeled with DyLight Fluor 633 and with DyLight Fluor 488, respectively. Tracer amounts of the labeled peptides were included in each transfection group. At 2 h post-transfection, the cells were fixed and analyzed by confocal microscopy. The results, shown in Figure 6B, confirmed the successful delivery of uORF peptides into intact A549 cells using the peptide transfection reagent.
The inhibitory activities of the uORF peptides on EPHX1 translation were assessed in A549 cells using SDS-PAGE analysis. The uORF 1 and uORF2 peptides were transfected both together and separately. At 6 h post-transfection, equivalent amounts of total cell protein were subjected to gel electrophoresis and analyzed for EPHX1 expression level. The levels of EPHX1 protein were clearly reduced in the uORF peptide transfected cells, unlike that of cells transfected with scrambled peptides (Fig. 6C). The level of inhibition was comparable to that produced by cycloheximide. Together, the results demonstrate that the E1-b′ derived uORF peptides were successfully delivered into the cells and that their presence markedly inhibited expression of endogenous EPHX1 protein.
Inhibition of EPHX1 protein translation by uORF peptides in wheat-germ extracts
The inhibitory function of the uORF peptides on EPHX1 protein synthesis was further examined in a plant-derived wheat germ in vitro transcription/translation system, programmed with the EPHX1 E1-b plasmid construct. Varying amounts of the respective uORF and scrambled peptides were titrated into the reactions, followed by fractionation by SDS-PAGE and visualization of the [35S]-methionine-labeled EPHX1 protein products by autoradiography. The results obtained were quite similar to those using the rabbit reticulocyte lysate system described earlier; EPHX1 translation in the coupled in vitro transcription/translation system wheat-germ extracts was markedly inhibited by the addition of the uORF1 and uORF2 peptides (Fig. 7). The uORF1 peptide inhibited EPHX1 protein translation in a dose-dependent manner, while uORF1 scrambled peptide was ineffective. Of interest, both the uORF2 peptide and the uORF2 scrambled peptide inhibited EPHX1 translation. However, a parallel experiment using a control peptide of similar length as uORF2 but with different amino acid composition had no effect on EPHX1 translation (data not shown), similar to the results presented in Figure 4F. These experiments were also repeated using wheat germ. In vitro translation extracts programmed with EPHX1 E1-b mRNA and highly similar results were obtained (data not shown). These findings imply that the uORF peptides interfere with the function of conserved translational machinery components. Furthermore, the translational interference contributed by the uORF peptides exhibits both amino acid sequence and composition/charge dependency.
FIGURE 7.

uORF peptides inhibit EPHX1 protein translation in wheat-germ extract. (A,C) Wheat-germ extract was programmed with full-length EPHX1 E1-b plasmid construct and in vitro transcription/translation occurred in the presence of [35S]-methionine. Different concentrations of uORF peptides and scrambled peptides were titrated into the wheat-germ extract system. (B,D) Quantification of EPHX1 E1-b protein expression in the presence of varying peptide concentrations, relative to control (no peptide). The data shown depict means and S.D. values from two separate experiments. (Varying peptide concentrations vs. no peptide; [*] P < 0.05; [**] P < 0.01; [***] P < 0.001; Student’s t-test.)
The N-terminal amino acid sequence of the E1-b′ uORF1 peptide is critical for its inhibitory function
It was envisioned that further mechanistic studies might be facilitated by use of tagged peptides. An E1-b′ uORF1 peptide was synthesized with an N-terminal biotin tag and subsequently tested using in vitro translation reactions. Unexpectedly, the presence of the N-terminal tag completely abolished the translational inhibitory function of the uORF1 peptide (Fig. 8A). To assess this issue further, a quantitative luciferase assay experiment was conducted in which several constructs were created, incorporating either the wild type E1-b′ 5′ UTR or specific uORF1 mutations and positioned upstream of the translation start site for the firefly luciferase reporter (see Fig. 8B). The constructs were transfected into A549 cells and assessed for luciferase activity. The results (Fig. 8C) demonstrated that although the presence of the unmodified E1-b′ UTR construct retained its inhibitory function, disruption of the initiation sequence of either uORF1 (M1) or uORF2 (M2) significantly restored luciferase activities, similar to the results presented earlier assessing these modifications within the context of an in vitro translation system (Fig. 2). Combined disruption of both of the uAUG1 and uAUG2 initiation codons resulted in the most striking increase in luciferase expression, about four to five times higher than the levels detected with the wild-type E1-b′ UTR sequence. Perhaps most interesting, missense mutations at the first few N-terminal amino acids of the uORF1 peptide (first N-term Mut) significantly restored luciferase activity to levels comparable to those achieved with the M1 construct, which is incapable of producing the inhibitory uORF1 peptide. Other missense mutations of the uORF1 peptide did not significantly modify the measured luciferase activities. These results are consistent with the in vitro translation data shown in Figure 8A, strongly suggesting that the N-terminal amino acid sequences of the uORF1 peptide are functionally critical in the inhibition scheme.
FIGURE 8.

The N-terminal amino acid sequence of E1-b′ uORF1 peptide is critical for its inhibitory function. (A) Rabbit reticulocyte lysates were programmed with full-length EPHX1 E1-b mRNA transcript, and in vitro translation occurred in the presence of [35S]-methionine with the addition of uORF peptides, control sEH peptide (40 µM, final concentration), or just water solvent and increasing concentration of Ahx-Biotin-tagged uORF1 peptide (40 µM, 80 µM, or 160 µM, final concentration). (B) Schematic showing the chimeric EPHX1-luciferase construct. E1-b′ 5′ UTR was cloned and replaced the luciferase 5′-leader sequence in the pGL3 control vector using the TK promoter. Amino acid coding sequences of wild-type E1-b′ uORF1 peptide and various mutation constructs were subsequently included: First N-terminal mutation, second N-terminal mutation, overlapping region (OPR) 6-amino-acid, and 2-amino-acid mutations. (C) Chimeric EPHX1-luciferase constructs were transfected into A549 cells. Renilla reniformis luciferase reporter (pRL-CMV) was used as an internal control. Luciferase assays were performed 24 h after transfection. M1, M2, and M12 constructs are chimeric EPHX1-luciferase constructs that contain mutated noninitiating start codons of uORF1, uORF2, or both, respectively. The data shown depict means and SD values derived from six separate experiments, each performed in triplicate. (Mutations of E1-b′ vs. E1-b′; [*] P < 0.05; [**] P < 0.01; [***] P < 0.001; Student’s t-test.)
E1-b′ transcript expression in human tissues
Quantitative RT-PCR analyses were conducted on total RNA isolated from a pool of 20 different human tissues. Of the total EPHX1 transcripts detected in most tissues, the E1-b′ transcript was expressed at a relatively low level, representing <10% of the total EPHX1 mRNA level. Previous studies have indicated that the highest levels of EPHX1 mRNA are detected in the liver; however, the E1-b′ transcript levels were preferentially expressed in the ovary (Fig. 9). We assessed the potential correlative relationship between E1-b′ transcript levels with that of EPHX1 protein expression across a sampling of ovary tissues obtained from different human donors. The characteristics of these tissue samples are described in Figure 10A. The results, shown in Figure 10, B and C, demonstrated a large interindividual variation among donors with respect to EPHX1 protein level as well as the measured levels of the E1-b′ and total EPHX1 transcripts. No correlation was apparent between the assessed protein expression and with the respective mRNA levels. These findings suggest the involvement of additional post-transcriptional regulation control of EPHX1 expression in ovary, similar to earlier reports of discordance of EPHX1 protein and mRNA levels in human liver and lung (Omiecinski et al. 1994).
FIGURE 9.

Quantification of EPHX1 E1-b′ transcript levels in human tissues. Real-time PCR was performed with RNA obtained from 20 human tissues; each RNA sample represents a pool of at least three individuals. The levels of the E1-b′ transcript and total EPHX1 (mEH) transcripts were quantified by absolute quantification based on standard curves determined using plasmid DNA templates, and the data are presented individually in the top and middle panels, respectively. The expression level of the E1-b′ transcript is presented as copy number per 1 ng of total RNA. (Lower panel) A combined data set, presented in log scale. The E1-b′ mRNA transcript is expressed significantly higher in ovary in comparison to other tissues (P < 0.001).
FIGURE 10.

Interindividual variation in EPHX1 transcript and protein expression profiles in human ovary tissues. (A) Characteristics of human ovary tissue samples. (B) E1-b′ and mEH mRNA expressions of different human tissue samples were measured relative to those of sample 4217. Data shown represent means and SD values for each sample performed in duplicate. The study was repeated twice. (C) Equal amounts of total proteins prepared from various human ovary tissues were fractionated on an SDS-PAGE gel and analyzed for mEH protein expression using anti-EPHX1 antibody. β-Actin serves as loading control.
DISCUSSION
Microsomal epoxide hydrolase contributes critical catalytic functions in the hydrolysis of many chemical epoxides, with resulting metabolites varying in their electrophilic character and bioreactivity (Fretland and Omiecinski 2000). In human tissues, EPHX1 is expressed tissue-selectively, with high levels detectable in tissues such as liver, ovary, testes, and lung (Liang et al. 2005). A primate-specific far upstream promoter has been identified that is the predominant driver of EPHX1 transcription across tissues, whereas a proximal promoter immediately upstream of the gene’s coding region is selectively active in liver tissues (Liang et al. 2005). In this study, a novel EPHX1 encoding transcript, termed E1-b′, was identified and characterized. Striking features of the E1-b′ transcript are its derivation from the far upstream E1-b promoter region, its preferential expression in human ovary tissues, and the presence of two upstream AUG codons within its 5′-UTR region. The uORFs are predicted to encode short peptides of 26 and 17 amino acids in length, respectively, and the experimental results described here demonstrate that these uORFs contribute to unique regulatory control of the translation process.
To briefly review, the hypothesis that the uAUGs of the EPHX1 E1-b′ transcript function to inhibit ribosome initiation at the main EPHX1 start codon was supported by data generated in mutational studies where the upstream AUGs of the E1-b′ uORFs were converted into GCAs, either individually or in combination. The results of these experiments demonstrated that the translation efficiency of the downstream main AUG is dependent on the two upstream ORFs (Fig. 2). Using synthetic E1-b′ uORF peptides, in vitro translation assays, and peptide-delivery methodology, we confirmed the inhibitory function of E1-b′ uORF peptides on EPHX1 translation (Figs. 4, 6). Mechanistically, uORF peptides did not contribute ribonucleolytic activity or impact protein degradation pathways (Fig. 5). The E1-b′ uORF peptides also retained their inhibitory function in a plant-derived in vitro translation system (Fig. 7), suggesting that the E1-b′ uORF-encoded nascent peptides function by disrupting activities of highly conserved features of the translational machinery. Furthermore, NH2-terminal modifications of the uORF1 or uORF2 peptides largely compromised their translational inhibitory function in transfected mammalian cells, within in vitro translation systems (Fig. 2) and in luciferase gene expression studies (Fig. 8). Together, these data strongly indicate that the NH2-terminal amino acid sequences of the uORF peptides play an important mechanistic role in directing translational inhibition. Interestingly, overexpression of E1-b′ in the presence of either the E-1 or E1-b mRNAs resulted in dramatic decreases in resulting EPHX1 expression relative to levels that otherwise typify these respective transcripts. When frameshifted E1-b′ uORF or M12 mutated constructs were tested in cultured cells, these inhibitory activities were lost, indicating that the E1-b′ uORFs contribute functional roles in inhibiting EPHX1 translation through trans suppression as well as by cis-regulatory mechanisms (Fig. 3).
Many studies have now delineated important roles for uORFs and their respectively encoded peptides as translational regulators (Lovett and Rogers 1996; Morris and Geballe 2000; Sachs and Geballe 2006). In these respects, ∼40% of mammalian mRNA sequences contain AUG trinucleotides upstream of the main open reading frame (Peri and Pandey 2001; Crowe et al. 2006). These open reading frames often exhibit certain levels of sequence conservation (Churbanov et al. 2005) and may encode functional polypeptides (Pendleton et al. 2005). It is noteworthy that in database scans the sequences of the E1-b′ encoded 26-amino-acid and 17-amino-acid peptides do not exhibit significant homology to any previously reported uORF peptide (data not shown). The regulation described for uORF peptides is exerted generally at the initiation phase of protein synthesis. In the ribosome scanning model, ribosomes initiate translation at the first upstream AUG encountered. Although leaky scanning and reinitiation of translation past an uORF may occur, the final translation efficiencies may be greatly reduced by the presence of uAUGs. Compelling studies in both prokaryotes and eukaryotes have demonstrated that translation initiation from uORFs may contribute to translation inhibition of the downstream main ORF (Imataka et al. 1994; Brown et al. 1999; Kozak 2005), and that these processes are often tightly controlled (Audic and Hartley 2004). In this context, results from cancer research investigations have demonstrated that the inefficient translation of uORFs is a regulatory mechanism used to limit expression of oncogene products that are required at low level but otherwise harmful if overexpressed. For example, the oncogene mdm2 is transcribed from two alternative promoters yielding L-mdm2, which contains two uORFs, and S-mdm2, which lacks these uORFs (Brown et al. 1999; Jin et al. 2003; Kozak 2005). L-mdm2 is the dominant transcript in normal human cells where the oncoprotein MDM2 is inefficiently produced. However, in tumor cells, elevated MDM2 levels result from a switch in promoter usage causing the up-regulation of the shorter mdm2 and thereby enhancing the translation of MDM2 ∼20-fold (Landers et al. 1997). Perhaps in certain cells, under stress situations imposed by the generation of epoxides, modulation of EPHX1 enzyme activity levels may be effected by a switch from E1-b′ gene promoter usage to the closely aligned E1-b promoter, which, in turn, may result in the generation of the shorter and simpler 5′-UTR transcript possessing higher translational capacity.
Studies in both eukaryotes and prokaryotes have demonstrated that uORF peptides can cause ribosomal stalling by various mechanisms, including interference with the peptidyl transferase center activity (Gu et al. 1994; Lovett and Rogers 1996), thereby inhibiting translation termination by preventing peptidyl-tRNA hydrolysis (Janzen et al. 2002) or by causing arrest of elongating or terminating ribosomes in response to a cellular signal (Luo et al. 1995; Wang and Sachs 1997; Hood et al. 2009). It is also noteworthy that studies have implicated the role of highly basic peptides in impeding translation, possibly by causing conformational changes in rRNA (Lovett and Rogers 1996). Since the E1-b′ uORF peptides contain highly basic amino acid content, this property may potentially explain the inhibitory function exhibited by the E1-b′ uORF peptides.
The relatively high levels of the E1-b′ transcript detected in human ovary was notable (Fig. 9) and may have implication in both estrogen metabolism and ovarian cytoprotection. For example, a previous study using a competitive chemical inhibitor to inhibit EPHX1 activity suggested the involvement of EPHX1 in estrogen production (Hattori et al. 2000). We attempted an shRNA approach that effectively down-regulated EPHX1 expression in human granulosa KGN cells, but in apparent contradiction to the cited report, our results indicated that decreased EPHX1 expression correlated with a higher level of estrogen secretion in these cells (data not shown). In either case, the results point to a potential involvement of EPHX1 in the metabolism of endogenous estrogen epoxide derivatives, a concept that is also consistent with an earlier report demonstrating that estroxide was a substrate for EPHX1 (Vogel et al. 1982). It is interesting to speculate that a high level of ovarian EPHX1 expression may provide a protective function in oocytes from effects of exposure to potentially reactive epoxides produced during steroidogenesis. The selective expression of the EPHX1 E1-b′ transcript in ovarian tissues may contribute to EPHX1 post-transcriptional regulation via inherent translational inhibitory activity associated with the E1-b′ uORF peptides.
In summary, despite sharing common coding region sequence within exons 2–9, there exists remarkable diversity among the three distinct EPHX1 mRNA variants, E1, E1-b, and E1-b′. Each of the transcripts demonstrates distinct tissue-specific expression profiles and each possesses a unique 5′ UTR. Here, we identified and characterized the E1-b′ EPHX1 transcript, and demonstrate that unlike the previously identified EPHX1 mRNAs, the E1-b′ transcript is expressed at highest levels in ovary, likely adopts a complex stem–loop structure in its 5′-UTR domain, and further, contains two uAUGs that encode peptides of 26 and 17 amino acids, respectively. These uAUG features in the E1-b′ transcript direct the translation of E1-b′ with significantly lower efficiency than either the E1 or E1 transcripts. The contribution of the EPHX1 E1-b′ transcript is significant, both with respect to the cis-inhibitory effects on translation contributed by its 5′ UTR, as well as the apparent trans-inhibitory effects on the translation process more generally. It is possible that these short uORF peptides may influence the active translational microenvironment and perhaps contribute a control mechanism operating independently of transcription and directly involved in the regulation of EPHX1 expression in human tissues.
MATERIALS AND METHODS
Materials
Unless indicated otherwise, all chemicals were purchased from Sigma-Aldrich, and all cell culture consumables were purchased from VWR Scientific. Primers for PCR were purchased from Integrated DNA Technologies. uORF peptides were synthesized by GenScript.
Cell culture
The human kidney cell lines, 293A and 293T, the human hepatoma cell line HepG2, HepG2-C3A, and the human lung carcinoma cell line A549 were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 0.15% sodium bicarbonate, 0.1 mM nonessential amino acids, 50 units/mL penicillin G, and 50 μg/mL streptomycin (all from Invitrogen Life Technologies). All cells were maintained at 37°C with 5% CO2.
Rolling circle amplification–rapid amplification of cDNA ends (RCA-RACE)
The rapid amplification of cDNA ends method (Polidoros et al. 2006) was used to simultaneously analyze the EPHX1 5′ and 3′ UTRs. RNA (2 µg) was reverse-transcribed into first-strand cDNA in a reaction containing 0.5 µg of oligo(dT)-adaptor primer [5′-GGCCACGCGTCGACTAGTAC(T)18-3′] phosphorylated at the 5′ end, 0.5 mM dNTPs, 10 mM dithiothreitol (DTT), 1× first-strand buffer (Invitrogen), and 200 units of Moloney murine leukemia virus (MMLV) reverse transcriptase (Invitrogen). The mixture was incubated for 1 h at 37°C followed by heat inactivation of reverse transcriptase for 15 min at 70°C. The RNA strand was then removed by the addition of 1 μL of RNase H (Invitrogen), incubated for 20 min at 37°C, and purified using the QIAquick PCR purification kit (QIAGEN). The purified cDNA (16 µL) was circularized using 1 μL of CircLigase (Epicentre Biotechnologies) for 1 h at 60°C followed by inactivation of the enzyme for 10 min at 80°C. The circularized cDNA was then amplified in an RCA reaction using the Φ29 DNA polymerase and random primers. The 50 µL of RCA mixture contained the following: 10 µL of circularized cDNA, 1 mM dNTPs, 200 µg/mL bovine serum albumin (BSA), 1× Φ29 DNA polymerase reaction buffer (New England Biolabs), 1 µL of Φ29 DNA polymerase, and 10 µM random hexamers modified by the addition of two phosphothioate linkages on the 3′ to confer resistance to the Φ29 exonuclease activity. Using the serially diluted RCA reaction as a template, PCR was carried out using AccuPOL DNA polymerase and EPHX1 forward (5′-TGGCAAAGTTTCCTCTTTGTCCCG-3′) and reverse (5′-ATGGAGGCCTGGAAAGGAAGTTCT-3′) primers. The PCR products were sequenced with specific E1, E1-b, or E1-b′ primers using a CEQ 8000 Genetic Analysis System (Beckman Coulter).
Plasmids
RNA was isolated from cultures of primary human hepatocytes using TRIzol Reagent (Invitrogen), and total RNA was converted to cDNA using the High Capacity cDNA Archive Kit (Applied Biosystems/Life Technologies). E1-b FP, E1 FP, E1-b′ FP, and EPHX1 RP were used for PCR amplification of full-length EPHX1 E1-b, E1, and E1-b′ transcripts, respectively. An additional round of PCR amplification was used to introduce the KpnI (5′) and BamHI (3′) restriction enzyme sites to the ends of E1-b, E1, and E1-b′ transcripts. Purified products were separated by agarose gel electrophoresis, and DNA fragments were purified using a QIAGEN gel purification kit (QIAGEN). Purified fragments were ligated into the KpnI/BamHI sites of pcDNA3.1+ and transformed into DH5α ultracompetent cells (Invitrogen). Colonies were screened by PCR to confirm the presence of the insert. Positive clones were grown overnight in 2-mL cultures, purified, and sequenced using a CEQ 8000 Genetic Analysis System (Beckman Coulter). Mutation in the 5′ UTR of E1-b′ transcript was performed using full-length E1-b′ as template and the provided primers by a two-step PCR protocol. Specifically, in Mut1 and Mut2 constructs, Mut1 FP/Mut1 RP and Mut2 FP/Mut2 RP were used to mutate start codon AUGs of uORF1 and uORF2, respectively, to noninitiating codon GCAs. In the Mut1+2 construct, both E1-b′ uAUGs are mutated to GCAs. In Del-50, and Del-110 constructs, the first 50 or 110 nucleotides of the E1-b′ transcript was correspondingly removed using Del-50 and Del-110 primers. In the STOP-1 construct, uORF1’s stop codon UGA was replaced with UGC, using STOP1 FP/STOP 1 RP primers. Finally, in the uORF1-fs construct, the uORF2 start codon (AUG) is mutated into a noninitiating codon (GCA) and uORF1 was frameshifted to produce a single 18-amino-acid uORF peptide of distinct sequence, using two primer pairs, FS1a FP/FS1a RP and FS1b FP/FS1b RP. The DNA primers used in PCR amplification and the predicted uORF1-fs peptide sequence are provided in the Supplemental Tables. Prior to transfection, plasmids were prepped using the Quantum Prep Plasmid Maxiprep Kit (Bio-Rad).
Protein expression in human cell lines
One day prior to transfection, cells were plated in six-well plates at a density of ∼400,000 cells/well. Cells were transfected with 2 μg of EPHX1 expression plasmid using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer’s protocol. Whole-cell lysates were generated 24 h post-transfection by sonication in RIPA buffer (50 mM Tris-HCl at pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA). Following centrifugation, the supernatant was stored at −20°C for subsequent analysis.
In vitro transcription and translation
For the EPHX1 transcript expression study (Figs. 1C, 2, 3A), plasmid DNA of EPHX1 variants (E1, E1-b, and E1-b′) and the mutated versions thereof, which contain the full-length EPHX1 cDNA sequence, were used as templates for in vitro transcription/translation in the TnT T7 Quick coupled reticulocyte lysate (Promega) according to the manufacturer’s protocol. Specifically, a total of 1 µg of plasmid DNA was transcribed/translated in the presence of [35S]-methionine in a 50-µL reaction. The reaction was incubated for 90 min at 37°C. Five microliters of reaction mix was separated in a pre-cast 10% Tris-HCl gel (Bio-Rad); the gel was subsequently dried and exposed to film overnight at −80°C.
For in vitro transcription and translation studies with peptide additions (Figs. 4, 7), the full-length EPHX1 E1-b plasmid construct was used as template in the TnT T7 Quick coupled reticulocyte lysate and the TnT T7 Coupled Wheat Germ Extract System (Promega). Various peptide concentrations were titrated to each reaction tube containing reaction master mix supplemented with 0.25 µg of plasmid DNA and followed by [35S]-methionine addition to a final reaction volume of 12.5 µL. For in vitro translation studies with peptide additions (Figs. 5A, 8A), rabbit reticulocyte lysate (nuclease treated; Promega) was programmed with 2 µg of 32P-labeled or unlabeled full-length EPHX1 E1-b mRNA supplemented with different peptide concentrations in a total volume of 50 µL. The reaction was performed as described previously.
For the protein degradation assay, rabbit reticulocyte lysate (nuclease treated; Promega) programmed with 2 µg of full-length EPHX1 E1-b mRNA in a total volume of 50 µL was translated for 45 min in the presence of [35S]-methionine at 30°C. The translation reactions were stopped by incubating the mixture with cycloheximide (500 ng/µL, final concentration) for 15 min at 30°C. 40 µM of various peptides or water (controls) were subsequently added to reaction mixtures and incubated for 60 min at room temperature. Translated protein was fractionated on pre-cast 10% gels.
Western immunoblotting
Equal amounts (10 µg) of total cell lysate were loaded on a 10% SDS-polyacrylamide gel. After separation, proteins were transferred to a polyvinylidene fluoride membrane. Detection of EPHX1 proteins was performed after membranes were incubated with EPHX1 peptide-directed polyclonal antibody (Hassett et al. 1997), followed by incubation with HRP-conjugated goat anti-rabbit antibody (Bio-Rad). Proteins were visualized using the Lumi-Light Western blotting substrate (Roche). Detection of GAPDH or β-actin using a GAPDH antibody (Sigma-Aldrich) or β-actin antibody (Santa Cruz Biotechnology), respectively, served as a loading control.
Lentiviral transduction
The E1-b′ construct and its M12 derivative, where uAUGs were mutated into noninitiating codon GCAs, were enzyme-digested out of pcDNA3.1+ vector and subcloned into the NheI/BamHI sites of pCDH1-MCS1-EF1-copGFP (pCDH1) cDNA lentivector (System Biosciences). Twenty-four hours prior to transfection, 293T cells were seeded in a 10-cm dish and grown to ∼70% confluence over the following day. On the day of transfection, 293T cells were washed with PBS to remove traces of serum and incubated in regular media supplemented with 10% HyClone FBS, 10 mM HEPES, 0.1 mM nonessential amino acids, and 1.5 g/L sodium bicarbonate. E1-b′, M12, or empty vector pCDH1 plasmids were cotransfected with pPACKH1 packaging plasmids into 293T cells using FuGENE 6 (Roche Applied Science) according to the manufacturer’s protocol. Pseudoviral supernatants harvested from packaging cells 48 h later were filtered through 0.2-µm Supor membranes (Acrodisc Syringe Filters; Pall Corporation) prior to infection of HepG2-C3A human hepatoma cells. GFP expression in target cells was visualized ∼48–72 h after infection using a Nikon inverted fluorescence microscope connected to a digital camera. SpotRT software was used to capture images as described previously (Zamule et al. 2008). Cells were subsequently harvested for protein analysis.
RNA stability assay
The full-length EPHX1 E1-b mRNA transcript was synthesized using RiboMAX Large Scale RNA Production System (Promega) and labeled with [α-32P]ATP and used to program rabbit reticulocyte lysate (nuclease treated; Promega) as described previously. In vitro translation was performed with unlabeled methionine in the presence or absence of 40 µM uORF1 or uORF 2 peptides. In vitro translation was performed for 0, 20, 40, or 60 min at 30°C, and the reactions were stopped by transferring the tubes to ice. RNA was then extracted from each reaction using TRIzol Reagent (Life Technologies), and precipitated RNA samples were fractionated on a 4% acrylamide/8 M urea gel, subsequently dried and exposed to film at −80°C with an intensifying screen.
Fluorescent labeling, transfection, and visualization of peptides
uORF peptides were labeled using DyLight Amine-Reactive Fluors (Thermo Fisher Scientific Inc.) according to the manufacturer’s protocol. Specifically, the uORF1 and the uORF2 peptides were labeled with DyLight Fluor 633 and DyLight Fluor 488, respectively, in 0.05 M sodium borate buffer at pH 8.5. Free dye was removed by dialyzing overnight at 4°C using a Slide-A-Lyzer G2 Dialysis Cassette (Thermo Fisher Scientific Inc.) with a 2K molecular weight cutoff. One day before transfection, 1 × 106 A549 cells were seeded in a 60-mm dish, and ∼24 h later the cells were washed with PBS and then transfected with 12 µg of peptide in Opti-MEM I Reduced Serum Media (Life Technologies) using the TurboFect Protein Transfection Reagent (Fermantas Inc.), according to the manufacturer’s protocol. After 2 h, transfection complexes were removed and cells were incubated in normal media. Cells were harvested at 6 h or 24 h post-transfection for protein analysis. For the peptide visualization experiment, 50,000 A549 cells were seeded in a Lab-Tek II Chamber Slide System 8-well glass slide (Nalge Nunc International Corp.). After 24 h, peptide transfection was performed as described previously, with 250 ng of labeled peptides as tracer and 750 ng of unlabeled peptides. After 2 h, the chambers were removed, and cells on glass slides were washed with PBS, fixed using 4% formaldehyde, stained with DAPI, and mounted using Crystal/Mount (Biomeda Corp.). Fluorescent images were captured using an Olympus Fluoview FV1000 confocal microscope.
Quantitative real-time PCR
Human tissue RNA was obtained from the FirstChoice Human total RNA survey panel (Ambion). Human ovary tissues were obtained from the Penn State Hershey Cancer Institute Tissue Bank. TRIzol Reagent (Life Technologies) was used to extract total RNA from the tissues. Total RNA was converted to cDNA using the High Capacity cDNA Archive Kit according to the manufacturer’s instructions. Real-time PCR was performed using Custom Taqman Gene Expression Products (Applied Biosystems) following the manufacturer’s standard protocol. The E1-b′ variant transcript was detected using the forward primer (5′-CGGCGGTGAAATGCACTTAATT-3′), reverse primer (5′- CACGGACGCGCATGAAAAT-3′) and probe (5′-CCAAGTCGGAACACTG-3′). 18S rRNA (Assay ID number Hs99999901_s1), total EPHX1 (Assay ID number Hs00164458_m1), and β-actin (Assay ID number Hs99999903_m1) were similarly detected using the Applied Biosystems Taqman Gene Expression Assay. Real-time RT-PCR data were analyzed using methods previously described (Olsavsky et al. 2007; Page et al. 2007). Plasmids containing cloned E1-b or E1-b′ full-length sequences were diluted to create standard curves ranging from 30 copies to 3 × 107 copies. DNA samples of known target sequence were used to verify the specificity of the assays.
Preparation of EPHX1-luciferase constructs
The chimeric EPHX1-Luciferase reporters were constructed such that the unique EPHX1 5′ UTRs were positioned directly upstream of the luciferase reporter gene. The E1-b′ 5′ UTRs were amplified using the forward primers HindIII E1-b′ and the reverse primer EPHX1-LucRP such that the amplicon contained a HindIII site on the 5′ end and a segment of the luciferase gene with an NarI site at the 3′ end. The fragment was then digested with HindIII and NarI and cloned into the pGL3 vector (Promega) such that the E1-b′ 5′ UTR replaced the luciferase 5′ UTR. The QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene) was used to introduce different mutations to the wild-type construct. DNA primers used for the mutagenesis are provided in Supplemental Table 1.
Luciferase reporter assay
One day prior to transfection, 200,000 A549 cells were plated in each well of a 24-well plate. Lipofectamine 2000 (Invitrogen) was used to deliver 0.8 µg of wild-type or mutated chimeric EPHX1 E1-b′-luciferase constructs and 0.05 µg of R. reniformis luciferase reporter (pRL-CMV) according to the manufacturer’s protocol. Transfection of each construct was performed in triplicates. Twenty-four hours after transfection, cells were harvested for luciferase reporter assay using the Dual-Glo Reporter Assay (Promega) and Veritas Microplate Luminometer (Turner Biosystems).
Secondary structure modeling
The secondary structures of EPHX1 E1-b′ mRNA 5′ UTRs were modeled using the GeneQuest module of Lasergene version 6 software (DNASTAR).
Statistical analysis
All statistical analyses were performed using GraphPad Prism v4.00 for Windows (GraphPad Software Inc.). A Student’s t-test or ANOVA followed by post hoc analyses were performed. Results were considered significant when P < 0.05.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by a USPHS grant from the National Institute of Environmental Health Sciences, ES016358, to C.J.O. The authors gratefully acknowledge the critical research discussions contributed by Dr. Elizabeth M. Laurenzana, Shengzhong Su, Dr. Tao Chen, and Denise M. Coslo. The authors also acknowledge the Huck Institutes of the Life Sciences’ Microscopy and Cytometry Facility and Staff for the assistance with confocal microscopy.
REFERENCES
- Abdel-Rahman SZ, Ammenheuser MM, Omiecinski CJ, Wickliffe JK, Rosenblatt JI, Ward JB Jr 2005. Variability in human sensitivity to 1,3-butadiene: Influence of polymorphisms in the 5′-flanking region of the microsomal epoxide hydrolase gene (EPHX1). Toxicol Sci 85: 624–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audic Y, Hartley RS 2004. Post-transcriptional regulation in cancer. Biol Cell 96: 479–498 [DOI] [PubMed] [Google Scholar]
- Brown CY, Mize GJ, Pineda M, George DL, Morris DR 1999. Role of two upstream open reading frames in the translational control of oncogene mdm2. Oncogene 18: 5631–5637 [DOI] [PubMed] [Google Scholar]
- Buterin T, Hess MT, Luneva N, Geacintov NE, Amin S, Kroth H, Seidel A, Naegeli H 2000. Unrepaired fjord region polycyclic aromatic hydrocarbon-DNA adducts in ras codon 61 mutational hot spots. Cancer Res 60: 1849–1856 [PubMed] [Google Scholar]
- Churbanov A, Rogozin IB, Babenko VN, Ali H, Koonin EV 2005. Evolutionary conservation suggests a regulatory function of AUG triplets in 5′-UTRs of eukaryotic genes. Nucleic Acids Res 33: 5512–5520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller JK, Fritz P, Zanger UM, Siegle I, Eichelbaum M, Kroemer HK, Murdter TE 2001. Distribution of microsomal epoxide hydrolase in humans: An immunohistochemical study in normal tissues, and benign and malignant tumours. Histochem J 33: 329–336 [DOI] [PubMed] [Google Scholar]
- Crowe ML, Wang XQ, Rothnagel JA 2006. Evidence for conservation and selection of upstream open reading frames suggests probable encoding of bioactive peptides. BMC Genomics 7: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fretland AJ, Omiecinski CJ 2000. Epoxide hydrolases: Biochemistry and molecular biology. Chem Biol Interact 129: 41–59 [DOI] [PubMed] [Google Scholar]
- Gu Z, Harrod R, Rogers EJ, Lovett PS 1994. Anti-peptidyl transferase leader peptides of attenuation-regulated chloramphenicol-resistance genes. Proc Natl Acad Sci 91: 5612–5616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenthner TM, Cai D, Wallin R 1998. Co-purification of microsomal epoxide hydrolase with the warfarin-sensitive vitamin K1 oxide reductase of the vitamin K cycle. Biochem Pharmacol 55: 169–175 [DOI] [PubMed] [Google Scholar]
- Hassett C, Lin J, Carty CL, Laurenzana EM, Omiecinski CJ 1997. Human hepatic microsomal epoxide hydrolase: Comparative analysis of polymorphic expression. Arch Biochem Biophys 337: 275–283 [DOI] [PubMed] [Google Scholar]
- Hassett C, Laurenzana EM, Sidhu JS, Omiecinski CJ 1998. Effects of chemical inducers on human microsomal epoxide hydrolase in primary hepatocyte cultures. Biochem Pharmacol 55: 1059–1069 [DOI] [PubMed] [Google Scholar]
- Hattori N, Fujiwara H, Maeda M, Fujii S, Ueda M 2000. Epoxide hydrolase affects estrogen production in the human ovary. Endocrinology 141: 3353–3365 [DOI] [PubMed] [Google Scholar]
- Hood HM, Neafsey DE, Galagan J, Sachs MS 2009. Evolutionary roles of upstream open reading frames in mediating gene regulation in fungi. Annu Rev Microbiol 63: 385–409 [DOI] [PubMed] [Google Scholar]
- Imataka H, Nakayama K, Yasumoto K, Mizuno A, Fujiikuriyama Y, Hayami M 1994. Cell-specific translational control of transcription factor BTEB expression—role of an upstream AUG in the 5′-untranslated region. J Biol Chem 269: 20668–20673 [PubMed] [Google Scholar]
- Janzen DM, Frolova L, Geballe AP 2002. Inhibition of translation termination mediated by an interaction of eukaryotic release factor 1 with a nascent peptidyl-tRNA. Mol Cell Biol 22: 8562–8570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin XP, Turcott E, Englehardt S, Mize GJ, Morris DR 2003. The two upstream open reading frames of oncogene mdm2 have different translational regulatory properties. J Biol Chem 278: 25716–25721 [DOI] [PubMed] [Google Scholar]
- Kozak M 1989. Circumstances and mechanisms of inhibition of translation by secondary structure in eukaryotic messenger-RNAs. Mol Cell Biol 9: 5134–5142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M 2005. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361: 13–37 [DOI] [PubMed] [Google Scholar]
- Landers JE, Cassel SL, George DL 1997. Translational enhancement of mdm2 oncogene expression in human tumor cells containing a stabilized wild-type p53 protein. Cancer Res 57: 3562–3568 [PubMed] [Google Scholar]
- Li X, Hu Z, Qu X, Zhu J, Li L, Ring BZ, Su L 2011. Putative EPHX1 enzyme activity is related with risk of lung and upper aerodigestive tract cancers: A comprehensive meta-analysis. PLoS ONE 6: e14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SH, Hassett C, Omiecinski CJ 2005. Alternative promoters determine tissue-specific expression profiles of the human microsomal epoxide hydrolase gene (EPHX1). Mol Pharmacol 67: 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd DR, Hanawalt PC 2000. p53-dependent global genomic repair of benzo[a]pyrene-7,8-diol-9,10-epoxide adducts in human cells. Cancer Res 60: 517–521 [PubMed] [Google Scholar]
- Lovett PS, Rogers EJ 1996. Ribosome regulation by the nascent peptide. Microbiol Rev 60: 366–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Freitag M, Sachs MS 1995. Translational regulation in response to changes in amino acid availability in Neurospora crassa. Mol Cell Biol 15: 5235–5245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata M, Kudo G, Lee YH, Yang TJ, Gelboin HV, Fernandez-Salguero P, Kimura S, Gonzalez FJ 1999. Targeted disruption of the microsomal epoxide hydrolase gene. Microsomal epoxide hydrolase is required for the carcinogenic activity of 7,12-dimethylbenz[a]anthracene. J Biol Chem 274: 23963–23968 [DOI] [PubMed] [Google Scholar]
- Morris DR, Geballe AP 2000. Upstream open reading frames as regulators of mRNA translation. Mol Cell Biol 20: 8635–8642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JW, Morisseau C, Hammock BD 2005. Epoxide hydrolases: Their roles and interactions with lipid metabolism. Prog Lipid Res 44: 1–51 [DOI] [PubMed] [Google Scholar]
- Olsavsky KM, Page JL, Johnson MC, Zarbl H, Strom SC, Omiecinski CJ 2007. Gene expression profiling and differentiation assessment in primary human hepatocyte cultures, established hepatoma cell lines, and human liver tissues. Toxicol Appl Pharmacol 222: 42–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omiecinski CJ, Aicher L, Swenson L 1994. Developmental expression of human microsomal epoxide hydrolase. J Pharmacol Exp Ther 269: 417–423 [PubMed] [Google Scholar]
- Page JL, Johnson MC, Olsavsky KM, Strom SC, Zarbl H, Omiecinski CJ 2007. Gene expression profiling of extracellular matrix as an effector of human hepatocyte phenotype in primary cell culture. Toxicol Sci 97: 384–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton LC, Goodwin BL, Solomonson LP, Eichler DC 2005. Regulation of endothelial argininosuccinate synthase expression and NO production by an upstream open reading frame. J Biol Chem 280: 24252–24260 [DOI] [PubMed] [Google Scholar]
- Peri S, Pandey A 2001. A reassessment of the translation initiation codon in vertebrates. Trends Genet 17: 685–687 [DOI] [PubMed] [Google Scholar]
- Polidoros AN, Pasentsis K, Tsaftaris AS 2006. Rolling circle amplification-RACE: A method for simultaneous isolation of 5′ and 3′ cDNA ends from amplified cDNA templates. BioTechniques 41: 35–40 [DOI] [PubMed] [Google Scholar]
- Raaka S, Hassett C, Omiecinski CJ 1998. Human microsomal epoxide hydrolase: 5′-flanking region genetic polymorphisms. Carcinogenesis 19: 387–393 [DOI] [PubMed] [Google Scholar]
- Rappaport SM, Yeowell-O’Connell K, Bodell W, Yager JW, Symanski E 1996. An investigation of multiple biomarkers among workers exposed to styrene and styrene-7,8-oxide. Cancer Res 56: 5410–5416 [PubMed] [Google Scholar]
- Sachs MS, Geballe AP 2006. Downstream control of upstream open reading frames. Genes Dev 20: 915–921 [DOI] [PubMed] [Google Scholar]
- Vogel BU, Bentley P, Oesch F 1982. Endogenous role of microsomal epoxide hydrolase. Ontogenesis, induction inhibition, tissue distribution, immunological behaviour and purification of microsomal epoxide hydrolase with 16α, 17α-epoxyandrostene-3-one as substrate. Eur J Biochem 126: 425–431 [PubMed] [Google Scholar]
- Wang Z, Sachs MS 1997. Ribosome stalling is responsible for arginine-specific translational attenuation in Neurospora crassa. Mol Cell Biol 17: 4904–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Liang SH, Weyant DM, Lazarus P, Gallagher CJ, Omiecinski CJ 2009. The expression of human microsomal epoxide hydrolase is predominantly driven by a genetically polymorphic far upstream promoter. J Pharmacol Exp Ther 330: 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamule SM, Strom SC, Omiecinski CJ 2008. Preservation of hepatic phenotype in lentiviral-transduced primary human hepatocytes. Chem Biol Interact 173: 179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.