

The DNA end-binding factor Ku binds to double-stranded DNA without sequence specificity, a property crucial for its broad biological roles in DNA rearrangement and DNA repair. Curiously, Ku also binds to some yeast telomerase RNAs to mediate RNP nuclear localization and telomere maintenance. Building from a previous demonstration that the DNA- and RNA-binding activities of Ku are mutually exclusive, the authors investigate the specificity of Ku RNA-binding activity in biochemical detail. In contrast to DNA binding, RNA binding has notable sequence and structure specificity. The delineation of Ku RNA-binding site preferences expands knowledge of protein–RNA recognition and allows the authors new insight into the generality of Ku RNA-binding activity.
Keywords: Ku heterodimer, RNA binding, telomerase, footprinting, chemical modification
Abstract
Most nucleic acid-binding proteins selectively bind either DNA or RNA, but not both nucleic acids. The Saccharomyces cerevisiae Ku heterodimer is unusual in that it has two very different biologically relevant binding modes: (1) Ku is a sequence-nonspecific double-stranded DNA end-binding protein with prominent roles in nonhomologous end-joining and telomeric capping, and (2) Ku associates with a specific stem–loop of TLC1, the RNA subunit of budding yeast telomerase, and is necessary for proper nuclear localization of this ribonucleoprotein enzyme. TLC1 RNA-binding and dsDNA-binding are mutually exclusive, so they may be mediated by the same site on Ku. Although dsDNA binding by Ku is well studied, much less is known about what features of an RNA hairpin enable specific recognition by Ku. To address this question, we localized the Ku-binding site of the TLC1 hairpin with single-nucleotide resolution using phosphorothioate footprinting, used chemical modification to identify an unpredicted motif within the hairpin secondary structure, and carried out mutagenesis of the stem–loop to ascertain the critical elements within the RNA that permit Ku binding. Finally, we provide evidence that the Ku-binding site is present in additional budding yeast telomerase RNAs and discuss the possibility that RNA binding is a conserved function of the Ku heterodimer.
INTRODUCTION
Telomerase is the enzyme that synthesizes telomeric DNA repeats to cap chromosome ends in eukaryotic cells. Telomerase is a ribonucleoprotein complex, and the conserved core enzyme is comprised of a reverse transcriptase (TERT) and an RNA component (TER) (Greider and Blackburn 1987; Lingner et al. 1997). TER contains an internal template, which is used by TERT to catalyze the addition of telomeric DNA repeats to chromosome termini during the late stages of semiconservative DNA replication. The core enzyme is sufficient for catalysis in vitro. However, additional protein cofactors are necessary in vivo to mediate essential functions such as nuclear localization, recruitment of telomerase to the telomere, and stimulation of telomerase repeat addition processivity (Egan and Collins 2012; Nandakumar and Cech 2013).
The telomerase core in the budding yeast Saccharomyces cerevisiae consists of the large 1.1-kb TLC1 RNA and Est2 (Ever shorter telomeres 2), the yeast TERT (Singer and Gottschling 1994; Lingner et al. 1997). TLC1 is present at ∼30 copies per haploid cell, which is less than the number of telomeric ends (Mozdy and Cech 2006). Consistent with limiting telomerase, only a subset of telomeres are extended by a few repeats each cell cycle, with preferential extension at telomeres with shorter tracts (Teixeira et al. 2004). Structurally, TLC1 contains three long helical arms that emanate from a conserved pseudoknot (Dandjinou et al. 2004; Lin et al. 2004; Zappulla and Cech 2004). Est2 binds to the central pseudoknot and template region, and each of the arms serves as a scaffold for one of the protein cofactors that comprise the holoenzyme (Zappulla and Cech 2004). The terminal arm contains the binding site for the Sm7 complex, another arm contains the Est1-binding site, and the arm extending from the template boundary helix contains the Ku-binding site (KBS) (Seto et al. 1999, 2002; Peterson et al. 2001).
The Ku heterodimer is comprised of two subunits that share the same overall topology, namely, an N-terminal α/β domain followed by a β-barrel domain and a variable C-terminal arm (Walker et al. 2001). The structural basis for the recognition of duplex DNA by Ku is illustrated in the cocrystal structure of human Ku bound to DNA (Walker et al. 2001). The two β-barrel domains of each subunit associate at a large dimerization interface. A preformed ring structure is formed by two loops protruding from each subunit to bridge the interface. DNA binds in the central cavity of the ring and makes sequence-independent electrostatic contacts with the surfaces of both subunits adjacent to the dimerization interface. In yeast, both Ku subunits are ∼70 kDa and show sequence similarity with the subunits of the human Ku heterodimer (Feldmann et al. 1996). The Ku70 subunit, which faces toward the DNA terminus, is crucial for NHEJ function, and the Ku80 subunit, which faces away from the terminus, maintains telomeric silencing functions (Bertuch and Lundblad 2003; Ribes-Zamora et al. 2007). In yeast, knockout of the Ku heterodimer contributes to telomere shortening and elongated 3′ overhangs as a result of increased Exo1 access to the C-strand (Boulton and Jackson 1996; Gravel et al. 1998; Bertuch and Lundblad 2004; Vodenicharov et al. 2010). Ku’s well-established role as a multifaceted DNA end-binding protein raises questions about how and why it binds to the TLC1 RNA.
The association of Ku with TLC1 was originally identified in an overexpression screen for disruption of telomeric silencing, and deletion analysis localized the KBS to a 48-nt stem–loop at the distal end of the template boundary arm (Peterson et al. 2001). The terminal KBS (nt 288–312) is predicted to form a hairpin with a two-base bulge at the center. The fold of the terminal KBS is retained in subsequent structural modeling of TLC1, but the conformation of the helix adjacent to the KBS varies among the proposed structures (Dandjinou et al. 2004; Zappulla and Cech 2004). Disruption of the KBS structure perturbs Ku binding both in vivo and in vitro, resulting in a telomere shortening phenotype (Peterson et al. 2001; Stellwagen et al. 2003).
Ku has been proposed to serve as a bridging factor to recruit telomerase to the telomere during G1 of the cell cycle, and thereby help to facilitate telomerase action during late S-phase (Peterson et al. 2001; Stellwagen et al. 2003; Fisher et al. 2004). Ku-dependent recruitment is thought to act in concert with the established Est1-Cdc13 recruitment mechanism (Evans and Lundblad 1999; Fisher et al. 2004). Loss of Ku or TLC1 results in a substantial decrease in de novo telomere addition upon DNA damage, adding support to the Ku-dependent telomerase recruitment hypothesis (Myung et al. 2001; Stellwagen et al. 2003). Notably, deletion of the KBS within TLC1 does not affect telomere capping or telomeric silencing (Peterson et al. 2001; Vodenicharov et al. 2010).
More recent experiments indicate that the Ku–TLC1 interaction may play a more complex role in telomerase recruitment than originally thought. Fluorescent in situ hybridization experiments demonstrate that the deletion of the KBS or either of the Ku subunits results in decreased TLC1 nuclear localization, explaining in part the telomere shortening phenotype (Gallardo et al. 2008; Pfingsten et al. 2012). Live cell imaging experiments indicate that telomerase only transiently associates with telomeres during G1 (Gallardo et al. 2011). Additionally, Ku binds the KBS RNA and duplex DNA in a mutually exclusive and competitive manner in vitro (Pfingsten et al. 2012). These experiments call into question the hypothesis that Ku serves as a simple bridging factor to recruit telomerase. Nonetheless, the insertion of an additional KBS into the TLC1 RNA results in a telomere hyperelongation phenotype, indicating that the Ku–TLC1 interaction may contribute to telomere homeostasis in capacities beyond nuclear localization (Zappulla et al. 2011). For example, the Ku–TLC1 interaction is also thought to contribute to anchoring of the telomerase to the nuclear envelope (Schober et al. 2009).
In spite of the clear Ku–TLC1 association in S. cerevisiae, it is unclear whether Ku’s role as a protein cofactor for telomerase is conserved among other eukaryotes. Deletion of YKU70 in a S. paradoxus strain with long telomeres results in a telomere shortening phenotype, providing indirect evidence for a TLC1–Ku interaction (Liti et al. 2009b). KBSs are proposed to exist in both sensu stricto and sensu lato Saccharomycotina, and a putative KBS that varies in both hairpin size and sequence appears to be present in the Candida glabrata TER (Chappell and Lundblad 2004; Kachouri-Lafond et al. 2009). Knockout of YKU80 in Kluyveromyces lactis does not result in a telomere-shortening phenotype, and structural modeling suggests that K. lactis TER does not contain a KBS (Brown et al. 2007; Carter et al. 2007; Kabaha et al. 2008). Outside of the Saccharomycotina clade, KBSs have not been detected in the TERs of the fission yeast Schizosaccharomyces pombe or the filamentous ascomycete Neurospora crassa (Webb and Zakian 2008; Qi et al. 2012). Although the precise Ku-binding sites have not been identified, Ku is reported to bind to both human TER and specific TER isoforms in Arabidopsis thaliana (Ting et al. 2005, 2009; Cifuentes-Rojas et al. 2012) (see also Chai et al. 2002).
Ku also appears to bind to a host of other cellular RNAs. Prior to DNA damage, human Ku resides in the nucleolus and associates with a host of RNA-binding proteins in an RNA-dependent manner; following DNA damage, Ku disperses throughout the nucleus and incorporates into DNA damage complexes (Adelmant et al. 2012). Immunoprecipitation experiments demonstrate that Ku interacts with a pool of RNAs in HeLa nuclear extracts (Zhang et al. 2004). Ku is reported to bind the HIV TAR element stem–loop with high affinity in vitro (Kaczmarski and Khan 1993). Thus, mounting evidence suggests that RNA binding may be a conserved function of the Ku heterodimer.
In order to better understand the RNA-binding function of Ku, we set out to characterize the interaction between Ku and the TLC1 KBS. We first identify the site of Ku binding on the TLC1 KBS with nucleotide resolution. We next provide evidence that the KBS secondary structure folds into an unusual bulged hairpin. Through mutagenesis of the KBS, we identify the critical determinants for Ku binding and define a minimal KBS motif. Finally, we provide evidence that KBSs are likely retained in several Saccharomyces TERs.
RESULTS
Ku interacts with the terminal stem–loop of the template boundary arm
To identify the site of Ku binding on the TLC1 RNA with nucleotide resolution, we carried out phosphorothioate footprinting experiments. Protein contacts on an RNA molecule can be identified by protection of phosphorothioate linkages from chemical cleavage by iodine (Schatz et al. 1991; Rudinger et al. 1992). In vitro-transcribed RNAs, with randomly incorporated phosphorothioate nucleotide analogs, were subjected to iodine-mediated cleavage in the presence or absence of the purified Ku heterodimer (Fig. 1A). The cleavage of a number of phosphorothioate linkages was reduced in the presence of Ku (Fig. 1B). The most stable protections were clustered in the terminal loop of the KBS RNA at nt C300, A302, A303, and U306. Protections in the terminal loop had roughly twofold reductions in cleavage throughout the duration of the time course (Fig. 1C). In contrast, less stable protections extended from the loop into the adjacent stems surrounding the AU bulge. For example, nt U290, A294, G295, and G311 all had greater than twofold reductions in cleavage at 10 sec, but not 10 min (Fig. 1C).
FIGURE 1.

Phosphorothioate footprinting of the Ku–KBS RNA interaction. (A) Coomassie-stained SDS-PAGE analysis of the Ku heterodimer purified from S. cerevisiae. (B) Ku-dependent protection from iodine-mediated cleavage of a TLC1 KBS RNA construct transcribed in the presence of A/U or C/G phosphorothioate NTPs. Cleavage times are denoted above the gel and lasted for 10 sec, 34 sec, and 10 min. Seq indicates the sequencing lanes, and –I2 is a no-iodine control. Protected nucleotides are denoted by arrows. (C) Heatmaps of the fold-change in band intensity on a log2 scale are plotted on the secondary structure of the TLC1 KBS. KBS numbering corresponds to the numbering for full-length TLC1 RNA.
To further test the interaction of Ku with the terminal stem–loop of the KBS, nuclease V1 footprinting was carried out on the Ku–KBS complex (Supplemental Fig. 1A). Nuclease V1 cleaves duplexed or structured RNAs in a sequence-nonspecific manner to release products with a 3′ hydroxyl and a 5′ phosphate (Ehresmann et al. 1987). The KBS was most strongly protected by Ku from V1 cleavage from nt 284–310 (Supplemental Fig. 1B). We note that V1 nuclease gives a bigger and less-resolved footprint than iodine, as expected, because an enzyme is large and much more subject to steric hindrance than a small-molecule probe. Given the overlap between the iodine and V1 nuclease footprinting, it is likely that the protections observed here are due to direct protein contacts. Nevertheless, it must be kept in mind that changes in iodine reactivity may be due to dynamics in the RNA–protein contacts or protein-dependent changes in the structure of the RNA (Rose and Weeks 2001; Webb et al. 2001). Our results agree with previous in vivo mutagenesis studies that identified the AU bulge and surrounding base pairs as important for the Ku–TLC1 interaction (Peterson et al. 2001) and show that the interaction extends into the terminal loop.
The TLC1 KBS forms a hairpin with a bulge motif
In order to experimentally test the previously predicted terminal KBS secondary structure (Peterson et al. 2001; Dandjinou et al. 2004; Zappulla and Cech 2004), chemical modification experiments were carried out to identify unpaired nucleotides. TLC1 KBS RNA was treated with either dimethyl sulfate (DMS) or 1-cyclohexyl-3-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate (CMCT). Accessible imino groups at position 1 on adenosine and position 3 on cytosine can be methylated by DMS, and accessible imino groups at position 3 of uracil and position 1 of guanosine can be modified by CMCT (Ehresmann et al. 1987). Modifications can be detected as stops in a reverse-transcription reaction 1 nt before the site of modification (Inoue and Cech 1985).
Treatment of the KBS RNA with DMS resulted in a number of strong stops in the reverse-transcription reaction. Modification occurred at adenosines 302–304 in the terminal loop of the KBS RNA (Fig. 2A,C). Likewise, treatment of the KBS with CMCT modified U301 and U293 (Fig. 2B,D). Nucleotides in the putative stem immediately adjacent to the terminal loop were not heavily modified upon treatment with CMCT or DMS, given that the band intensities at these positions were within one standard deviation of the mean band intensity. The modification pattern of bases in the penultimate stem (nt 288–291 and 309–312) showed more variability than the terminal stem, suggesting a more complex motif. Additionally, stops corresponding to weak modification (less than one standard deviation above the mean band intensity) of U301 and U287 were observed in the DMS data. DMS is not known to react with uracil, and U287 is not modified by CMCT, so the meaning of these data is unknown. Collectively, the strong modification of the terminal loop and low reactivity of the majority of nucleotides in the putative stems are consistent with the hypothesis that nt 288–312 form a stem–loop.
FIGURE 2.
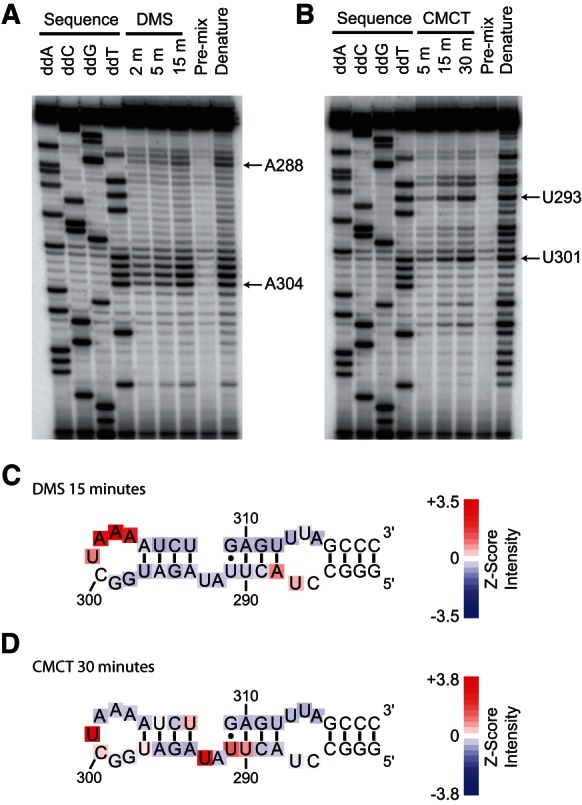
Testing the secondary structure model of the KBS by chemical modification. (A) DMS and (B) CMCT modification experiments. Modifications were detected as stops, one nucleotide prior to the site of modification, by reverse transcription. DMS modifies the N-1 of A and N-3 of unpaired C bases. CMCT modifies the N-3 of U and N-1 of unpaired G bases. Sequencing lanes generated by reverse transcription of the unmodified RNA in the presence of di-deoxy NTPs are denoted above the lane. RNA was allowed to react with DMS for 2, 5, or 15 min prior to quenching, and CMCT modification was carried out for 5, 15, or 30 min prior to quenching. Pre-mix controls consisted of the addition of RNA to the quench solution prior to addition of either DMS or CMCT. Lanes labeled as Denature denote RNA modification at 85°C for 30 sec. Selected A’s and U’s are indicated by arrows. (C) DMS modification at 15 min or (D) CMCT modification at 30 min plotted on the KBS secondary structure as a heatmap of the Z-score of band intensity for each position.
As mentioned above, the modification pattern for nt 291–293 did not appear to be consistent with a simple two-base bulge structure. U293 showed very high reactivity toward CMCT (more than three standard deviations above mean band intensity), while A292 showed virtually no reactivity toward DMS (below the mean band intensity). U291 had a moderate band intensity, greater than one standard deviation above the mean, while U290 and U308 were weakly reactive with band intensities that were above average (Fig. 2C,D). The modification data suggest that this portion of the KBS may fold into a specific structural motif. Varying chemical reactivity has been observed in the 2-nt bulge of domain V in the group II intron (Costa et al. 1998), which can likely be explained by the formation of a base-triple including one of the nucleotides in the bulge (Keating et al. 2010). Factors such as ion-binding sites or water-mediated hydrogen bonds may contribute to the protection of single-stranded nucleotides as well (Leontis and Westhof 1998). These modification data are consistent with previous secondary structure predictions in which the TLC1 KBS forms a hairpin; additionally, our data suggest that the hairpin contains a noncanonical bulge motif.
Ku from S. cerevisiae binds to the KBSs of noncognate TERs
An alignment of TLC1 sequences from 34 S. cerevisiae strains and 25 S. paradoxus strains, in addition to single TLC1 sequences from S. kudriavzevii, S. cariocanus, S. bayanus, S. pastorianus, S. mikatae, S. arboricola, and S. castellii demonstrated that the KBS is highly conserved from nt 289–311 (S. cerevisiae numbering, Supplemental Fig. 2A,B). Variation between the Saccharomyces spp. occurred in the predicted terminal loop (e.g., larger loops for S. bayanus and S. castellii). The length of the stem preceding the AU bulge varies between 3 and 4 bp (e.g., S. castellii compared with S. cerevisiae), and one A-to-G transition can be observed in the presumptive AU bulge (cf. S. cerevisiae and S. arboricola). Overall, the high degree of sequence conservation in the KBS relative to the weak conservation in other portions of this arm of TLC1 (Zappulla and Cech 2004) suggests that the RNA sequence is critical for structure and for recognition by Ku.
To verify the importance of conserved sequence in the KBS, affinity measurements were made using Ku from S. cerevisiae and the putative noncognate KBSs from selected sensu stricto strains, a sensu lato strain S. castellii, and the ascomycete K. lactis (Fig. 3A,B). Phylogenomic analyses indicate that S. paradoxus and S. cerevisiae are very closely related organisms (Fitzpatrick et al. 2006); correspondingly, the KBSs from these species show 100% identity from nt 288–312. Consistent with the importance of the conservation of the terminal KBS, Ku from S. cerevisiae bound the noncognate S. paradoxus KBS with an apparent Kd similar to that of the cognate interaction (Fig. 3B). Conversely, K. lactis is an outgroup to both the sensu stricto and sensu lato yeasts and is not predicted to have a KBS (Fitzpatrick et al. 2006; Brown et al. 2007). As expected, Ku from S. cerevisiae had a severely diminished affinity for the distal stem–loop of the template boundary arm from K. lactis.
FIGURE 3.

Binding analysis of S. cerevisiae Ku with noncognate KBSs. (A) Representative native gel-shift assays. Increasing concentrations of S. cerevisiae Ku were incubated with trace amounts of either the S. cerevisiae or S. bayanus KBS RNA. Ku–KBS complexes were resolved on a native gel to separate bound from free KBS. (B) Proposed secondary structures for putative KBSs from other budding yeasts and dissociation constants of S. cerevisiae Ku for the putative noncognate KBSs. Also shown are the percent amino acid identities between Ku from S. cerevisiae and Ku from other budding yeasts.
Noncognate KBSs with intermediate sequence conservation revealed additional determinants of Ku recognition. Apparent Kd’s from S. cerevisiae, S. bayanus, and S. castellii KBSs revealed that the size of the terminal loop, but not the sequence, is important for recognition. In S. castellii, the terminal loop of the KBS increased from 7 to 11 nt and changed in sequence, but had only a slight decrease in affinity for the S. cerevisiae Ku (Fig. 3B). In contrast, the S. bayanus KBS terminal loop increased from 7 to 24 nt and had an ∼50-fold reduction in affinity (Fig. 3A,B). The length of the penultimate stem is either three or four base pairs and appears to have a modest effect on affinity. In the case of the S. arboricola KBS, there is an A-to-G transition at position 292 in the bulge and a shortening of the stem preceding the bulge (Fig. 3). Both changes resulted in a moderate reduction in affinity for S. cerevisiae Ku. Collectively, the binding studies with TLC1 KBS homologs demonstrate that the size of the terminal loop and conservation of the AU bulge and flanking helices are important for recognition by Ku from S. cerevisiae. These data also provide evidence that the KBS may be a conserved feature of the TERs in both sensu stricto and sensu lato yeasts.
Terminal stem length and bulge motif are critical for Ku recognition
Binding assays with mutant S. cerevisiae KBS constructs were carried out to further elucidate the elements critical for Ku binding (Fig. 4A; Table 1). The complete set of mutant KBS constructs is shown in Supplemental Figure 4. Due to the high number of uridines in the conserved region of the KBS, we reasoned that mutants would have a high propensity to improperly fold and thereby artificially increase the Kd. To try to identify potentially misfolded mutants, and hence exclude them from our study, RNA secondary structures were analyzed by mFold. Then to experimentally confirm the mFold predictions and verify that sequence changes did not cause RNAs to adopt unanticipated conformations, we carried out V1 nuclease digests of selected mutants (Supplemental Fig. 3A). We found that when the lowest free energy structure for a mutant was misfolded (i.e., did not maintain the mFold-predicted 4 bp stems on either side of the bulge), the mutant RNA typically misfolded as judged by V1 nuclease digests (Supplemental Fig. 3B,C). Although our chemical modification data suggest that the native KBS may contain a more complex bulge motif than is represented by the predicted mFold secondary structure, we found mFold a useful tool for eliminating mutants prone to misfolding.
FIGURE 4.

Determining the minimum requirements for Ku–KBS RNA binding by mutagenesis. (A) Binding analysis of selected RNA mutants. Trace amounts of TLC1 KBS constructs were complexed with increasing concentrations of purified Ku heterodimer and run on native gels. The fraction of KBS bound was quantitated and plotted as a function of the Ku concentration to determine the dissociation constant for each of the mutants. Representative curves reflect the data normalized to the wild-type measurements for that set of replicates as described in Materials and Methods. (B) A minimum consensus for a KBS RNA. Nucleotides shown have a greater than fivefold impact on binding affinity, while circles represent nucleotides that can be mutated with little or no effect on binding. R indicates that a purine is tolerated at position 292. Nucleotides or circles strongly protected by Ku at the 10-sec time point are colored red, and nucleotides that were not protected are shown as open circles.
TABLE 1.
Binding data for mutant KBS RNAs

Previous models of the S. cerevisiae KBS secondary structure differ slightly in the conformation of the helices formed by nt 276–287 and 313–323 (Peterson et al. 2001; Dandjinou et al. 2004; Zappulla and Cech 2004). We tested two KBS constructs predicted to fold into these secondary structures (Supplemental Fig. 4; cf. Zappulla: TLC1-96 and Dandjinou: TLC1-56). We found that Ku bound both constructs with the same affinity (Fig. 3; Table 1). The construct with the predicted Zappulla fold (TLC1-96) bound Ku with a Kd of 1.3 ± 0.5 nM, and the construct with the predicted Dandjinou (TLC1-56) fold bound with a Kd of 1.13 ± 0.02 nM. In addition, both the sequence and predicted folds for the noncognate S. paradoxus and S. castellii KBSs differed from that of S. cerevisiae KBS, yet both noncognate KBSs bound S. cerevisiae Ku with only slightly diminished affinities. In sum, these data support the conclusion that the specific sequence or structure of the template boundary arm preceding the terminal hairpin is not a factor in Ku recognition, but that the terminal conserved region of the KBS (nt 289–311) is the critical Ku recognition element.
Considering the terminal hairpin loop, KBSs with sequence changes in the terminal loop bound Ku with near wild-type affinity (mutants 1 and 2) (Fig. 4A; Table 1). In contrast, mutant 16, which shortened the terminal loop to four bases, had a moderate fivefold reduction in affinity. These data were consistent with the findings of the noncognate binding studies, which indicated that the size of the terminal loop was important for high-affinity interactions with Ku. Deletions of internal loops and bulges outside of the conserved region of the KBS (nt 289–311) had minor effects on Ku recognition (mutants 6 and 7).
Our structure-probing data suggested that the conserved portion of the KBS formed a hairpin with a noncanonical bulge motif. In order to test this prediction, we made a series of mutants to produce compensatory base-pair substitutions within the helix. We reasoned that compensatory mutations in the helix should have little or no impact, provided that both the KBS structure and potential KBS–Ku contacts were preserved. Base-pair substitutions at three of the four base pairs in the terminal helix and at base-pair A288:312U in the penultimate helix had very little impact on KBS affinity for Ku (mutants 3, 21, 22, and 26) (Fig. 4A; Table 1; Supplemental Fig. 4). In contrast, mutations of base pairs surrounding the bulge at positions U290:310A, U291:309G, and A294:308U had moderate to severe reductions in affinity (∼5–60-fold, see mutants 12, 17–20, 25). To test whether the length of the helix between the AU bulge and terminal loop is important, the stem was lengthened by 2 or 4 bp while preserving the native KBS sequence immediately adjacent to the bulge. Lengthening of the stem resulted in increasingly drastic reductions in Ku affinity (mutants 23 and 24).
To determine the importance of the bulge motif for Ku association, a series of mutants was made to probe the size and sequence requirements within the bulge. Deletion of both bulge nucleotides resulted in a severe reduction in affinity (>230-fold, see mutant 5), indicating that this bulge is essential for proper Ku binding (Fig. 4A; Table 1; Supplemental Fig. 3A). Other mutations within the bulge had variable effects. For example, reduction of the AU bulge to 1 nt also resulted in a severe binding defect (mutant 28). On the other hand, mutations that altered the sequence at positions 292 or 293 resulted in much less severe binding defects. Inversion of nt 292 and 293 resulted in a sixfold reduction in affinity, while the transversion U293A had no impact on KBS–Ku binding (mutants 8 and 29, respectively). Finally, increasing the size of the bulge by the addition of adenosines resulted in moderate reductions in affinity (mutants 30 and 31), indicating that increases in bulge size were tolerated more than decreases in bulge size. We conclude that the terminal helix length and the nucleotides in and surrounding the noncanonical bulge motif (nt 290–294, 308–310) are crucial in mediating the KBS–Ku interaction.
Based on the results of the mutagenesis studies, noncognate binding assays, and footprinting experiments, we propose a minimum consensus for the KBS (Fig. 4B). Mutations resulting in greater than fivefold reductions in affinity are identified as positions in the stem–loop that are critical for Ku binding. The sequence of the two base pairs preceding the bulge, and the first base pair following the bulge, is important for recognition by Ku or the overall fold of the hairpin. The ability to substitute but not delete bulged nucleotides argues for a specific fold being important for Ku binding, but sequence-dependent contacts within the bulge cannot be entirely dismissed. The nucleotide identity at position 292 appears to be marginally important, while position 293 can be mutated without significantly impacting affinity (mutant 29). Based strictly on our mutagenesis, an adenosine appears to be important for Ku recognition; however, previous mutagenesis studies indicate that a guanosine is tolerated much better than a cytosine at this position in vivo (Peterson et al. 2001). With the exception of the compensatory mutations listed above, mutations that preserved helical structure, changed loop sequence, or disrupted internal loops had little effect on affinity and were deemed not to be critical determinants of Ku binding. In summary, we can now more precisely define the KBS for yeast Ku as a hairpin containing a noncanonical bulge motif that is capped by a 4-bp stem and terminal loop with 7–11 nt.
DISCUSSION
Previous studies identified a 48-nt hairpin as important for the TLC1–Ku interaction in vivo in S. cerevisiae (Peterson et al. 2001; Stellwagen et al. 2003). Here we extend these studies with footprinting, which indicates where Ku physically interacts with the KBS RNA, and with mutational analysis, which quantifies the contributions of individual RNA structural features to the binding energy. We identify a noncanonical bulge motif within the highly conserved portion of the KBS, and show that spacing between the bulge and terminal loop is critical for Ku recognition. Additionally, we provide direct evidence that S. cerevisiae Ku is able to recognize several noncognate KBSs. We suggest that the KBS is a conserved element within both sensu stricto and sensu lato Saccharomyces species and discuss the possibility that Ku recognizes additional cellular RNAs.
Phosphorothioate footprinting shows that the Ku interaction is restricted to 25 nt in the KBS and identifies putative contacts at nucleotide resolution. The contacts are in the terminal loop and stems on either side of the AU bulge. The footprint encompasses the portion of the KBS that is highly conserved between putative noncognate KBSs, and the noncanonical bulge motif that is particularly susceptible to mutation. The time-dependent contacts in the stem may be the result of transient KBS–protein interactions or Ku-dependent changes in the KBS structure (Rose and Weeks 2001; Webb et al. 2001). Although we cannot definitively distinguish between these possibilities, the complex was equilibrated prior to footprinting and any Ku-induced changes in local KBS structure to promote binding likely occurred prior to iodine-mediated cleavage. We favor a model in which Ku directly interacts with the terminal loop as well as the stems on either side of the bulge, as supported by the V1 nuclease protection data (Supplemental Fig. 1).
Our footprinting data show that Ku interacts with the highly conserved portion of the KBS (nt 289–311). Covariations to support the RNA secondary structure model are not present among currently identified yeast KBSs. Therefore, we utilized chemical probing experiments, which provided support for the proposed bulged hairpin structure. In addition, the modification pattern suggested that the KBS adopts a non-Watson–Crick helical fold from positions 290 to 293. The structure of this bulge motif cannot be predicted solely from the modification pattern. Some possibilities for the protection of A292, but not U293, include the formation of a base-triple including A292, intercalation of bases into the helical stack, ion-coordination, or water-mediated hydrogen bonding (Costa et al. 1998; Leontis and Westhof 1998). Further investigation will be necessary to distinguish between these possibilities. The importance of this bulge is underscored by the detrimental effect that mutations have in this region of the RNA, as discussed below.
Our direct binding data are consistent with previous in vivo studies, which show that disruption of the proposed KBS hairpin secondary structure perturbs the Ku–TLC1 genetic interaction (Peterson et al. 2001). Mutations in and surrounding the bulge region have the greatest impact on KBS function in vivo (Peterson et al. 2001). Because mutations in the terminal loop have a small impact on KBS function (Peterson et al. 2001) and binding (mutants 1, 2, and 4, this study), contacts between Ku and the terminal loop of the KBS are likely to be sequence nonspecific and may involve the RNA backbone. Mutations in and directly adjacent to the bulge have a strong impact both in vivo (Peterson et al. 2001) and in vitro (positions 291 and 292, this study). Our data suggest that mutations to the two terminal base pairs adjacent to the loop do not significantly affect the ability of the KBS to bind Ku (mutant 3); in agreement, mutations at these positions do not seem to perturb the KBS–Ku interaction in vivo. In contrast, our data show that binding is not compromised in mutant 21 (G295A:C307U); however, there appears to be a minor impact on KBS function for this mutant in vivo (Peterson et al. 2001). Finally, we show a moderate binding defect when making compensatory mutations at the U290:310A base pair. In vivo, mutations at this position do not have a detrimental impact. The differences in the impact of mutations to the KBS may be a reflection of the different assays used to monitor the Ku–KBS interaction. The in vivo analysis measures the ability of the mutant KBS to disrupt telomeric silencing in the context of overexpression, while we measure the impact on Ku–KBS binding equilibria in vitro.
The identification of KBSs competent for specific association with Ku in TERs of several Saccharomyces species adds to the growing evidence that RNA binding is a conserved function of the Ku heterodimer. Ku associates with cellular RNAs in human cells (Zhang et al. 2004; Ting et al. 2009; Adelmant et al. 2012). Ku interacts with noncoding RNAs such as TERRA and Y-RNAs (Zhang et al. 2011; Pfeiffer and Lingner 2012). RNA aptamers have been selected to bind human Ku with high affinity (Yoo and Dynan 1998). The association of human Ku with HIV-1 TAR RNA provides a specific example of another hairpin that is competent for binding Ku (Kaczmarski and Khan 1993). The TAR hairpin shows little or no sequence similarity with the TLC1 KBS; however, they share similar secondary structures and both are able to compete with double-stranded DNA for Ku binding (Kaczmarski and Khan 1993; Pfingsten et al. 2012). The TAR hairpin has a 6-nt terminal loop that is separated from a 3-nt bulge by an intervening 4-bp helix. The lack of sequence similarity between these RNAs may reflect differences between the human and S. cerevisiae Ku heterodimers; however, the conserved bulge hairpin with specific spacing requirements suggests that Ku recognizes RNAs with a conserved fold. We therefore favor a model in which Ku recognizes the hairpin structure rather than making sequence-specific contacts. One possibility is that the KBS has the ability to mimic a B-form DNA helix and thereby associate with Ku’s preformed ring; B-DNA mimicry by RNAs to associate with proteins has been previously observed (Reiter et al. 2008; Bullock et al. 2010). The identification of additional Ku-binding RNAs and an increased understanding of how Ku recognizes these hairpins will certainly shed light on how this nucleic acid-binding protein mediates a myriad of distinct cellular functions.
MATERIALS AND METHODS
KBS constructs and in vitro transcription of RNAs
TER sequences and alignments for representative Saccharomycotina were obtained from the Telomerase Database (Podlevsky et al. 2008); TER sequences from additional Saccharomyces strains and species were manually added to the alignment using the program BioEdit V7.1.3.0. S. cerevisiae and S. paradoxus sequences were obtained by BLAST from the Saccharomyces genome resequencing project (Liti et al. 2009a). The S. arboricola putative TER sequence was recovered by BLAST from the whole-genome assembly (GCA_000292725.1) using the S. mikatae TER as a query sequence.
For a complete list of RNA constructs used see Supplemental Figure 4. RNAs were designed and checked for misfolding using mFold version 2.3; RNAs were folded at 30°C (Zuker 2003). Mutant RNA constructs were prepared by quick-change mutagenesis (Agilent) of the previously reported TLC1-96 KBS construct (Pfingsten et al. 2012). The TLC1 KBS constructs used for phosphorothioate footprinting and mutagenesis experiments were prepared by T7 in vitro transcription from PCR products as previously described (Pfingsten et al. 2012). The TLC1 KBS construct used for the chemical modification experiments, as well as putative Ku arms from other Saccharomyces TERs, were made by annealing and cloning complementary oligos containing the putative Ku arm sequence, a T7 promoter, and FokI site. The RNAs were prepared by T7 polymerase run-off transcription from the FokI linearized plasmid. All in vitro-transcribed RNAs were purified on a denaturing 10% polyacrylamide gel. Prior to 32P-labeling RNAs, calf intestinal alkaline phosphatase (Roche) was used to remove the 5′ phosphate. RNAs were then 5′ labeled with [γ-32P]ATP (Perkin Elmer) and T4 polynucleotide kinase (NEB) according to the manufacturer’s protocol. 5′-labeled RNAs were then repurified on a denaturing 10% polyacrylamide gel.
Purification of the Ku heterodimer
Protein constructs and purification were previously described (Pfingsten et al. 2012) or had the following modifications. Cells were grown to a final density of 1–4 × 107 cells/mL. Lysate was passed over a 5-mL HisTrap HP column (GE Healthcare). Bound protein was eluted first in shallow gradient ranging from 20 to 80 mM imidazole over 10 column volumes (CV) and second in a steep gradient from 80 to 500 mM imidazole over five CV. The elution buffer also contained 50 mM Tris (pH 8.0) and 500 mM NaCl. Fractions containing Ku were dialyzed overnight into a buffer containing 50 mM Tris (pH 8.0), 50 mM NaCl, and 2 mM DTT. Ku was then exchanged over a HiTrap Q HP column (GE Healthcare) under the same buffer conditions used for dialysis. Bound protein was eluted from the Q column in a shallow gradient ranging from 50 mM to 1 M NaCl over 10 CV and then in a steep gradient ranging from 1 to 2 M NaCl over five CV. Fractions containing Ku were then recaptured on a 1-mL HisTrap HP column. The nickel column was then directly connected to a Superdex 200 (GE Healthcare), and protein was injected from the nickel column onto the sizing column using 4 mL of 500 mM imidazole. Running buffer contained 50 mM Tris (pH 8.0), 500 mM NaCl, and 2 mM DTT. Fractions containing Ku were then combined, dialyzed, and frozen as previously described (Pfingsten et al. 2012).
Phosphorothioate footprinting assays
A ratio of 1 mM NTPs to 0.05 mM ATP [αS] and UTP [αS] or CTP [αS] and GTP [αS] (Sp diastereomers, Glen Research) was used to generate phosphorothioate substituted RNAs. The nucleotide analogs were randomly incorporated into the TLC1-96 RNA construct using T7 polymerase. The RNAs were end-labeled and purified as described above. Prior to the footprinting reaction, Ku protein was buffer exchanged into a storage buffer lacking DTT (25 mM Tris at pH 7.0, 20% glycerol, 200 mM NaCl, 0.5 mM EDTA, and 5 mM MgCl2). This storage buffer was also used as a 2X stock to achieve the final buffer and salt conditions for the footprinting reactions.
Final footprinting reactions contained 5′-32P labeled phosphorothioate substituted TLC1 KBS RNA, 1X storage buffer, 0.05 μg of yeast tRNA (Sigma), 135 nM Ku, and 0.2 mM I2 (dissolved in 100% ethanol). RNAs were annealed in the presence of all reaction components, with the exception of Ku and I2, by heating to 85°C for 1 min, followed by snap cooling on ice. Annealed RNAs were then split between tubes containing Ku or an equal volume of storage buffer and complexed at room temperature for 20 min. Footprinting reactions were initiated by the addition of I2, and reactions were subsequently quenched at various time points in an equal volume of 100 mM β-mercaptoethanol. The no-I2 controls were treated identically, but the RNA was removed prior to I2 addition. Sequencing reactions were prepared by carrying out I2-mediated cleavage of the substituted RNAs at 65°C for 2 min. Reactions were precipitated by the addition of 2 vol of RNase inactivation/precipitation buffer (Life Technologies), precipitated overnight, and washed once with 70% ethanol. RNAs were then suspended in equal volumes of H2O and 2X formamide loading dye (0.5X TBE, 93.5% formamide, 30 mM EDTA, 0.5% xylene cyanol and bromophenol blue). Samples were heated to 85°C for 1 min, cooled on ice, and equal numbers of scintillation counts for each sample were loaded onto a 10% polyacrylamide sequencing gel. Gels were dried and visualized by Phosphorimagery (GE Healthcare). The computer program SAFA was used to align the gel and calculate band intensities in order to determine the level of protection (Das et al. 2005). SAFA-calculated band intensities were normalized to the total number of counts per lane. The fold-change in band intensity upon addition of Ku was calculated with the normalized values. Intensities were plotted onto the secondary structure on a log2 scale.
Nuclease V1 footprinting assays
RNAs for footprinting reactions were prepared by in vitro transcription as described above. Final footprinting reactions contained 5′-32P-labeled TLC1 KBS RNA, V1 footprinting buffer (15 mM Tris at pH 8.0, 12% glycerol, 130 mM NaCl, 5 mM MgCl2, 1.6 mM DTT), 0.05 μg of yeast tRNA (Sigma), 1.08 μM Ku, and 0.005 or 0.0013 units of RNase V1 (Life Technologies) diluted in Ku storage buffer (25 mM Tris at pH 8.0, 20% glycerol, 200 mM NaCl, 0.5 mM EDTA, and 2 mM DTT) prior to initiation of the reaction. RNAs were annealed as described for the footprinting reactions, and Ku–KBS complexes were reconstituted for 30 min at room temperature prior to the addition of nuclease V1. Digestions were carried out at room temperature for 10 min and then quenched in RNase inactivation/precipitation buffer (Life Technologies). RNAs were precipitated and processed as described above for the phosphorothioate footprinting reactions. The alkaline hydrolysis ladder was prepared by incubating 5′-32P-labeled TLC1 KBS RNA and 2 μg of yeast tRNA (Sigma) in 50 mM sodium bicarbonate (pH 9.2) at 85°C for 4 min. The RNase T1 ladder reactions contained 5′-32P-labeled TLC1 KBS RNA, 2 μg of yeast tRNA (Sigma), 1X RNA sequencing buffer (Life Technologies), and 0.1 unit of RNase T1 (Life Technologies). RNAs were heated to 55°C for 1 min prior to the addition of RNase T1, and then incubated at 55°C for 3 min. RNase T1 and hydrolysis ladders were quenched in 1X formamide loading dye as described above. Samples were prepared, run on sequencing gels, and quantitated as described for phosphorothioate footprinting.
Chemical modification assays
Chemical modification and subsequent reverse transcription experiments were carried out as previously described (Zaug and Cech 1995; Brunel and Romby 2000), with minor modifications. Final reactions contained ∼1 μg of TLC1-38 KBS primer extension RNA (2.7 μM), 150 mM NaCl, 25 mM HEPES (pH 8.0), and 5 mM MgCl2, and either 90 mM DMS (Sigma, diluted in 100% ethanol) or 18 mg/mL CMCT (Sigma, dissolved in H2O). RNAs were annealed as described for the footprinting experiments in the absence of DMS or CMCT. Modification reactions were initiated by the addition of either DMS or CMCT, and reactions were quenched at various time points. For the premix negative control reactions, annealed RNA was added to the appropriate quench solution prior to adding DMS or CMCT. Denature controls consisted of modifying the RNA with the appropriate chemical reagent for 30 sec at 85°C. DMS modification was quenched by addition of 9 vol of DMS quench buffer consisting of 90 mM β-Mercaptoethanol, 333 mM NaOAC (pH 5.2), and 20 μg of glycogen (Roche). The CMCT quench buffer was identical to the DMS quench buffer except that it lacked β-mercaptoethanol. Quenched reactions were then precipitated by the addition of 2.5 vol of ice-cold ethanol. RNAs were washed once with 70% ethanol and then resuspended in H2O.
Final reverse transcription reactions contained ∼0.2 μg of modified RNA, trace amounts of 32P-labeled primer, 4 mM dNTPs, AMV reverse transcriptase (NEB), and 1X AMV buffer (NEB). Primers were annealed to RNA in 1X AMV buffer by heating to 85°C for 2 min, followed by snap cooling on ice. Reverse-transcription reactions were initiated by the addition of an equal volume of mix containing the dNTPs, 1X AMV buffer, and the reverse transcriptase. Primer extension reactions were allowed to proceed at 37°C for 40 min. Sequencing reactions were carried out with the same reverse-transcription conditions described above and the addition of a single ddNTP to a final concentration of 0.5 mM. Reverse-transcription reactions were quenched by the addition of 2X formamide loading dye and heating to 95°C for 5 min. Reactions were loaded onto a 10% sequencing gel and the gel was dried and band intensities were calculated as described for the phosphorothioate footprinting experiments. Modification levels were calculated by normalizing the SAFA calculated band intensities to the total counts per lane. The mean and standard deviation of the band intensities in a lane were used to calculate a z-score for each band in the lane.
Determination of dissociation constants
Dissociation constants for the mutant and additional putative yeast KBS RNAs were measured as previously described (Pfingsten et al. 2012). Briefly, trace amounts of KBS RNA were incubated with Ku at room temperature for 1 h in a buffer containing 21 mM HEPES (pH 7.5), 6 mM Tris (pH 8), 150 mM NaCl, 16% glycerol, 5 mM MgCl2, 1 mM EDTA, 25 μg/mL tRNA, 0.1 mg/mL BSA, and 1 mM DTT. Ku-bound KBS RNAs were separated from free KBS RNA on a native 4%–20% acrylamide TBE gel (Life Technologies); gel boxes were placed in ice for the electrophoretic run. All measurements represent the average of three independent replicates. Mutant Kd values were normalized to the wild-type Kd for a given set of experimental replicates by multiplying the measured mutant Kd by the ratio of the measured wild-type Kd over the average wild-type Kd for all experiments. All measured wild-type KBS Kd values were within 2.5-fold of the average wild-type Kd reported in Table 1.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the members of the Cech laboratory for helpful discussions, to Jayakrishnan Nandakumar for the critical reading of this manuscript, and Art Zaug for technical advice. T.R.C. is an investigator of the HHMI.
REFERENCES
- Adelmant G, Calkins AS, Garg BK, Card JD, Askenazi M, Miron A, Sobhian B, Zhang Y, Nakatani Y, Silver PA, et al. 2012. DNA ends alter the molecular composition and localization of Ku multicomponent complexes. Mol Cell Proteomics 11: 411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertuch AA, Lundblad V 2003. The Ku heterodimer performs separable activities at double-strand breaks and chromosome termini. Mol Cell Biol 23: 8202–8215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertuch AA, Lundblad V 2004. EXO1 contributes to telomere maintenance in both telomerase-proficient and telomerase-deficient Saccharomyces cerevisiae. Genetics 166: 1651–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP 1996. Identification of a Saccharomyces cerevisiae Ku80 homologue: Roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res 24: 4639–4648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown Y, Abraham M, Pearl S, Kabaha MM, Elboher E, Tzfati Y 2007. A critical three-way junction is conserved in budding yeast and vertebrate telomerase RNAs. Nucleic Acids Res 35: 6280–6289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel C, Romby P 2000. Probing RNA structure and RNA-ligand complexes with chemical probes. Methods Enzymol 318: 3–21 [DOI] [PubMed] [Google Scholar]
- Bullock SL, Ringel I, Ish-Horowicz D, Lukavsky PJ 2010. A′-form RNA helices are required for cytoplasmic mRNA transport in Drosophila. Nat Struct Mol Biol 17: 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SD, Iyer S, Xu J, McEachern MJ, Astrom SU 2007. The role of nonhomologous end-joining components in telomere metabolism in Kluyveromyces lactis. Genetics 175: 1035–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai W, Ford LP, Lenertz L, Wright WE, Shay JW 2002. Human Ku70/80 associates physically with telomerase through interaction with hTERT. J Biol Chem 277: 47242–47247 [DOI] [PubMed] [Google Scholar]
- Chappell AS, Lundblad V 2004. Structural elements required for association of the Saccharomyces cerevisiae telomerase RNA with the Est2 reverse transcriptase. Mol Cell Biol 24: 7720–7736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Rojas C, Nelson AD, Boltz KA, Kannan K, She X, Shippen DE 2012. An alternative telomerase RNA in Arabidopsis modulates enzyme activity in response to DNA damage. Genes Dev 26: 2512–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Christian EL, Michel F 1998. Differential chemical probing of a group II self-splicing intron identifies bases involved in tertiary interactions and supports an alternative secondary structure model of domain V. RNA 4: 1055–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandjinou AT, Levesque N, Larose S, Lucier JF, Abou Elela S, Wellinger RJ 2004. A phylogenetically based secondary structure for the yeast telomerase RNA. Curr Biol 14: 1148–1158 [DOI] [PubMed] [Google Scholar]
- Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB 2005. SAFA: Semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA 11: 344–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan ED, Collins K 2012. Biogenesis of telomerase ribonucleoproteins. RNA 18: 1747–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehresmann C, Baudin F, Mougel M, Romby P, Ebel JP, Ehresmann B 1987. Probing the structure of RNAs in solution. Nucleic Acids Res 15: 9109–9128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans SK, Lundblad V 1999. Est1 and Cdc13 as comediators of telomerase access. Science 286: 117–120 [DOI] [PubMed] [Google Scholar]
- Feldmann H, Driller L, Meier B, Mages G, Kellermann J, Winnacker EL 1996. HDF2, the second subunit of the Ku homologue from Saccharomyces cerevisiae. J Biol Chem 271: 27765–27769 [DOI] [PubMed] [Google Scholar]
- Fisher TS, Taggart AK, Zakian VA 2004. Cell cycle-dependent regulation of yeast telomerase by Ku. Nat Struct Mol Biol 11: 1198–1205 [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DA, Logue ME, Stajich JE, Butler G 2006. A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol Biol 6: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo F, Olivier C, Dandjinou AT, Wellinger RJ, Chartrand P 2008. TLC1 RNA nucleo-cytoplasmic trafficking links telomerase biogenesis to its recruitment to telomeres. EMBO J 27: 748–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo F, Laterreur N, Cusanelli E, Ouenzar F, Querido E, Wellinger RJ, Chartrand P 2011. Live cell imaging of telomerase RNA dynamics reveals cell cycle-dependent clustering of telomerase at elongating telomeres. Mol Cell 44: 819–827 [DOI] [PubMed] [Google Scholar]
- Gravel S, Larrivee M, Labrecque P, Wellinger RJ 1998. Yeast Ku as a regulator of chromosomal DNA end structure. Science 280: 741–744 [DOI] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH 1987. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 51: 887–898 [DOI] [PubMed] [Google Scholar]
- Inoue T, Cech TR 1985. Secondary structure of the circular form of the Tetrahymena rRNA intervening sequence: A technique for RNA structure analysis using chemical probes and reverse transcriptase. Proc Natl Acad Sci 82: 648–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabaha MM, Zhitomirsky B, Schwartz I, Tzfati Y 2008. The 5′ arm of Kluyveromyces lactis telomerase RNA is critical for telomerase function. Mol Cell Biol 28: 1875–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachouri-Lafond R, Dujon B, Gilson E, Westhof E, Fairhead C, Teixeira MT 2009. Large telomerase RNA, telomere length heterogeneity and escape from senescence in Candida glabrata. FEBS Lett 583: 3605–3610 [DOI] [PubMed] [Google Scholar]
- Kaczmarski W, Khan SA 1993. Lupus autoantigen Ku protein binds HIV-1 TAR RNA in vitro. Biochem Biophys Res Commun 196: 935–942 [DOI] [PubMed] [Google Scholar]
- Keating KS, Toor N, Perlman PS, Pyle AM 2010. A structural analysis of the group II intron active site and implications for the spliceosome. RNA 16: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontis NB, Westhof E 1998. The 5S rRNA loop E: Chemical probing and phylogenetic data versus crystal structure. RNA 4: 1134–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Ly H, Hussain A, Abraham M, Pearl S, Tzfati Y, Parslow TG, Blackburn EH 2004. A universal telomerase RNA core structure includes structured motifs required for binding the telomerase reverse transcriptase protein. Proc Natl Acad Sci 101: 14713–14718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR 1997. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276: 561–567 [DOI] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, et al. 2009a. Population genomics of domestic and wild yeasts. Nature 458: 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Haricharan S, Cubillos FA, Tierney AL, Sharp S, Bertuch AA, Parts L, Bailes E, Louis EJ 2009b. Segregating YKU80 and TLC1 alleles underlying natural variation in telomere properties in wild yeast. PLoS Genet 5: e1000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdy AD, Cech TR 2006. Low abundance of telomerase in yeast: Implications for telomerase haploinsufficiency. RNA 12: 1721–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, Chen C, Kolodner RD 2001. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature 411: 1073–1076 [DOI] [PubMed] [Google Scholar]
- Nandakumar J, Cech TR 2013. Finding the end: Recruitment of telomerase to telomeres. Nat Rev Mol Cell Biol 14: 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SE, Stellwagen AE, Diede SJ, Singer MS, Haimberger ZW, Johnson CO, Tzoneva M, Gottschling DE 2001. The function of a stem-loop in telomerase RNA is linked to the DNA repair protein Ku. Nat Genet 27: 64–67 [DOI] [PubMed] [Google Scholar]
- Pfeiffer V, Lingner J 2012. TERRA promotes telomere shortening through exonuclease 1-mediated resection of chromosome ends. PLoS Genet 8: e1002747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfingsten JS, Goodrich KJ, Taabazuing C, Ouenzar F, Chartrand P, Cech TR 2012. Mutually exclusive binding of telomerase RNA and DNA by Ku alters telomerase recruitment model. Cell 148: 922–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlevsky JD, Bley CJ, Omana RV, Qi X, Chen JJ 2008. The telomerase database. Nucleic Acids Res 36 (Database issue): D339–D343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Li Y, Honda S, Hoffmann S, Marz M, Mosig A, Podlevsky JD, Stadler PF, Selker EU, Chen JJ 2012. The common ancestral core of vertebrate and fungal telomerase RNAs. Nucleic Acids Res 41: 450–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter NJ, Maher LJ III, Butcher SE 2008. DNA mimicry by a high-affinity anti-NF-κB RNA aptamer. Nucleic Acids Res 36: 1227–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribes-Zamora A, Mihalek I, Lichtarge O, Bertuch AA 2007. Distinct faces of the Ku heterodimer mediate DNA repair and telomeric functions. Nat Struct Mol Biol 14: 301–307 [DOI] [PubMed] [Google Scholar]
- Rose MA, Weeks KM 2001. Visualizing induced fit in early assembly of the human signal recognition particle. Nat Struct Biol 8: 515–520 [DOI] [PubMed] [Google Scholar]
- Rudinger J, Puglisi JD, Putz J, Schatz D, Eckstein F, Florentz C, Giege R 1992. Determinant nucleotides of yeast tRNAAsp interact directly with aspartyl-tRNA synthetase. Proc Natl Acad Sci 89: 5882–5886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz D, Leberman R, Eckstein F 1991. Interaction of Escherichia coli tRNASer with its cognate aminoacyl-tRNA synthetase as determined by footprinting with phosphorothioate-containing tRNA transcripts. Proc Natl Acad Sci 88: 6132–6136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober H, Ferreira H, Kalck V, Gehlen LR, Gasser SM 2009. Yeast telomerase and the SUN domain protein Mps3 anchor telomeres and repress subtelomeric recombination. Genes Dev 23: 928–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto AG, Zaug AJ, Sobel SG, Wolin SL, Cech TR 1999. Saccharomyces cerevisiae telomerase is an Sm small nuclear ribonucleoprotein particle. Nature 401: 177–180 [DOI] [PubMed] [Google Scholar]
- Seto AG, Livengood AJ, Tzfati Y, Blackburn EH, Cech TR 2002. A bulged stem tethers Est1p to telomerase RNA in budding yeast. Genes Dev 16: 2800–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer MS, Gottschling DE 1994. TLC1: Template RNA component of Saccharomyces cerevisiae telomerase. Science 266: 404–409 [DOI] [PubMed] [Google Scholar]
- Stellwagen AE, Haimberger ZW, Veatch JR, Gottschling DE 2003. Ku interacts with telomerase RNA to promote telomere addition at native and broken chromosome ends. Genes Dev 17: 2384–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira MT, Arneric M, Sperisen P, Lingner J 2004. Telomere length homeostasis is achieved via a switch between telomerase-extendible and -nonextendible states. Cell 117: 323–335 [DOI] [PubMed] [Google Scholar]
- Ting NS, Yu Y, Pohorelic B, Lees-Miller SP, Beattie TL 2005. Human Ku70/80 interacts directly with hTR, the RNA component of human telomerase. Nucleic Acids Res 33: 2090–2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting NS, Pohorelic B, Yu Y, Lees-Miller SP, Beattie TL 2009. The human telomerase RNA component, hTR, activates the DNA-dependent protein kinase to phosphorylate heterogeneous nuclear ribonucleoprotein A1. Nucleic Acids Res 37: 6105–6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodenicharov MD, Laterreur N, Wellinger RJ 2010. Telomere capping in non-dividing yeast cells requires Yku and Rap1. EMBO J 29: 3007–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JR, Corpina RA, Goldberg J 2001. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412: 607–614 [DOI] [PubMed] [Google Scholar]
- Webb CJ, Zakian VA 2008. Identification and characterization of the Schizosaccharomyces pombe TER1 telomerase RNA. Nat Struct Mol Biol 15: 34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AE, Rose MA, Westhof E, Weeks KM 2001. Protein-dependent transition states for ribonucleoprotein assembly. J Mol Biol 309: 1087–1100 [DOI] [PubMed] [Google Scholar]
- Yoo S, Dynan WS 1998. Characterization of the RNA binding properties of Ku protein. Biochemistry 37: 1336–1343 [DOI] [PubMed] [Google Scholar]
- Zappulla DC, Cech TR 2004. Yeast telomerase RNA: A flexible scaffold for protein subunits. Proc Natl Acad Sci 101: 10024–10029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappulla DC, Goodrich KJ, Arthur JR, Gurski LA, Denham EM, Stellwagen AE, Cech TR 2011. Ku can contribute to telomere lengthening in yeast at multiple positions in the telomerase RNP. RNA 17: 298–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaug AJ, Cech TR 1995. Analysis of the structure of Tetrahymena nuclear RNAs in vivo: Telomerase RNA, the self-splicing rRNA intron, and U2 snRNA. RNA 1: 363–374 [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Schlott B, Gorlach M, Grosse F 2004. DNA-dependent protein kinase (DNA-PK) phosphorylates nuclear DNA helicase II/RNA helicase A and hnRNP proteins in an RNA-dependent manner. Nucleic Acids Res 32: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang AT, Langley AR, Christov CP, Kheir E, Shafee T, Gardiner TJ, Krude T 2011. Dynamic interaction of Y RNAs with chromatin and initiation proteins during human DNA replication. J Cell Sci 124 (Pt 12): 2058–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.