

The telomerase RNA subunit has a biologically essential function but surprisingly divergent primary sequence and overall secondary structure. Here, Chen and coworkers contribute yet another surprise about telomerase RNA structure and function conservation. The authors first establish a general method for telomerase RNA identification in organisms where the RNA is not possible to predict from the genome sequence using bioinformatics. Next, they demonstrate that a stem–loop motif previously considered essential for multicellular organism telomerase RNA function is, in fact, dispensable for purple sea urchin telomerase enzyme activity.
Keywords: telomere, telomerase RNA, ribonucleoprotein, next-generation sequencing
Abstract
Telomerase is a ribonucleoprotein (RNP) enzyme essential for telomere maintenance and chromosome stability. While the catalytic telomerase reverse transcriptase (TERT) protein is well conserved across eukaryotes, telomerase RNA (TR) is extensively divergent in size, sequence, and structure. This diversity prohibits TR identification from many important organisms. Here we report a novel approach for TR discovery that combines in vitro TR enrichment from total RNA, next-generation sequencing, and a computational screening pipeline. With this approach, we have successfully identified TR from Strongylocentrotus purpuratus (purple sea urchin) from the phylum Echinodermata. Reconstitution of activity in vitro confirmed that this RNA is an integral component of sea urchin telomerase. Comparative phylogenetic analysis against vertebrate TR sequences revealed that the purple sea urchin TR contains vertebrate-like template-pseudoknot and H/ACA domains. While lacking a vertebrate-like CR4/5 domain, sea urchin TR has a unique central domain critical for telomerase activity. This is the first TR identified from the previously unexplored invertebrate clade and provides the first glimpse of TR evolution in the deuterostome lineage. Moreover, our TR discovery approach is a significant step toward the comprehensive understanding of telomerase RNP evolution.
INTRODUCTION
Telomerase is a specialized reverse transcriptase (RT) essential for the synthesis of telomeric DNA repeats onto the ends of linear chromosomes. Functioning as a ribonucleoprotein (RNP) enzyme, telomerase consists of two essential core components: the catalytic telomerase reverse transcriptase (TERT) protein and telomerase RNA (TR) that provides the template for DNA synthesis (Blackburn and Collins 2011). TERT is highly conserved, with the central domain constituting the active site and an N-terminal domain that binds specifically to the TR (Bley et al. 2011; Mason et al. 2011). In contrast, TR is extremely divergent in size, sequence, and structure (Podlevsky et al. 2008; Podlevsky and Chen 2012).
Ciliate, yeast, and vertebrate TRs show no sequence homology or structural similarity apart from the universal pseudoknot structure. Following the identification of Neurospora crassa and 72 additional filamentous fungal TRs, a second highly conserved element, P6/6.1, was identified within fungal and vertebrate TRs in the CR4/5 (vertebrate) and three-way-junction (fungal) domains (Qi et al. 2013). However, the critical loop of P6.1 element is not conserved in closely related budding yeasts. Furthermore, these universally conserved structural elements have yet to be found within the recently identified Arabidopsis TR (Cifuentes-Rojas et al. 2011). The telomerase holoenzyme comprises a myriad of accessory proteins that vary dramatically among species by binding species-specific structural domains within TR. These accessory proteins are essential for regulating telomerase biogenesis. Understanding the evolution of telomerase structure and function requires the identification of TR sequences from all major clades. However, the massive divergence in TR sequence hinders TR discovery from unexplored phylogenetic groups by conventional molecular and bioinformatics approaches.
Molecular biology approaches for TR identification have relied heavily on telomerase enzyme purification (Greider and Blackburn 1989; Leonardi et al. 2008; Webb and Zakian 2008; Cifuentes-Rojas et al. 2011; Qi et al. 2013). However, telomerase purification is not feasible for the vast majority of organisms due to the low abundance of the enzyme or the lack of genetic tools necessary for expressing recombinant affinity-tagged proteins. Even within species amenable to telomerase purification, this methodology is often complex, requiring multiple purification steps and compatible telomerase activity assays to follow telomerase activity throughout purification.
Bypassing enzyme purification from cells, PCR amplification from genomic DNA has been successfully applied for TR identification from a large number of vertebrate species (Chen et al. 2000). Based on the human TR sequence, degenerate PCR primers targeted the conserved pseudoknot and CR4/5 regions. This method is not effective for certain species, such as teleost fishes and some reptiles, where the primer targeting sites are not well conserved. Alternatively, PCR amplification using primers targeting syntenic protein genes flanking the TR gene has been used to identify TRs from Sensu Stricto Saccharomyces species (Dandjinou et al. 2004). However, these PCR-based methodologies are most adept for a group of species in which an initial TR sequence has been identified previously. In place of PCR amplifying from genomic DNA, RT-PCR amplifying the TR transcript from total RNA with primers targeting the template sequence predicted from telomeric DNA repeat sequence has been used to identify TR transcripts from a number of Candida yeast species that contain relatively long TR templates (Gunisova et al. 2009). Other molecular methods including nucleic acid hybridization and/or gene-knockout library screening have thus far been applied exclusively in yeasts for TR identification (McEachern and Blackburn 1995; Hsu et al. 2007; Kachouri-Lafond et al. 2009). These other molecular approaches are generally limited to organisms with small genomes and established genetic tools, and cannot be readily applied to other groups of species.
Recent advances in genome sequencing have made BLAST, basic local alignment search tool, a rather straightforward means for searching TR homologs within closely related species (Altschul et al. 1990; Qi et al. 2013). Augmenting BLAST with position-specific weight matrices (PWM) has been shown to enhance degenerate TR sequence searches in teleost fish (Xie et al. 2008). While PWM allows for more degenerate search patterns, a sufficient number of well-aligned sequences are required to generate the weight matrices for nucleotide probability at each position (Mozig et al. 2007). Thus, this methodology is most applicable for TR identification from species diverged from a closely related group for which numerous sequences have previously been identified.
To date, no TR has been identified from invertebrates despite their evolutionary proximity to vertebrates. Here we report the identification of the first invertebrate TR from Strongylocentrotus purpuratus (purple sea urchin) using a novel approach that combines in vitro TR enrichment from total RNA, next-generation sequencing, and an optimized computational screening pipeline. Initial characterization of the sea urchin TR found putative deuterostome-specific structures and has provided important insights into the evolution of vertebrate TR structure by defining ancestral structural features.
RESULTS
Cloning TR from invertebrate species has been hindered by the low abundance of telomerase enzyme in cells, as well as the size, sequence, and structural divergence of TR. To overcome these obstacles and create a simple and straightforward system for identifying TRs, we have developed a novel approach that uses in vitro enrichment of the TR from total RNA, next-generation sequencing, and bioinformatics screening. In this study, we aimed to identify the TR from S. purpuratus, a model organism that has a sequenced genome and is closely related to vertebrates. The multiple-step TR-enrichment strategy we developed is based on the assumptions that invertebrate TR would have a 5′-trimethylguanosine (TMG) cap as well as preferentially be bound by the TERT protein in vitro. These are two common features of vertebrate and yeast TRs (Seto et al. 1999; Jady et al. 2004; Webb and Zakian 2008). A cDNA library generated from the enriched RNA was then subjected to next-generation sequencing, followed by in silico assembly of sequencing reads, screening for putative template sequences, and removal of known or hypothetical gene candidates.
TR enrichment was performed in three sequential steps: anti-TMG immuno-precipitation (IP), Terminator exonuclease treatment and TERT co-IP of RNA (Fig. 1). In the first step of TR enrichment, 12 mg of input total RNA isolated from purple sea urchin gonads produced 5 µg of TMG-capped RNA. Next, this TMG-capped RNA was treated with Terminator, a 5′-monophosphate-dependent exonuclease (Epicentre). The TMG-capped RNAs were resistant to Terminator exonuclease degradation while copurified ribosomal RNAs (rRNAs), which contain a 5′-monophosphate, were degraded. The final step of TR enrichment was TERT co-IP of RNA. A recombinant S. purpuratus TERT (SpuTERT) protein with a streptavidin-binding peptide (SBP) tag was in vitro synthesized in the rabbit reticulocyte lysate (RRL) and affinity purified by streptavidin Sepharose beads. The bead-bound SBP-SpuTERT protein was treated with Micrococcal nuclease to remove copurified RRL nucleic acids as well as the SpuTERT expression vector prior to incubation with the TMG-capped exonuclease-treated RNA under a lysate-free condition (Fig. 1). The SpuTERT-bound RNAs were extracted for cDNA library construction.
FIGURE 1.

Schematic of three-step SpuTR enrichment from total RNA. TMG-capped (red) RNA was purified from total RNA by anti-TMG IP and was then treated with 5′-monophosphate-specific exonuclease (orange) to remove ribosomal RNA. Recombinant SBP-tagged SpuTERT (blue) synthesized in vitro was affinity purified from RRL by streptavidin Sepharose beads and incubated with the TMG-capped exonuclease-treated RNA.
Next-generation sequencing of the cDNA library generated over 9,000,000 50-bp reads (Fig. 2). The reads were then analyzed in silico with a bioinformatics pipeline composed of three major steps: mapping onto the reference genome, searching for putative templates, and removing known genes. First, sequencing reads that could map onto the rabbit genome were removed, and the remaining sequencing reads were then mapped onto the purple sea urchin draft genome (assembly version 2.5), generating 181,881 independent loci. These mapped loci were then screened for putative TR template sequences, generating 1173 putative template loci. The putative template for S. purpuratus TR (SpuTR) was predicted to be 8–18 nt in length and a circular permutation of 5′-CCCUAA-3′ based on the telomeric DNA repeat sequence of 5′-TTAGGG-3′ in purple sea urchin (Sinclair et al. 2007). Finally, removing sequences corresponding to known or hypothetical genes resulted in a final set of 654 candidates (Fig. 2).
FIGURE 2.
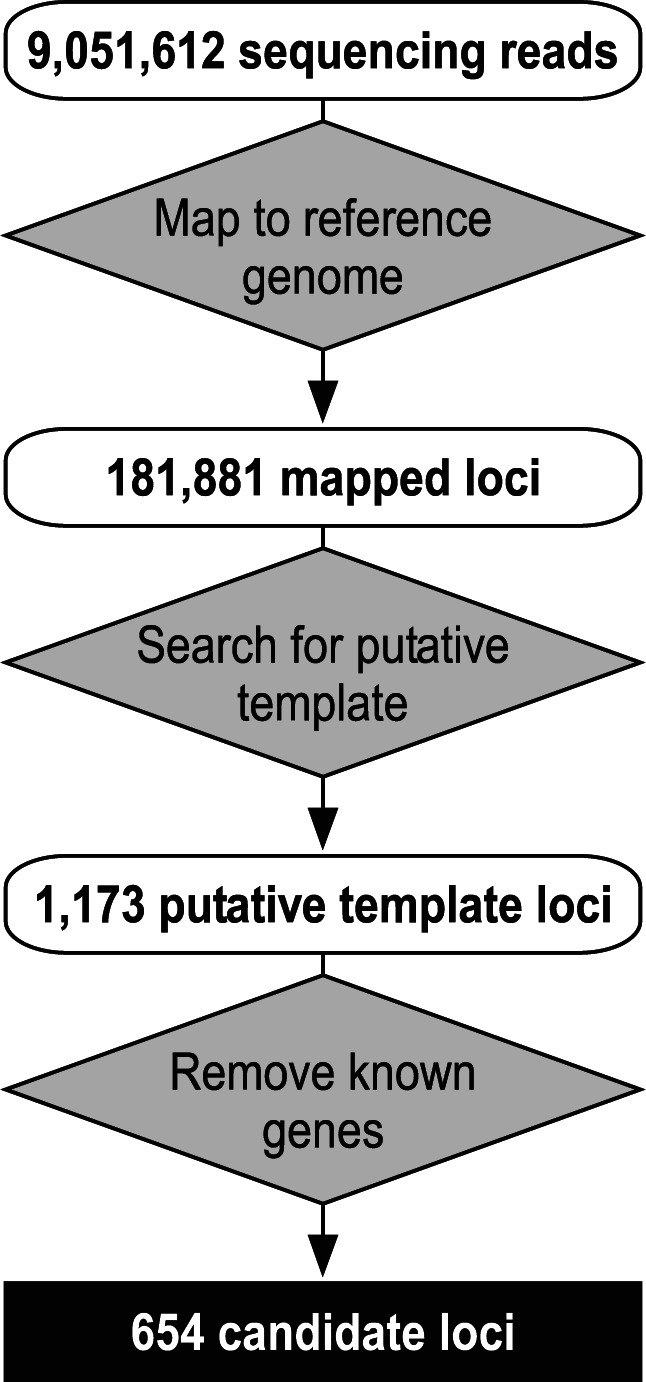
Computational screening for SpuTR candidates. Sequencing reads (9,051,612) were mapped onto the purple sea urchin reference genome. Mapped loci (181,881) were screened for putative template sequences, resulting in 1173 putative template loci. Putative template loci matching known or hypothetical genes were discarded, resulting in 654 candidate loci. Inputs (white rounded boxes), processes (gray diamonds), and final output (black square) are depicted.
The top five loci were ranked by the highest coverage per base of mapped reads and further analyzed for a potential pseudoknot structure and a long open-reading frame (ORF) sequence (Fig. 3A). The pseudoknot is a universal feature of TR, and the lack of a long ORF is an indicator of a noncoding RNA gene. We devised an algorithm, herein named TR-PK-finder, to detect TR pseudoknot characteristics based on conserved sequence motifs and base-pairing potentials (Supplemental Fig. S1; see Materials and Methods). TR-PK-finder predicted that both candidates #1 and #2 contain a potential pseudoknot structure (Fig. 3A). Similar to known TR structures, the predicted pseudoknot in candidate #1 is located 5′-proximal, starting at position 59 of the 522-nt mapped sequence. Moreover, the region with the pseudoknot structure had the highest sequencing read coverage of the candidate #1 locus, consistent with the pseudoknot domain functioning as a TERT-binding site (Fig. 3B). In contrast, the pseudoknot in candidate #2 is located 3′-proximal and had low-sequencing read coverage. Finally, ORF analysis suggested that candidate #1 is a noncoding RNA and candidate #2 is a putative protein-coding mRNA (Fig. 3A).
FIGURE 3.

Characterization of top-ranking candidate loci. (A) Ranking, average read coverage, number of sequencing reads, length of mapped loci, predicted template, genomic location, and presence of putative pseudoknot structure (Ψ-knot) or open-reading frame (ORF) are listed. Ranking is based on the average read coverage calculated by (Read-number × Read-length/Locus-length). (B) Patterns of read coverage for candidates #1 and #2. The locations of the predicted template (red) and the putative pseudoknot structure (green) are depicted.
To validate candidate #1 as a TR, we reconstituted telomerase activity by combining in vitro-transcribed candidate #1 RNA with in vitro-synthesized SpuTERT protein in RRL. The direct primer-extension assay detected activity from assembled SpuTERT and candidate #1 RNA, producing apparent telomeric products with the characteristic 6-nt ladder pattern (Fig. 4A). Reactions lacking either SpuTERT or candidate #1 RNA, or with RNase A-treatment, failed to produce products (Fig. 4A, lanes 1–3). To discern telomerase activity from nonspecific polymerase activity, we utilized four telomeric DNA primers with circularly permuted sequences annealing to different positions on the TR template (Fig. 4A). Extension by telomerase with these circular-permutated primers generated offset ladder patterns in a template-dependent manner (Fig. 4A, lanes 4–7). Taken together, these results unequivocally affirmed candidate #1 RNA as the S. purpuratus telomerase RNA (SpuTR).
FIGURE 4.

Validation and characterization of purple sea urchin TR. (A) Reconstitution of telomerase activity. (Left) Sequence of the putative template (open box) with the annealing position of four circular permuted telomeric DNA primers (a–d) shown. Predicted primer-extended products (lowercase, shaded gray) are depicted and aligned with the putative template. (Right) Direct primer-extension assay of telomerase reconstituted in vitro from synthesized SpuTERT and T7 transcribed candidate #1 RNA (522 nt). An RNase A-treated reaction is included as a control (lane 3). A 32P-end-labeled 18-mer oligonucleotide is added as the loading control (l.c.) to the reaction prior to ethanol precipitation of the products. Numbers to the right indicate nucleotides added to the primer. (B) Restriction map of the genomic region surrounding the SpuTR gene. The predicted sizes of the restriction digestion products are depicted (dashed arrows). (C) Southern blot analysis of the SpuTR gene. Genomic DNA digested with either NcoI or NdeI was probed with a riboprobe targeting the SpuTR gene. The approximate size of the restriction digestion products is shown to the right.
We then characterized the gene structure and transcript of SpuTR. The copy number of the SpuTR gene in the genome was determined by Southern blot. Genomic DNA digested by either NcoI or NdeI generated a single band of ∼5.3 and 3.3 kb, respectively (Fig. 4B,C). The single band from two different restriction digestions indicated that SpuTR is a single-copy gene. Furthermore, BLAST failed to identify any homologous sequences of the SpuTR gene within the purple sea urchin genome. The size of the full-length SpuTR transcript was determined by 5′- and 3′-RACE to be 535 nt (Supplemental Fig. S2). Interestingly, 5′-RACE analysis indicated that the 5′-end of SpuTR is heterogeneous, as additional 5′-RACE clones had 5′-ends at positions ranging from −5 to +2 compared with our initial SpuTR clone (Supplemental Fig. S2). Sequence analysis of additional independent SpuTR clones generated by PCR from genomic DNA revealed numerous single nucleotide polymorphisms (SNPs) and a single insertion (Supplemental Fig. S2). However, this is not too surprising as sea urchin is known to have 4%–5% SNPs, which is an order of magnitude higher than human (Britten et al. 1978).
The efficiency of each TR enrichment step was evaluated by the abundance of TR in each of the RNA samples (Fig. 5). RNA samples were extracted from each of the three sequential purification steps, as well as the input total RNA, and analyzed by next-generation sequencing together with our bioinformatics pipeline (Fig. 2). Remarkably, the TMG-IP step enriched SpuTR by almost 40-fold compared with the total RNA, based on the average coverage per base of the SpuTR sequence. The second enrichment step, Terminator exonuclease-treatment, only augmented the TR abundance by 2.7-fold. Unexpectedly, the final step, TERT co-IP, did not significantly increase the read coverage of the SpuTR sequence (Fig. 5). This step, however, massively depleted other RNAs, such as the U2 snRNA, within the TERT co-IP RNA sample (Supplemental Fig. S4). Moreover, the TERT co-IP step significantly reduced the number of candidate loci from 4564 in exonuclease-treated TMG-capped RNA to 606 in the TERT co-IP RNA (Fig. 5).
FIGURE 5.

Efficacy of individual TR enrichment steps. Total RNA and RNA samples from each of the three sequential TR enrichment steps (anti-TMG IP, exonuclease-treatment, and TERT-binding) were analyzed by Illumina next-generation (Next-Gen) sequencing. Read coverage for SpuTR in each sample and fold enrichment of coverage between samples (curved arrows) are indicated. Ranking the SpuTR locus and the number of template-containing loci were determined by bioinformatics screening and shown for each sample. Sequencing reads corresponding to the SpuTERT expression vector contaminated from the RRL reactions were removed prior to bioinformatics screening.
A secondary structure model for sea urchin TR was inferred from the sequence alignment of the SpuTR against vertebrate TRs. A high degree of sequence conservation was found in the template-pseudoknot and the H/ACA domains (Fig. 6A). In contrast, no sequence or structure corresponding to the vertebrate CR4/5 domain was identified in the SpuTR sequence. Based on sequence and structural homology with vertebrate TRs, a secondary structure model of SpuTR was established for specifically the template-pseudoknot and H/ACA domains (Fig. 6B). In the template-pseudoknot domain, SpuTR contained a unique additional helix, herein named P1.1, located between helix P1 and the template region. The remaining helices in the template-pseudoknot domain are universally conserved between purple sea urchin and vertebrates and were named similarly. The H/ACA domain of SpuTR included three motifs: Box H, Box ACA, and the CAB box, which are universally conserved in vertebrate TR, aside from teleost fish TR that lacks an obvious CAB box (Xie et al. 2008). While lacking sequence or structural homology with vertebrate TRs, the central region of the sea urchin TR was predicted by the mfold program (Zuker 2003) to fold into two long helices, herein named P4.1 and P4.2, stemming from helix P4 (Fig. 6B).
FIGURE 6.

Sequence alignment and secondary structure model of SpuTR. (A) SpuTR sequence aligned against six vertebrate TR sequences. Multiple-sequence alignment was performed in the program, BioEdit, using the ClustalW algorithm. Manual adjustment was performed based on highly conserved regions and moieties. Base-pairings are shaded within template-pseudoknot (violet) and H/ACA (cyan) domains and >80% identity shaded (black). The central 286 nt failed to align with vertebrate TR sequences and was omitted. (B) Secondary structure model of SpuTR. The sequence for the template-pseudoknot (violet) and H/ACA (cyan) domains are shown. Outline of two putative helices (P4.1 and P4.2) predicted by mfold is shown in the central domain. Nucleotides universally conserved among SpuTR and vertebrate TRs are colored (red).
To determine structural elements essential for telomerase activity within the urchin TR central domain, we analyzed the SpuTR central domain by serial truncations (Fig. 7A). Various central domain fragments and a template-pseudoknot (T-PK) fragment were assembled in trans with recombinant SpuTERT in RRL to reconstitute activity. Telomerase activity was assessed by the direct primer-extension assay (Fig. 7B; Supplemental Fig. S3). The T-PK RNA fragment (6–185 nt) alone produced 33% activity compared with T-PK with the P4/P4.1/P4.2 (186–456 nt) RNA fragments (7B, lanes 1,2). The T-PK and P4.2 fragments reconstituted telomerase activity similar to the full-length TR (Supplemental Fig. S3). The analysis of individual regions of the central domain indicated that the P4.2 putative helix (312–420 nt) is essential, while the P4.1 putative helix (191–291 nt) appears dispensable for rescuing telomerase activity (Fig. 7B, lanes 3,4). Serial truncations of P4.2 identified the smallest RNA fragment, P4.2C (332–400 nt), which retained full activity, while P4.2D (337–394 nt) only partially rescued activity, and P4.2E (343–385 nt) did not stimulate activity (Fig. 7B, lanes 5–8).
FIGURE 7.

Telomerase assay to determine the essential SpuTR fragments necessary for activity. (A) Outline of two putative helices forming the SpuTR central domain. Nucleotide numbers denote the 5′- and 3′-ends of the T7 transcribed RNA fragments, P4.1 (191–291 nt), P4.2 (312–420 nt), and five P4.2 truncated fragment (A–E), of the central domain (186–456 nt). (B) Direct primer-extension assay of telomerase reconstituted from various T7 transcribed SpuTR fragments and in vitro-synthesized SpuTERT. The SpuTR fragments included in each reaction are indicated above the gel. The primer-extension reactions were performed in the presence of radioactive [α-32P]dGTP and 1 µM telomeric primer (TTAGGG)3. A 32P-end-labeled 18-mer oligonucleotide was included as the loading control (LC). Numbers to the left indicate the number of nucleotides added to the primer. Relative activity (%) of each reaction was normalized to the reaction with the entire central domain (186–456 nt) and is indicated below the gel.
DISCUSSION
Phylogenetic studies of telomerase RNP structure and function require effective and efficient methods for identifying TR genes from all major lineages of eukaryotes. However, as one of the most divergent noncoding RNAs, TR cannot be readily identified from many key organisms with conventional molecular and bioinformatics techniques. Thus, we have established a novel TR discovery approach that leverages TR enrichment from total RNA by basic molecular techniques and massively parallel sequencing technology. We have validated our approach by successfully identifying the first TR sequence from an echinoderm, the purple sea urchin.
Our affinity-purification scheme relies on two common features of TRs, a 5′-TMG cap and TERT-affinity. The TMG-purification step is highly effective, as this step alone enriched SpuTR by nearly 40-fold and diminished the overly abundant rRNAs and mRNAs (Fig. 5). Since known TR identified from vertebrates and yeasts contain a 5′-TMG, this TMG-purification step presumably can be applied toward identifying TRs from numerous additional species. However, there are species such as ciliates and filamentous fungi that lack a TMG cap in TR. Identification of TR from these species would not be possible with a TMG-IP step and would require alternative enrichement approaches. The TERT-binding step requires cloning and expression of the TERT protein gene from the species of interest, which is rather straightforward due to the sequence conservation of the TERT protein and the large number of sequenced eukaryotic genomes available. While it did not signifigantly enrich for SpuTR in sequencing read coverage, the TERT co-IP step alone reduced the number of candidate loci from 4564 to only 606 and repositioned SpuTR to become the highest ranked candidate (Fig. 5). Therefore, the TERT co-IP will be a useful step for identifying TRs that lack 5′ TMG-cap.
Separate from PCR-based approcahes, our approach includes affinity purification of TMG-capped or TERT-bound RNAs that does not require any information from the TR sequence and thus avoids issues arising from the vast TR sequence divergence among clades. Our next-generation sequencing strategy also overcomes the constraints of template-targeting RT-PCR that often generate nonspecific products amplified from rRNA or other abundant RNA species that contain similar sequences. In comparison, massively parallel sequencing identifies essentially all RNA sequences in a given sample, dependent on the depth of coverage. Our computational screening pipeline requires only the telomere sequence from the species of interest for predicting possible template sequences. This template screening process is minimal and yet effective. In our search for sea urchin TR, this screening dramatically reduced the initial 181,881 loci down to 1173 template-containing loci, and finally to 654 after removing known and hypothetical genes (Fig. 2). Template searching also improved the ranking of SpuTR, based on read coverage, from the 415th candidate among the 181,881 mapped loci to the fourth among the 1173 template-containing candidates, prior to removing known and hypothetical genes (Fig. 2). Predictions of pseudoknot structure and ORF in these candidate sequences, while not essential, are additional filters that may facilitate TR discovery in certain species. Of the top five candidates, all but candidate #1 SpuTR can be eliminated based on the presence of a large ORF or the absence of a predicted pseudoknot (Fig. 3A). With the growing capacity of next-generation sequencing alongside the increasing number of sequenced eukaryotic genomes, our next-generation sequencing approach would expedite identification of TR from key organisms and facilitate the evolutionary study of telomerase structure and function.
The SpuTR structure contains the universal template-pseudoknot domain and the vertebrate-specific H/ACA domain. The secondary structure model for these two domains can be simply inferred from the sequence alignment of SpuTR with vertebrate TR sequences (Fig. 6). However, it is intriguing that the vertebrate CR4/5 could not be found within the closely related SpuTR, while distantly related filamentous fungal TRs preserved a vertebrate-like CR4/5 element. The CR4/5 element found in filamentous fungal TRs includes the P6.1 stem–loop, which is essential for reconstituting telomerase activity in vitro (Qi et al. 2013). The lack of a vertebrate-like CR4/5 domain, together with the basal in vitro activity reconstituted from the template-pseudoknot domain alone, suggests that SpuTR has diverged from the vertebrate-fungal lineage for this structural feature. The SpuTR template-pseudoknot domain contains a unique ancillary template-adjacent helix P1.1 with a function that has yet to be determined (Fig. 6). We suspect that this structure serves as a template boundary element, similar to the template boundary structure commonly seen in fungal TRs, in place of the P1 element found in vertebrate TRs (Tzfati et al. 2000; Chen and Greider 2003). Similar to vertebrate TRs, the SpuTR H/ACA domain contains the conserved motifs, Box H, Box ACA, and CAB box. The conservation of a CAB box in an invertebrate TR would indicate that teleost fish TRs have lost this important moiety during evolution (Xie et al. 2008). This provides the first evidence that the teleost TR evolved from requiring a CAB box for TR biogenesis rather than higher vertebrates acquiring this moiety.
The discovery of TR sequences outside of already established clades is essential for understanding telomerase RNP evolution and the divergence in structure, biogenesis pathways, and telomere maintenance mechanisms among eukaryotic lineages. Additional TR sequences will further define the diversity of species-specific moieties and reveal the underlying conserved and essential structures within newly explored groups of species. With the ever-expanding number of sequenced genomes, our system of TR discovery will identify new TR sequences from previously unexplored clades, which is the foundation for the comprehensive study on telomerase structure, function, and evolution.
MATERIALS AND METHODS
Isolation of total RNA
Gonad tissue was dissected from live purple sea urchin (Marine Research and Educational Products). Total RNA was isolated from gonads using TRI-Reagent (Molecular Research Center, Inc.) following the manufacturer’s instructions.
TMG immunoprecipitation of total RNA
Twelve milligrams of total RNA was incubated with 300 µg of anti-TMG (K121) monoclonal antibody (Santa Cruz Biotechnology) in 1X IP buffer (25 mM Tris-HCl at pH 8.0, 200 mM NaCl, and 0.2 mM EDTA) with 0.1 units/µL SUPERase-In RNase inhibitor (Ambion) at 4°C for 2 h. The mixture was then combined with 400 µL of Ultralink Immobilized Protein A/G gel slurry (Pierce) that was preblocked with 1 mg/mL of RNase-free BSA (Ambion) in 1X IP buffer at 4°C for 20 min with gentle agitation. The reaction was incubated at 4°C for 2 h with gentle agitation. The gel slurry was washed five times with 1X Wash buffer (25 mM Tris-HCl at pH 8.0, 300 mM NaCl, and 0.2 mM EDTA) and eluted with 1X SDS Elution buffer (25 mM Tris-HCl at pH 8.0, 200 mM NaCl, 5 mM EDTA, and 1% SDS) at room temperature for 10 min. RNA was isolated by acid phenol/chloroform extraction and ethanol precipitation.
Cloning, synthesis, and purification of SpuTERT
The SpuTERT cDNA (GenBank accession no. EF611988) was amplified from mRNA by RT-PCR and cloned into the pCITE-4a vector (Novagen). A 38-amino acid SBP or FLAG tag was appended to the N terminus of SpuTERT, generating pNSBP-SpuTERT and pNFLAG-SpuTERT, respectively (Keefe et al. 2001). The SpuTERT protein was synthesized in vitro in 0.5 mL of RRL using the TNT Quick Coupled Transcription/Translation System (Promega) following the manufacturer’s instructions. The RRL reaction was combined with 240 µL of streptavidin Sepharose beads (GE Healthcare) that were prewashed four times with 1X Wash buffer and incubated at room temperature for 16 h with gentle agitation. Beads were then washed three times with 1X Wash buffer and treated with 400 units of Micrococcal nuclease (Worthington) in 1X MNase buffer (50 mM Tris-HCl at pH 8.3, 5 mM CaCl2, and 1 mg/mL BSA) at room temperature for 40 min with gentle agitation. The beads were washed three times with 1X Wash buffer and stored in 20 µL of 1X RNA-binding buffer (50 mM Tris-HCl at pH 8.0, 100 mM KCl, 2 mM MgCl2, 2 mM DTT, 10 mM EGTA, 2 units/µL SUPERase-In, and 0.025% Tween 20).
Purification and next-generation sequencing of TERT-binding RNA
Five micrograms of TMG-capped RNA was first treated with 2 units of Terminator 5′-Monophosphate-Dependent Exonuclease (Epicentre) at 30°C for 1 h to remove rRNA copurified. RNA was isolated by acid phenol/chloroform extraction and ethanol precipitation. Exonuclease-treated RNA was then combined with affinity-purified SpuTERT protein on beads in 1X RNA-binding buffer, and incubated at room temperature for 20 min with gentle agitation. Beads were then washed three times with ice-cold 1X Wash buffer. TERT-bound RNA was eluted with 1X SDS Elution buffer, acid phenol/chloroform extracted, and ethanol precipitated. RNA quality was determined by a Bioanalyzer 2100 (Agilent Technologies, Inc.). A cDNA library was constructed from extracted TERT-bound RNA and a single-end 50-bp sequencing run on an Illumina Genome Analyzer performed by Cofactor Genomics.
Computational analysis of sequencing data
The sequencing data comprising 9,051,612 reads was mapped to reference genomes using Segemehl (Hoffmann et al. 2009). Contamination of rabbit RNA from RRL was minimized by removing sequencing reads mapped to the rabbit genome (Oryctolagus cuniculus, v.50, ENSEMBL). The remaining sequencing reads were mapped onto the purple sea urchin genome (S. purpuratus genome assembly, v.2.5, HGSC) (Sodergren et al. 2006). All sequence mapping required a minimum accuracy of 85% and a maximum seed E-value of 10. Each read was permitted up to 100 matches with an optimal score in the target genome. All suboptimal hits were discarded. Expression for ambiguous reads was corrected by hit normalization. Approximately 88% of the sequencing reads were mapped to the purple sea urchin genome.
Mapped loci were defined by genomic intervals mapped with sequencing reads. Covered genomic intervals separated by <200 nt were defined as a single loci. Mapped loci were then searched for putative template sequences with the program, RNABOB, developed by Sean Eddy (http://selab.janelia.org/software.html). Putative template sequences were defined as the circular permutations of repeated sequence “CCCUAA” from 8 to 18 nt in length and their reverse complementary sequences, 132 permutations in all.
Putative template loci matching known or hypothetical purple sea urchin gene sequences were discarded. Known and hypothetical gene sequences were defined to include the following: noncoding RNA sequences from Rfam, v.10.0 with BLAST parameters -p blastn -e 1e-4 (Altschul et al. 1990; Gardner et al. 2009), tRNA sequences from tRNAscan-SE with standard parameters (Lowe and Eddy 1997), rRNA sequences (GenBank accession nos. L28056 and AF212171), and known and hypothetical protein genes (NCBI) with BLAST parameters -p blastn -e 1e-10 (Altschul et al. 1990).
Recognition of telomerase pseudoknot
Mapped loci not matching known or hypothetical gene sequences were defined as candidate loci. Candidate loci were queried with the TR-PK-finder algorithm for detection of a putative pseudoknot structure based on features conserved in the vertebrate TR pseudoknot structure (Supplemental Fig. S1). Sequences were assessed with four quality scores t, a, c, and m. Scoring t and a for region S2 and S6, respectively, were determined by:
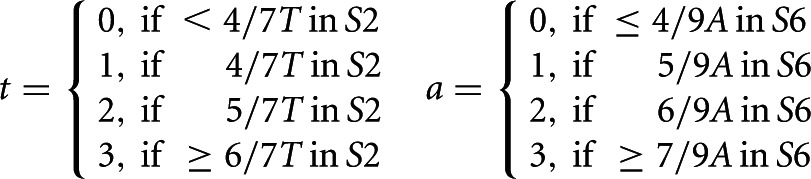 |
Scoring c for S4, was determined by an exact match to the described pattern, c = 1 or else c = 0. Values for t, a, and c were determined by RNABOB. Scoring for m as determined by the estimated thermodynamics of the pseudoknot fold. The program RNAfold (Hofacker et al. 1994) evaluated the free energy of two structures facilitating pseudoknot formation, A and B, and a disruptive structure C with folding energies EA, EB, and EC, respectively. The relative folding energy, e = (EA + EB)/EC, estimates the stability and thermodynamics of the pseudoknot fold. The relative folding energy was then converted into a qualitative score by e > 0.8, m = 2, 0.8 > e > 0.5, m = 1 or else m = 0. The final score for predicting the pseudoknot is the sum of t, a, c, and m. Threshold for a positive pseudoknot prediction was set at minimally 6 of 9.
Characterization and cloning of SpuTR
The 5′- and 3′-ends of SpuTR were determined by Rapid Amplification of cDNA Ends (RACE) using the FirstChoice RLM-RACE kit (Ambion). The full-length SpuTR gene was PCR amplified from genomic DNA that was isolated from sea urchin gonad tissue with DNAzol (Invitrogen). The initial full-length SpuTR clone was sequenced and deposited in GenBank (accession no. JQ684708).
SpuTR gene copy number was determined by Southern blot. Eight micrograms of genomic DNA was restriction digested to completion with either 50 units of NcoI (NEB) in 1X buffer 3 or 60 units of NdeI (NEB) in 1X buffer 4 with BSA at 37°C for 16 h. Digested DNA was extracted with phenol/chloroform, ethanol precipitated, resolved on a 0.8% agarose gel, and transferred onto a Hybond-XL nylon membrane (Amersham) following the manufacturer’s instructions. After prehybridization in ULTRAhyb solution (Ambion) for 30 min at 42°C, the membrane was hybridized for 16 h at 37°C with a riboprobe targeting SpuTR. The riboprobe was synthesized by the MAXIscript T7 kit (Ambion) following the manufacturer’s instructions and labeled internally with [α-32P]UTP (3000 Ci/mmol, 10 mCi/mL, PerkinElmer). The membrane was washed twice with 2X SSC (0.3 M NaCl and 30 mM sodium citrate) and 0.5% SDS for 20 min, and once with 0.2X SSC and 0.1% SDS for 40 min at 42°C. The membrane was exposed to a phosphor storage screen and analyzed with an FX Pro Molecular Imager (Bio-Rad).
Efficacy of SpuTR enrichment
Total RNA and RNA samples extracted from TMG-IP, exonuclease-treatment, and TERT-binding steps were used for cDNA library construction with the ScriptSeq v2 RNA-Seq Library Preparation Kit (Epicentre) following the manufacturer’s instructions. The cDNA libraries were constructed with ScriptSeq Index PCR Primers (Epicentre) and the indexed cDNA libraries were pooled for a single multiplexed single-end 100-bp sequencing run on an Illumina HiSeq 2000. The pooled sample was demultiplexed, and each population of sequencing reads was independently mapped. The mapped loci were searched for putative template sequences and known and hypothetical genes were removed, as previously described. The in vitro-synthesized SpuTERT protein was not treated with Micrococcal nuclease prior to binding exonuclease-treated TMG-IP RNA. Sequencing reads corresponding to the expression vector copurified with the SpuTERT were removed prior to computational analysis.
Reconstitution of sea urchin telomerase activity
The recombinant NFLAG-SpuTERT protein was synthesized in vitro with the TnT Quick Coupled Transcription/Translation System (Promega) following the manufacturer’s instructions, assembled with in vitro T7 transcribed SpuTR in RRL, and assayed for telomerase activity with the direct primer-extension assay. The direct primer-extension assay was performed with 6 µL of in vitro-reconstituted telomerase in a 20-µL reaction containing 1x PE buffer (50 mM Tris–HCl at pH 8.3, 2 mM DTT, 0.5 mM MgCl2, and 1 mM spermidine, 0.5 mM dTTP, 0.5 mM dATP, 2 µM dGTP, 0.165 µM [α-32P]dGTP [3000 Ci/mmol, 10 mCi/mL, PerkinElmer], and 1 µM telomeric primer (TTAGGG)3, (GTTAGG)3, (AGGGTT)3, or (GGTTAG)3, as denoted). The reactions were incubated at 30°C for 60 min and a 32P-end-labeled 18-mer DNA oligonucleotide was added to the reaction as a loading control. The reactions were terminated by phenol/chloroform extraction and ethanol precipitated. Products of telomerase reactions were resolved on a denaturing 8-M urea/10% polyacrylamide gel. The dried gel was exposed to a phosphor storage screen and analyzed with an FX Pro Molecular Imager (Bio-Rad).
DATA DEPOSITION
The complete sequences of SpuTERT mRNA and SpuTR determined in this study have been deposited in the GenBank under accession nos. EF611988 and JQ684708, respectively.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Jim Allen and members of the Chen lab for critical reading of the manuscript and helpful discussion. This work was supported by National Sciences Foundation Career Award MCB0642857 to J.J.-L.C., DFG grants GK 1384 and MA-5082/1 to M.M., DFG-SPP 1258 to P.F.S. The LIFE Leipzig Research Center for Civilization Diseases is supported by the European Regional Development Fund (ERDF) and the Free State of Saxony.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ 1990. Basic local alignment search tool. J Mol Biol 215: 403–410 [DOI] [PubMed] [Google Scholar]
- Blackburn EH, Collins K 2011. Telomerase: An RNP enzyme synthesizes DNA. Cold Spring Harb Perspect Biol 3: a003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bley CJ, Qi X, Rand DP, Borges CR, Nelson RW, Chen JJ-L 2011. RNA-protein binding interface in the telomerase ribonucleoprotein. Proc Natl Acad Sci 108: 20333–20338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ, Cetta A, Davidson EH 1978. The single-copy DNA sequence polymorphism of the sea urchin Strongylocentrotus purpuratus. Cell 15: 1175–1186 [DOI] [PubMed] [Google Scholar]
- Chen J-L, Greider CW 2003. Template boundary definition in mammalian telomerase. Genes Dev 17: 2747–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J-L, Blasco MA, Greider CW 2000. Secondary structure of vertebrate telomerase RNA. Cell 100: 503–514 [DOI] [PubMed] [Google Scholar]
- Cifuentes-Rojas C, Kannan K, Tseng L, Shippen DE 2011. Two RNA subunits and POT1a are components of Arabidopsis telomerase. Proc Natl Acad Sci 108: 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dandjinou AT, Lévesque N, Larose S, Lucier J-F, Abou Elela S, Wellinger RJ 2004. A phylogenetically based secondary structure for the yeast telomerase RNA. Curr Biol 14: 1148–1158 [DOI] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, et al. 2009. Rfam: Updates to the RNA families database. Nucleic Acids Res 37: D136–D140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH 1989. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 337: 331–337 [DOI] [PubMed] [Google Scholar]
- Gunisova S, Elboher E, Nosek J, Gorkovoy V, Brown Y, Lucier JF, Laterreur N, Wellinger RJ, Tzfati Y, Tomaska L 2009. Identification and comparative analysis of telomerase RNAs from Candida species reveal conservation of functional elements. RNA 15: 546–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P 1994. Fast folding and comparison of RNA secondary structures. Monatshefte für Chemie 125: 167–188 [Google Scholar]
- Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermuller J 2009. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol 5: e1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M, McEachern MJ, Dandjinou AT, Tzfati Y, Orr E, Blackburn EH, Lue NF 2007. Telomerase core components protect Candida telomeres from aberrant overhang accumulation. Proc Natl Acad Sci 104: 11682–11687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jady BE, Bertrand E, Kiss T 2004. Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body-specific localization signal. J Cell Biol 164: 647–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachouri-Lafond R, Dujon B, Gilson E, Westhof E, Fairhead C, Teixeira M 2009. Large telomerase RNA, telomere length heterogeneity and escape from senescence in Candida glabrata. FEBS Lett 583: 3605–3610 [DOI] [PubMed] [Google Scholar]
- Keefe AD, Wilson DS, Seelig B, Szostak JW 2001. One-step purification of recombinant proteins using a nanomolar-affinity streptavidin-binding peptide, the SBP-Tag. Protein Expr Purif 23: 440–446 [DOI] [PubMed] [Google Scholar]
- Leonardi J, Box JA, Bunch JT, Baumann P 2008. TER1, the RNA subunit of fission yeast telomerase. Nat Struct Mol Biol 15: 26–33 [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR 1997. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason M, Schuller A, Skordalakes E 2011. Telomerase structure function. Curr Opin Struct Biol 21: 92–100 [DOI] [PubMed] [Google Scholar]
- McEachern MJ, Blackburn EH 1995. Runaway telomere elongation caused by telomerase RNA gene mutations. Nature 376: 403–409 [DOI] [PubMed] [Google Scholar]
- Mozig A, Chen JJ-L, Stadler PF 2007. Homology search with fragmented nucleic acid sequence patterns. Lect Notes Comput Sci 4645: 335–345 [Google Scholar]
- Podlevsky JD, Chen JJ-L 2012. It all comes together at the ends: Telomerase structure, function, and biogenesis. Mutat Res 730: 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlevsky JD, Bley CJ, Omana RV, Qi X, Chen JJ-L 2008. The telomerase database. Nucleic Acids Res 36: D339–D343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Li Y, Honda S, Hoffmann S, Marz M, Mosig A, Podlevsky JD, Stadler PF, Selker EU, Chen JJ-L 2013. The common ancestral core of vertebrate and fungal telomerase RNAs. Nucleic Acids Res 41: 450–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto AG, Zaug AJ, Sobel SG, Wolin SL, Cech TR 1999. Saccharomyces cerevisiae telomerase is an Sm small nuclear ribonucleoprotein particle. Nature 401: 177–180 [DOI] [PubMed] [Google Scholar]
- Sinclair CS, Richmond RH, Ostrander GK 2007. Characterization of the telomere regions of scleractinian coral, Acropora surculosa. Genetica 129: 227–233 [DOI] [PubMed] [Google Scholar]
- Sodergren E, Weinstock GM, Davidson EH, Cameron RA, Gibbs RA, Angerer RC, Angerer LM, Arnone MI, Burgess DR, Burke RD, et al. 2006. The genome of the sea urchin Strongylocentrotus purpuratus. Science 314: 941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzfati Y, Fulton TB, Roy J, Blackburn EH 2000. Template boundary in a yeast telomerase specified by RNA structure. Science 288: 863–867 [DOI] [PubMed] [Google Scholar]
- Webb CJ, Zakian VA 2008. Identification and characterization of the Schizosaccharomyces pombe TER1 telomerase RNA. Nat Struct Mol Biol 15: 34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Mosig A, Qi X, Li Y, Stadler PF, Chen JJ-L 2008. Structure and function of the smallest vertebrate telomerase RNA from teleost fish. J Biol Chem 283: 2049–2059 [DOI] [PubMed] [Google Scholar]
- Zuker M 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.