

The miRNA profiles of samples with global miRNA decrease were analyzed using Affymetrix miRNA microarrays following the inducible genetic deletion of Dicer1. Surprisingly, up to one-third of deregulated miRNAs identified upon Dicer1 depletion were found to be up-regulated following standard robust multichip average (RMA) background correction and quantile normalization, indicative of normalization bias. These findings highlight the importance of miRNA microarray normalization for the detection of miRNAs that are truly differentially expressed.
Keywords: microRNA, miRNA microarray, normalization, Dicer1
Abstract
Recent studies have established that mutations or deletions in microRNA (miRNA) processing enzymes resulting in a global decrease of miRNA expression are frequent across cancers and can be associated with a poorer prognosis. While very popular in miRNA profiling studies, it remains unclear whether miRNA microarrays are suited or not to accurately detecting global miRNA decreases seen in cancers. In this work, we analyzed the miRNA profiles of samples with global miRNA decreases using Affymetrix miRNA microarrays following the inducible genetic deletion of Dicer1. Surprisingly, up to a third of deregulated miRNAs identified upon Dicer1 depletion were found to be up-regulated following standard robust multichip average (RMA) background correction and quantile normalization, indicative of normalization bias. Our comparisons of five preprocess steps performed at the probe level demonstrated that the use of cyclic loess relying on non-miRNA small RNAs present on the Affymetrix platform significantly improved specificity and sensitivity of detection of decreased miRNAs. These findings were validated in samples from patients with prostate cancer, where conjugation of robust normal-exponential background correction with cyclic loess normalization and array weights correctly identified the greatest number of decreased miRNAs, and the lowest amount of false-positive up-regulated miRNAs. These findings highlight the importance of miRNA microarray normalization for the detection of miRNAs that are truly differentially expressed and suggest that the use of cyclic loess based on non-miRNA small RNAs can help to improve the sensitivity and specificity of miRNA profiling in cancer samples with global miRNA decrease.
INTRODUCTION
MicroRNAs (miRNAs) are small RNAs of ∼22 nt involved in the translation control of complementary target messenger RNAs. miRNAs are processed in the nucleus from longer RNA polymerase II transcripts with hairpin-like secondary structures that are excised by the endonuclease Drosha to release miRNA precursors (pre-miRNAs) (Krol et al. 2010). Pre-miRNAs are subsequently exported to the cytoplasm through recruitment by exportin-5 (XPO5), where they are finally processed by a second endonuclease, Dicer1, which cleaves the loop structure of the hairpin and generates two separate ∼22-nt molecules (Krol et al. 2010). The resulting mature miRNA is recruited to a protein complex made up of several proteins, including TAR RNA-binding protein 2 (TARBP2) and Argonaute proteins, to direct regulation of sequence-specific messenger RNA (mRNA) sequences. Because mRNA target recognition by an miRNA only relies on complementarity with the 6–8 5′ nucleotides of the miRNAs, a single miRNA can affect several hundred target mRNAs (Gantier 2010). There are currently more than 2000 and more than 1200 mature miRNAs reported in human and mice, respectively (Griffiths-Jones 2010), but the function of most of these remains poorly defined.
Nevertheless, it is now well established that the translational control assured by miRNAs is crucial to most aspects of normal cellular function. Through the regulation of tumor suppressors and/or oncogenes, several miRNAs have now been shown to be directly involved in cancer development (e.g., miR-19b or miR-15a/16-1) (Mu et al. 2009; Aqeilan et al. 2010). miRNA expression is altered in most types of cancers (Melo and Esteller 2011), and early reports of miRNA profiling using PCR-based strategies demonstrated a prevalent global miRNA decrease in tumor samples of various origins (Lu et al. 2005; Gaur et al. 2007). It has recently been suggested that this global miRNA decrease in tumor samples could originate from genetic alterations affecting components of the miRNA biogenesis machinery (Dicer1, XPO5, and TARBP2). Indeed, 30% of human tumors analyzed in a study using 322 tumor samples (from breast, kidney, large intestine, liver, lung, ovary, pancreas, and stomach cancers) exhibited monoallelic loss of Dicer1 (Kumar et al. 2009), and 60% of ovarian cancers (from 111 patients) had decreased Dicer1 mRNA levels (Merritt et al. 2008). Similarly, studies in tumors with microsatellite instability, such as hereditary nonpolyposis colon cancer, apparently sporadic colorectal, gastric, or endometrial cancers, revealed an important proportion of monoallelic XPO5 (22.8% over 337 tumors) (Melo et al. 2010) or TARBP2 (26% in 282 tumors) (Melo et al. 2009) frameshift mutations resulting in decreased miRNA levels. Collectively, these findings establish that a significant proportion of tumors display a global miRNA decrease through the impairment of miRNA processing.
While a global miRNA decrease is relatively frequent in cancer samples, the majority of early microarray-based miRNA profiling studies have found relatively balanced proportions of miRNAs being up- and down-regulated in cancer (Volinia et al. 2006; Yanaihara et al. 2006). Given that the original reports of global miRNA decrease relied on PCR-based detection of miRNAs rather than microarray profiling (Lu et al. 2005; Gaur et al. 2007), we postulated that miRNA microarray profiling using methods carried over from mRNA microarrays could introduce a bias in the analysis of samples with global miRNA decrease. To determine whether miRNA microarray profiling could accurately identify samples with global miRNA decrease, we decided to analyze miRNA levels following Dicer1 deletion (Gantier et al. 2011, 2012), using a single-color miRNA microarray platform (Affymetrix).
The robust multichip average (RMA) algorithm is a standard method for background correcting, summarizing, and normalizing data from Affymetrix gene expression GeneChips (Irizarry et al. 2003). RMA background correction is achieved by modeling the intensity values in terms of signal plus noise (Irizarry et al. 2003; Bolstad 2004). The final step in the RMA algorithm is quantile normalization, which has the effect of forcing the distribution of normalized expression values to be identical for every microarray (Bolstad et al. 2003). This assumes, in effect, that the bulk of genes are not differentially expressed and that up and down expression changes are roughly balanced between experiment conditions. Although very popular for mRNA microarrays, the RMA algorithm does not make use of control probes and is not well suited to experiments showing global expression changes (Wu and Aryee 2010). The RMA background correction model was further developed to achieve more accurate estimates of the signal and noise components in the context of two-color microarrays (Ritchie et al. 2007; Silver et al. 2009) and was adapted to make use of control probes in the context of Illumina BeadChips (Ding et al. 2008; Shi et al. 2010b). In particular, a robust model-estimation strategy was developed to guard against the possibility that a minority of negative control probes represented real signal (Shi et al. 2010b).
Loess normalization is a popular method for two-color microarrays (Yang et al. 2002). Loess normalization has also been applied to one-channel microarrays by applying it cyclically to each possible pair of arrays (Bolstad et al. 2003). Unlike quantile normalization, loess normalization can be generalized to use unequal probe-weights (Smyth and Speed 2003). Probe-weighted loess normalization in conjunction with control probes was shown to be effective for normalizing two-color microarrays even in the presence of unbalanced global changes in gene expression (Oshlack et al. 2007).
In this article, we explore the effectiveness of probe-weighted cyclic loess for normalizing Affymetrix miRNA microarrays when a global change in expression is present. A potential advantage of this approach is that a variety of non-miRNA probes can be treated as invariant controls in order to stabilize the normalization curves. Comparison of five combinations of preprocessing steps performed at the probe level suggested that the use of cyclic loess relying on non-miRNA small RNAs could help to reduce detection of false-positive up-regulated miRNAs and improve detection of truly down-regulated miRNAs. These findings were validated in prostate cancer samples where miRNAs are preferentially down-regulated (Ozen et al. 2008). Our results suggest that the use of robust normal-exponential (normexp) background correction (Shi et al. 2010b) with probe-weighted cyclic loess normalization can help to reduce the incidence of false-positive up-regulated miRNAs and improve detection of truly down-regulated miRNAs during microarray normalization of cancer samples.
RESULTS
Generation of samples with global miRNA decrease
We generated samples with gradual global miRNA depletion relying on Dicerflox/flox × Cre/Esr1 mouse embryonic fibroblasts (MEFs) in which the majority of the second RNase III domain of Dicer1 can be deleted following 4-hydroxy-tamoxifen (OHT) treatment of the cells (Gantier et al. 2011). Having previously established that the intracellular levels of miRNAs were mostly affected by cell division in this model (Gantier et al. 2011), we conducted a series of cell passages following OHT-induced Dicer1 deletion to ensure active cell division over the course of the experiment (Fig. 1A). Preliminary experiments demonstrated that intracellular miRNA levels decreased from day 2 following OHT (data not shown). Cells were, therefore, collected on days 2, 3, 4, and 5 following OHT treatment, and validation of global miRNA decrease was assessed using TaqMan reverse transcription quantitative PCR (RT-qPCR) low density arrays for one set of samples (Fig. 1B). Two hundred and twenty-two miRNAs were found to be detected on day 2 (i.e., with a Cq ≤ 35). The overall distribution of miRNA expression for these 222 miRNAs showed a significant decrease in miRNA expression with time following OHT treatment. To confirm these results, we next analyzed the expression profile of four miRNAs by individual RT-qPCR assays of miR-125b, miR-21, miR-19b, and miR-155 (Fig. 1C). These results showed a significant decrease in miRNAs averaging ∼25% (ranging between 17% and 37% for the miRNAs analyzed) between days 2 and 3 following OHT treatment of the cells. Similarly, we observed an ∼64% decrease in miRNA levels (ranging between 58% and 70%) between days 2 and 4 following OHT-deletion of Dicer1. Importantly, the variability among the biological replicates was very low according to the RT-qPCR results. With the exception of miR-451, all mouse pre-miRNAs are dependent on Dicer1 to be processed into mature miRNAs (Cheloufi et al. 2010). Collectively, the results thereby indicate that these samples reproduced a global miRNA decrease, as can be observed in cancer samples.
FIGURE 1.
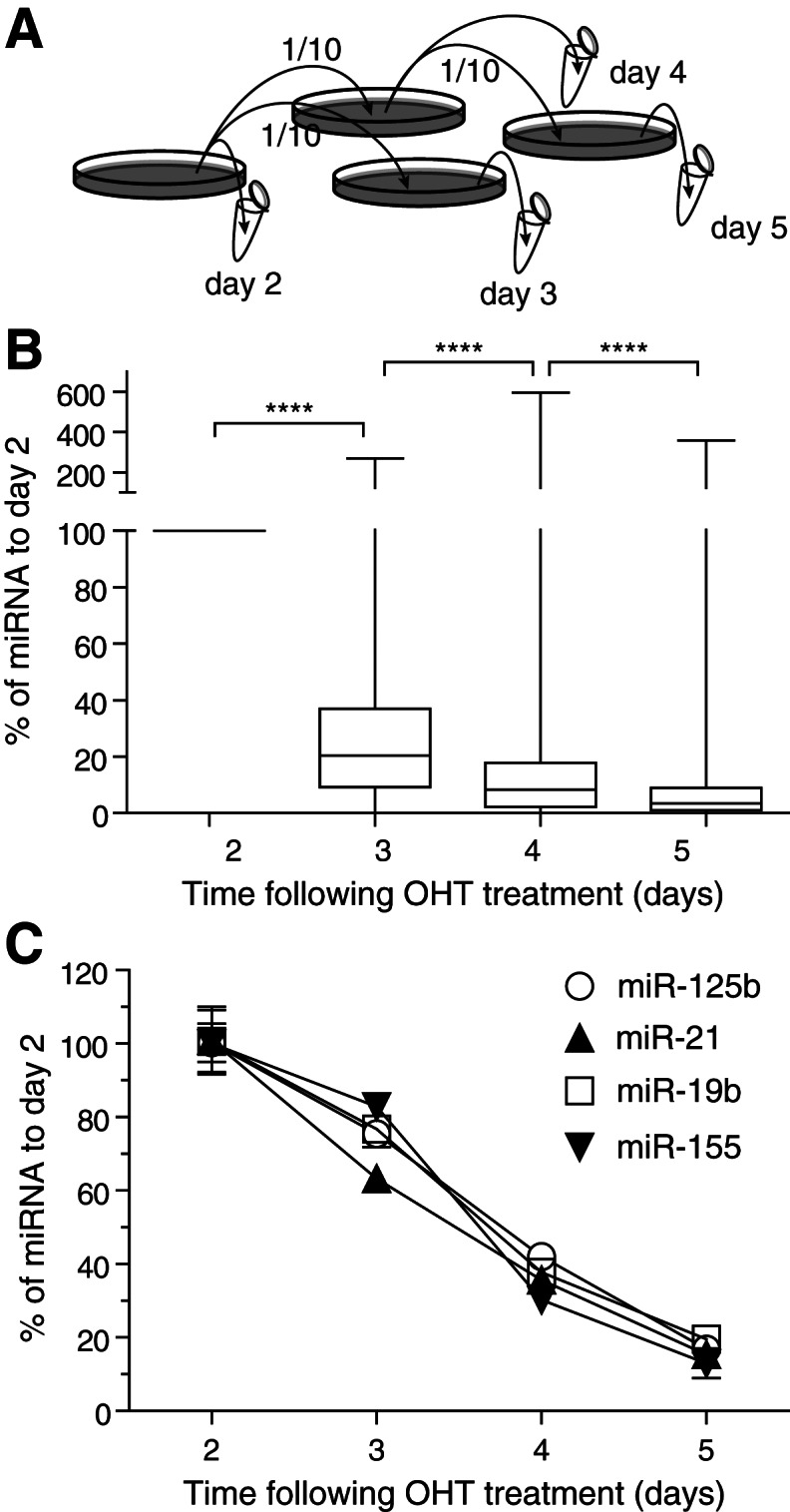
Gradual depletion of miRNAs following OHT treatment of the cells. (A) Schematic representation of the experimental setup. Dicerflox/flox × Cre/Esr1 MEFs were plated in 100-mm dishes, treated overnight with 500 nM OHT, and washed with fresh complete DMEM the next day (day 1). The cells were passaged with 1/10 splits on days 2 and 4 as depicted in the schematic. Cells were lysed, and total RNA was collected from each biological replicate set on days 2, 3, 4, and 5 as indicated. Three biological triplicates were collected for each time point. (B) Box and whiskers plots of 222 miRNAs detected on day 2, from one set of biological samples, by TaqMan low-density RT-qPCR arrays. Whiskers indicate the minimum to maximum values. The amplification data are given in Supplemental Table 1. To compensate for a skewed distribution, the data were log2-transformed prior to statistical analysis. One-tailed paired t-tests comparing the levels of each miRNA at different time points are shown. (****) P < 0.0001 (day 3 vs. day 2: P < 2.2 × 10−16; day 4 vs. day 3: P = 1.74 × 10−13; day 5 vs. day 4: P = 1.451 × 10−9). (C) Individual miRNA RT-qPCRs carried out on the samples generated in A confirm gradual depletion of select miRNAs. The results from biological triplicates normalized to the expression of snoRNA202 were reported to the average values for day 2. Error bars show the standard error of the mean (SEM).
miRNA microarray analysis with RMA background correction
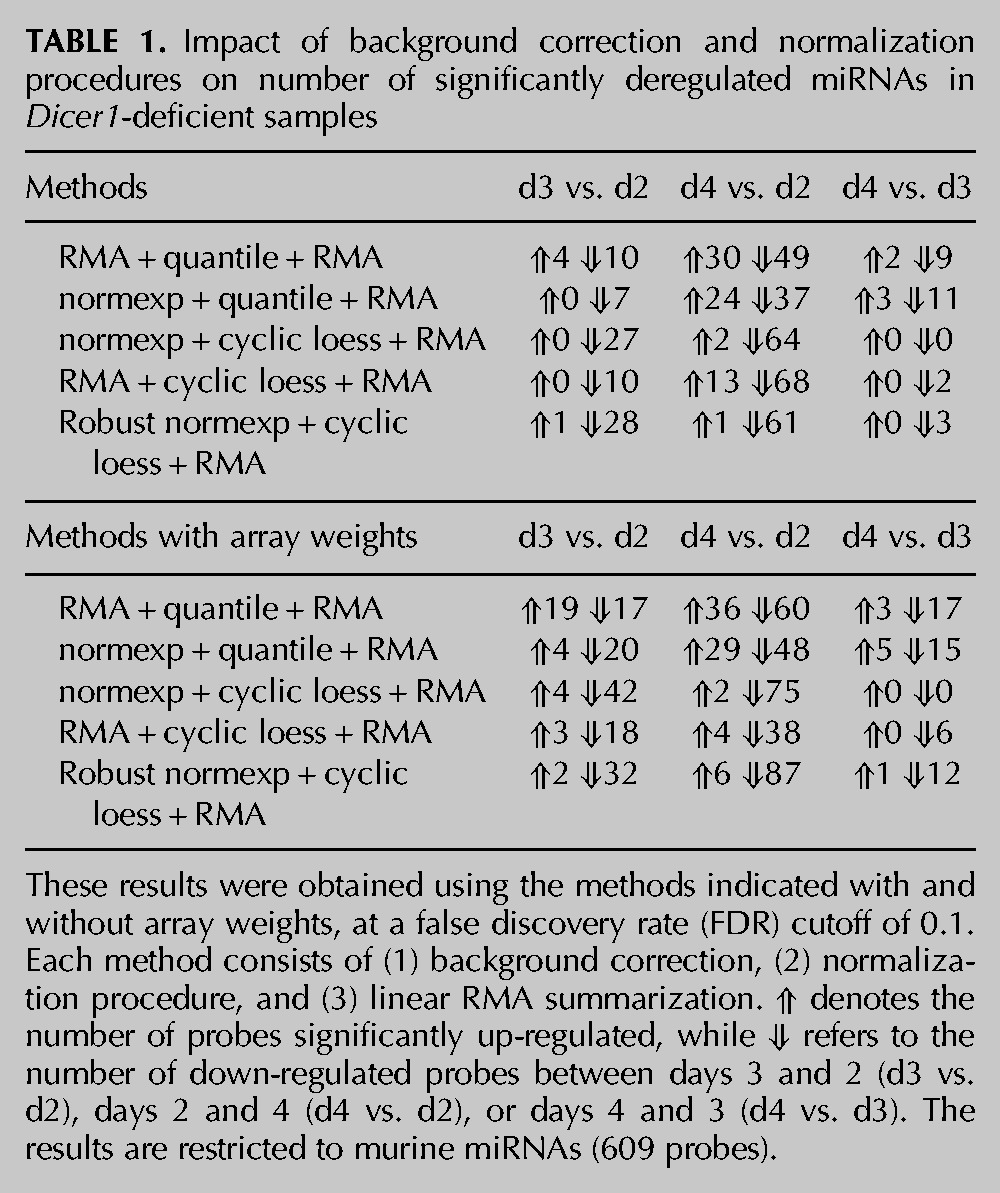
To investigate the ability of miRNA microarrays to detect global miRNA decrease, we next analyzed three sets of biological samples with gradual miRNA depletion, collected on days 2, 3, and 4, using Affymetrix GeneChip miRNA microarrays (previously analyzed for miRNA expression in Fig. 1C). Given that the average decrease in miRNA levels measured between days 2 and 4 was greater than twofold in individual RT-qPCR assays (Fig. 1C), we anticipated that most miRNAs expressed by the MEFs would display a significant reduction between these two time points. Quality control analyses of the arrays indicated important variations of averaged log2 intensities between the replicate arrays, warranting the need for array normalization (Fig. 2A). Importantly, an MA plot of the distribution of day 4/day 2 indicated a divergence of an important proportion of the points from log2 intensity ratio M = 0, which was particularly pronounced for the miRNA probes and the non-miRNA small RNA probes (Fig. 2B). Given previous reports that quantile normalization worked well for single-color miRNA microarrays (Rao et al. 2008; Zhao et al. 2010), we initially performed a standard RMA background correction/normalization. RMA relies on a model-based background correction, quantile normalization, log2 transformation, and probe-set summarization (Irizarry et al. 2003). It is a standard background correction/normalization procedure for Affymetrix GeneChip data and is recommended by Affymetrix for its miRNA microarray analyses, through the miRNA QC tool. Surprisingly, such normalization identified an important number of miRNAs to be up-regulated following Dicer1 deletion (Table 1; see RMA + quantile + RMA condition). Indeed, up to 38% (30 out of a total 79) of differentially expressed miRNAs were found to be up-regulated (when comparing miRNA levels between days 4 and 2). This was unexpected, given our previous results of global miRNA decrease in these samples by RT-qPCR (Fig. 1B,C). In addition, an MA plot of the distribution of day 4/day 2 following RMA background correction, quantile normalization, and RMA probe-set summarization did not help to identify further the expected global miRNA decrease (Fig. 2B,C; cf. the distribution of miRNA probes in black), thereby suggesting a bias of the microarray analyses in the identification of false-positive up-regulated miRNAs.
FIGURE 2.

Quality control of the raw data and MA plots. (A) Box plot of the raw PM log2 intensities for the 46,227 probes on each array, shown on log2 scale. The majority of the log2 intensities are low, and the interquartile range (IQR, which is the range of the box) is very narrow. (B) MA plot of the raw PM log2 intensity data of the array c at day 4 vs. the array b at day 2. (C,D) MA plots of the PM log2 intensity after RMA + quantile + RMA (C) or normexp + cyclic loess + RMA (D) normalization for the same arrays. In the MA plot, the y-axis represents the log2 intensity ratio (M:) between the two arrays (day 4 to day 2). The x-axis represents the average log2 intensity of the two arrays (A:). The colors represent the different types of probes.
TABLE 1.
Impact of background correction and normalization procedures on number of significantly deregulated miRNAs in Dicer1-deficient samples
Normexp background correction with quantile normalization
Because of the limited length of miRNAs (<25 nt), probes detecting miRNAs are designed to be directly complementary to the mature miRNA sequence. Consequently, the probe GC content is directly constrained to that of the miRNA. It is, therefore, expected that GC-rich miRNAs (or other RNAs) will have better affinity for the microarray probes and yield increased signal. To take this potential bias into account, the Affymetrix miRNA microarrays contain a set of GC control probes. Close examination of the 95 background control probe families of 8221 probes on the array (green dots in Fig. 2B–D) showed that these range from 17 to 25 nt long, with increasing GC content (for instance, ranging from 3 to 25 G/C for 25-nt-long control GC probes). Noteworthy is that each probe family is composed of non-miRNA random sequence variants with the same amount of GC. Analysis of the log2 intensity for these non-miRNA probe families confirmed a direct impact of the GC content on background intensities (Supplemental Fig. 1A). In addition, there was a significant correlation between the GC content of individual miRNAs and the raw intensities obtained, in that miRNAs with higher GC yielded increased signal (Supplemental Fig. 1B). These results suggested that control probes could be used for enhanced background correction. Ritchie et al. previously found normexp to be the best background correction method for two-color microarray data (Ritchie et al. 2007). The normexp method relies on the same normal plus exponential convolution model as the RMA algorithm. However, Ritchie et al. and Silver et al. replaced the kernel density parameter estimation method in RMA by a maximum-likelihood estimation in normexp (Ritchie et al. 2007; Silver et al. 2009). In addition, Shi et al. demonstrated that the use of normexp optimized the noise vs. bias trade-off in Illumina microarrays and developed normexp using control probes (Shi et al. 2010b). Although not directly accounting for the different GC content in different probes, the use of normexp relying on the GC control probe sets allowed us to take into account some of the effects of GC content on the background signal of the array. We next performed normexp background correction based on these control probe sets (Shi et al. 2010b), prior to quantile normalization, log2 transformation, and probe-set summarization. As shown in Table 1, the normexp background correction with use of the GC background control probe sets substantially reduced the number of false-positive up-regulated miRNAs to 0, between days 3 and 2, but only marginally improved the results between days 4 and 2, with 24 false-positive up-regulated miRNAs vs. 30 compared to the RMA background correction. This indicated that the control probe-based normexp was slightly better than RMA background correction at limiting the detection of false positives. Note that we refer to the control probe-based normexp when mentioning normexp in the rest of the paper.
Normexp background correction with cyclic loess normalization
After determining that normexp was a more suitable background correction method for analyses of microarrays with global miRNA decrease, we then considered the normalization method. Given the strong divergence of the points from M = 0 (M for log fold-change between samples) on MA plots previously calculated (Fig. 2B,C), we hypothesized that another normalization procedure, which does not assume equal distribution of up- and down-regulated probes, would be more appropriate. Risso et al. recently reported the use of a modified loess method, which allowed them to identify a strong prevalence of down-regulated miRNAs in samples that were previously shown to have equal proportions of up- and down-regulated miRNAs (Risso et al. 2009). However, that loess method (coined loessM as it relies on median intensities for normalization) is restricted to two-color microarrays and was, therefore, not suitable for our analyses. As an alternative, we decided to investigate whether cyclic loess for single-color microarrays (Bolstad et al. 2003) could help to limit the detection of false-positive up-regulated miRNAs, as seen with loessM for two-color arrays (Risso et al. 2009). Cyclic loess is a nonlinear method applied to the probe intensities from two separate arrays at a time, which helps to center the probe intensities around the M = 0 axis (Bolstad et al. 2003). Importantly, the loess curves can be tuned to give special weight to control probes or to probes expected to be invariant between experimental conditions (Oshlack et al. 2007). The technique of Oshlack et al. involves giving a small positive weight to all regular probes on the array but much higher weight to control probes (Oshlack et al. 2007). Different classes of probes can receive different weights depending on their reliability as invariant controls. Affymetrix miRNA microarrays contain about one-fifth of non-miRNA small RNA probes (small nucleolar RNAs—snoRNAs—comprising small Cajal body-specific RNAs, and C/D box and H/ACA box small RNAs), which are mostly independent of the miRNA processing pathway (Langenberger et al. 2013). We postulated that such non-miRNA probes from the Affymetrix platform could be used as invariant probes for cyclic loess. Accordingly, snoRNAs were given the highest weight (100), while miRNA probes were attributed a weight of 0.001, and all other probes (GC control, spike in, hybridization control, 5.8S rRNA) were given a weight of 1 for cyclic loess normalization. When combining normexp background correction with cyclic loess normalization based on snoRNAs, we saw a visible shift of down-regulated miRNA probes on the MA plot compared to the other non-miRNA small RNAs and control probes present on the array (Fig. 2D). This resulted in the near ablation of false-positive up-regulated miRNAs at each time point (Table 1). Critically, this effect was not detrimental to the overall number of probes differentially regulated, as seen with the results between quantile normalization and cyclic loess with a total of 61 and 66 differentially regulated miRNAs between days 4 and 2, respectively. However, cyclic loess gave the most down-regulated probes, with 64 between days 4 and 2 (vs. 37 for quantile normalization).
To determine the contribution of non-miRNA small RNA (snoRNA) probes in the reduction of false-positive up-regulated miRNAs, we next compared the effect of normexp background correction with cyclic loess normalization based on weights varying between 0 and 1000 for the snoRNA probes—the weight of miRNAs and other control probes being fixed at 0.001 and 1, respectively (Table 2). In accordance with an important contribution of snoRNA probes in the effect of cyclic loess, reduction of the snoRNA weight to 0 resulted in a large increase of false-positive up-regulated miRNAs (from two miRNAs for all other weights to 12 miRNAs for the weight of 0, between days 4 and 2). Nonetheless, even without taking the snoRNA probes into account (i.e., with a weight of 0), cyclic loess outperformed quantile normalization and identified half the amount of false up-regulated miRNAs between days 4 and 2 (12 vs. 24 miRNAs) (Tables 1 and 2).
TABLE 2.
Impact of non-miRNA probes on effect of cyclic loess normalization on number of significantly deregulated miRNAs in Dicer1-deficient samples
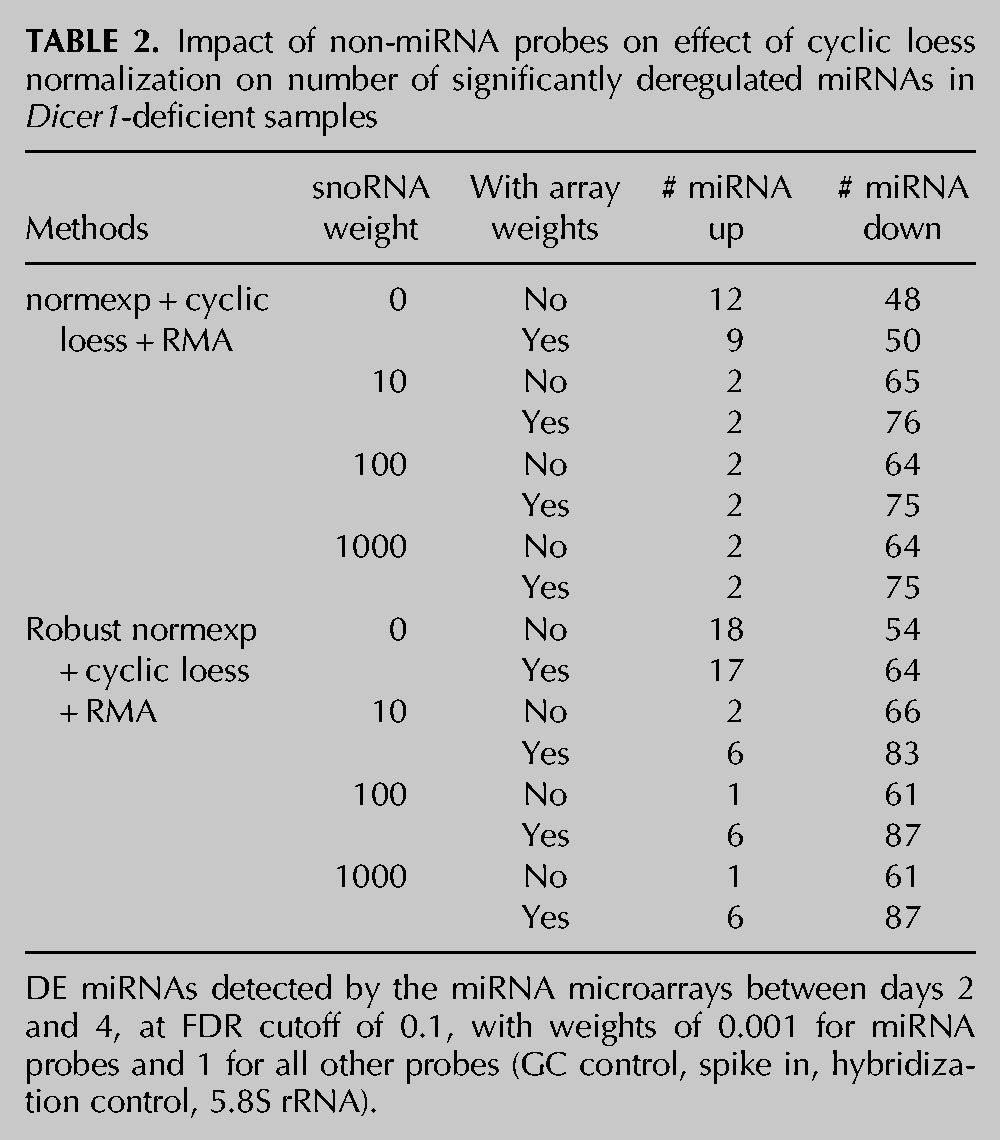
We also assessed the contribution of normexp background correction by comparing these results to RMA background correction followed by cyclic loess normalization. As seen with quantile normalization, normexp background correction performed better than RMA background correction, when combined with cyclic loess, by identifying fewer false-positive up-regulated miRNAs (2 vs. 13 between days 4 and 2 for normexp and RMA, respectively) but led to similar numbers of decreased miRNAs (64 vs. 68 between days 4 and 2 for normexp and RMA, respectively).
Robust normexp background correction with cyclic loess normalization and array weights
In order to investigate whether we could increase the sensitivity of our analyses, we next studied the impact of robust estimation on normexp background correction (Shi et al. 2010b). Robust estimation takes into account the possible cross-hybridization of control probes with miRNAs (Shi et al. 2010b). Box plots of the log2 intensities following normexp indicated a specific bias on certain arrays, which was prevented with the use of robust normexp (Fig. 3). Robust normexp and standard normexp background correction with cyclic loess normalization performed very similarly (Tables 1 and 2; cf. normexp and robust normexp lines). A multidimensional scaling plot of the arrays indicated that a significant difference remained between probes from replicate arrays of days 2 and day 4 (Fig. 4A) following robust normexp background correction with cyclic loess normalization and summarization. Based on the normalized and summarized miRNA data, we calculated the array quality weights with the design matrix allowing for compensation of variations observed between the arrays (Fig. 4B; Ritchie et al. 2006). The linear model fitted with array weights increased the number of significantly down-regulated probes with both standard and robust normexp background correction with cyclic loess normalization, with a more pronounced effect between days 3 and 2 and days 4 and 3 (where miRNA levels varied only modestly, according to the PCR data) (Table 1). However, array weights also increased the number of false-positive up-regulated miRNAs, with a lesser impact on standard normexp with cyclic loess normalization (Tables 1 and 2).
FIGURE 3.

The impact of robust estimation on normexp background correction. Box plots of log2 (A) raw intensities, (B) normexp background corrected intensity, and (C) robust normexp background corrected intensity. All 46,227 probes were used in these plots. After normexp background correction (B), the variability between the arrays is smaller, as the IQR in each array is smaller. However, two arrays, day 3 array c and day 4 array a, have abnormal higher intensities for both miRNA and GC background control probes (here called negative control probes—“neg. controls”) following normexp background correction. This does not agree with the RT-qPCR results shown in Figure 1C, which showed very small variations between the biological replicates. These two arrays have the largest log-variance from normexp model parameter estimates, suggesting that “robust normexp” could resolve these variations. Using robust normexp, the log-variance was estimated to be smaller than ordinary normexp, and the background corrected log2 intensities of each array were in similar ranges, with small IQR (C), indicating that robust normexp is more suitable than ordinary normexp.
FIGURE 4.

Examination of the relationship between samples and calculation of array quality weights, restricted to the mouse miRNA probe sets. (A) Multidimensional scaling (MDS) plot of the summarized microarray data following robust normexp background correction with cyclic loess normalization. This MDS plot shows the relationship between samples. Arrays day 2c and day 4a were not well grouped with arrays from the matching biological replicates, as indicated with the arrows. (B) Array quality weights were calculated using arrayWeightsSimple in limma, with or without considering the design matrix. The array weights calculated with the design matrix reflect the relationship between the samples seen in the MDS plot (A), with sample 2c and 4a having lower weights compared to 2a/2b and 4b/4c, respectively. These weights were used in the further comparisons of the normalization methods.
Evaluation of the accuracy of microarray analyses using RT-qPCR
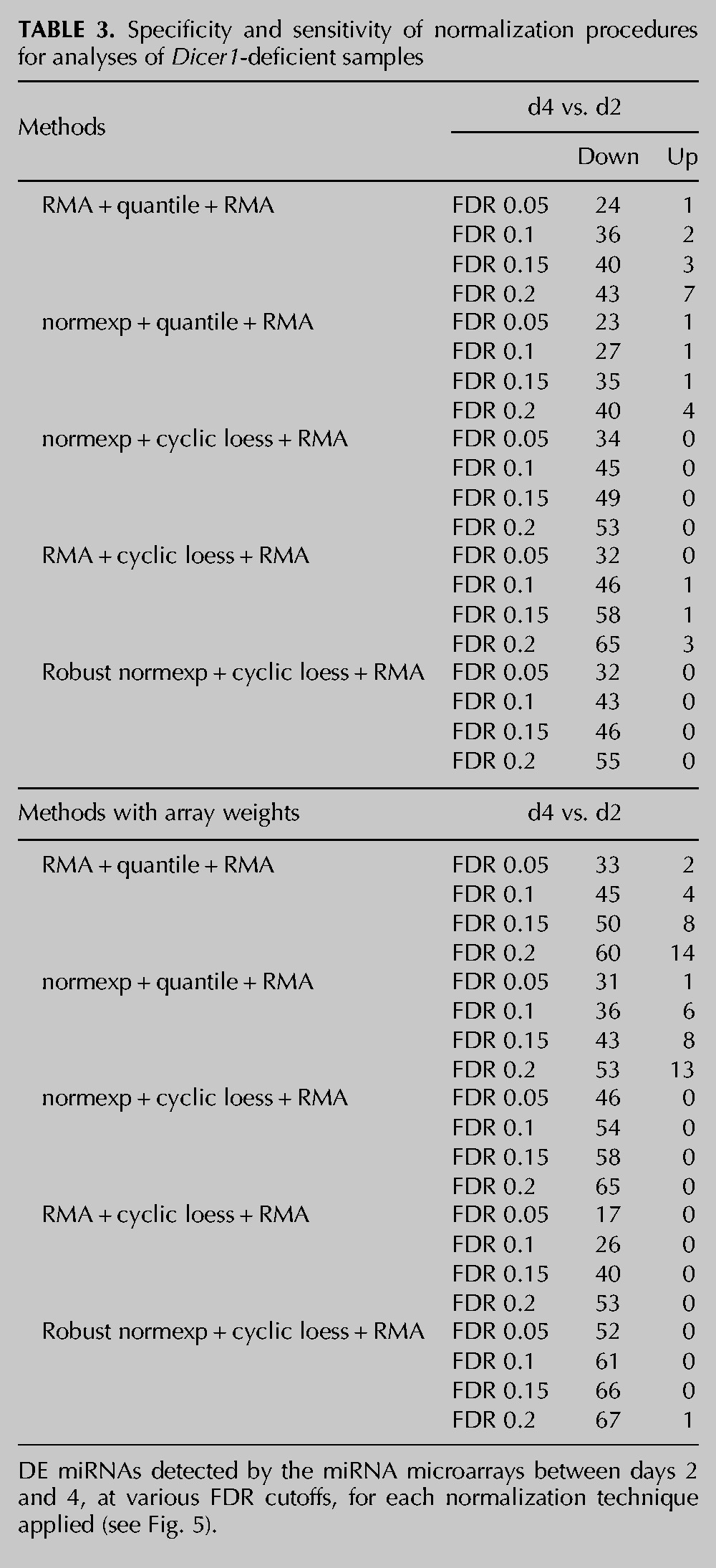
In order to define the accuracy of the microarray normalization analyses described above, we analyzed the overlap of predicted down-regulated miRNAs with 209 miRNAs (referred to as “truly expressed”) that we had previously identified to be down-regulated between days 2 and 4 by TaqMan RT-qPCR low-density array (Fig. 1B; Supplemental Table 1) and that were also present on the Affymetrix microarray platform. The associated results, summarized in Figure 5 and Table 3, confirmed that cyclic loess normalization procedures performed better than quantile normalization procedures at reducing the number of false-positive up-regulated miRNAs. The impact of array weights was particularly visible for low false discovery rate (FDR) cutoffs (0.05 and 0.1), where it strongly increased the number of true down-regulated miRNAs for robust and standard normexp + cyclic loess normalizations, while not increasing the number of false-positive miRNAs. Robust normexp performed marginally better than standard normexp for the detection of true down-regulated miRNAs at low false discovery rates. Collectively, these results suggest that robust normexp background correction with cyclic loess normalization and RMA summarization together with array weights is the most sensitive and specific normalization method for this platform.
FIGURE 5.
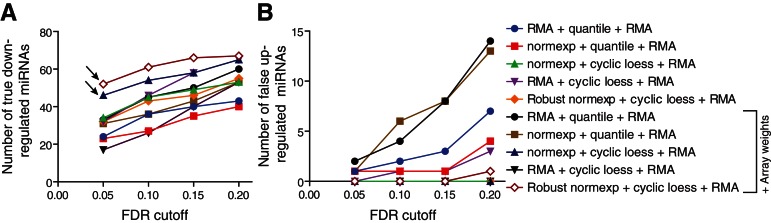
Assessment of true down-regulated and false up-regulated miRNAs in Dicer1-deficient samples. Curves showing the number of differentially expressed miRNAs detected by the miRNA microarrays between days 2 and 4, at various FDR cutoffs, for each normalization technique applied (see Table 3). The analyses shown are restricted to 209 miRNAs that were validated as true down-regulated miRNAs by TaqMan RT-qPCR arrays, also present on the Affymetrix platform (see Materials and Methods). The number of miRNAs confirmed to be significantly “true down-regulated” (A) and significantly “false up-regulated” (B), using the qPCR data as a reference, are given. The arrows highlight the better performance of normexp + cyclic loess + RMA and robust normexp + cyclic loess + RMA with array weights, which gives the highest amount of true down-regulated miRNAs at the most stringent FDR cutoffs of 0.05 and 0.1 (A), while giving a minimum of false up-regulated miRNAs (B).
TABLE 3.
Specificity and sensitivity of normalization procedures for analyses of Dicer1-deficient samples
Validation of the accuracy of robust normexp background correction with cyclic loess normalization and array weights on an independent data set
We next analyzed a published data set of Affymetrix miRNA microarrays from a cohort of 20 prostate cancer samples (and 20 matched normal tissues) (Wach et al. 2012) in order to further validate our approach using cancer samples in which miRNAs are preferentially down-regulated. Previous analyses of similar prostate cancer samples have indicated a prevalent global down-regulation of miRNAs in prostate cancer (Lu et al. 2005; Ozen et al. 2008). RNA-seq data from 10 normal and 10 prostate cancer pooled samples from the same group that reported the Affymetrix study (Szczyrba et al. 2010) were used as a reference for the identification of “truly differentially expressed” miRNAs (206 miRNAs were present in both Affymetrix and RNA-seq data sets). Noteworthy, our own analysis of the RNA-seq data from Szczyrba et al. (2010) also suggested a prevalent down-regulation of miRNAs in prostate cancers. We found that cyclic loess normalization methods preferentially detected down-regulated miRNAs, while quantile normalization methods favored up-regulated miRNAs (Table 4). Robust normexp background correction with cyclic loess and array weights allowed for the detection of the greatest amount of differentially expressed (DE) miRNAs with the minimum of false-positive DE miRNAs (compared to robust normexp and RMA background correction with quantile normalization) (Table 4; Fig. 6; Supplemental Table 2). Collectively, these results establish that the use of robust normexp background correction with cyclic loess and array weights can help to improve the sensitivity and specificity of miRNA profiling in cancer samples with global miRNA decrease.
TABLE 4.
Impact of background correction and normalization procedures on number of significantly deregulated miRNAs in prostate cancer samples
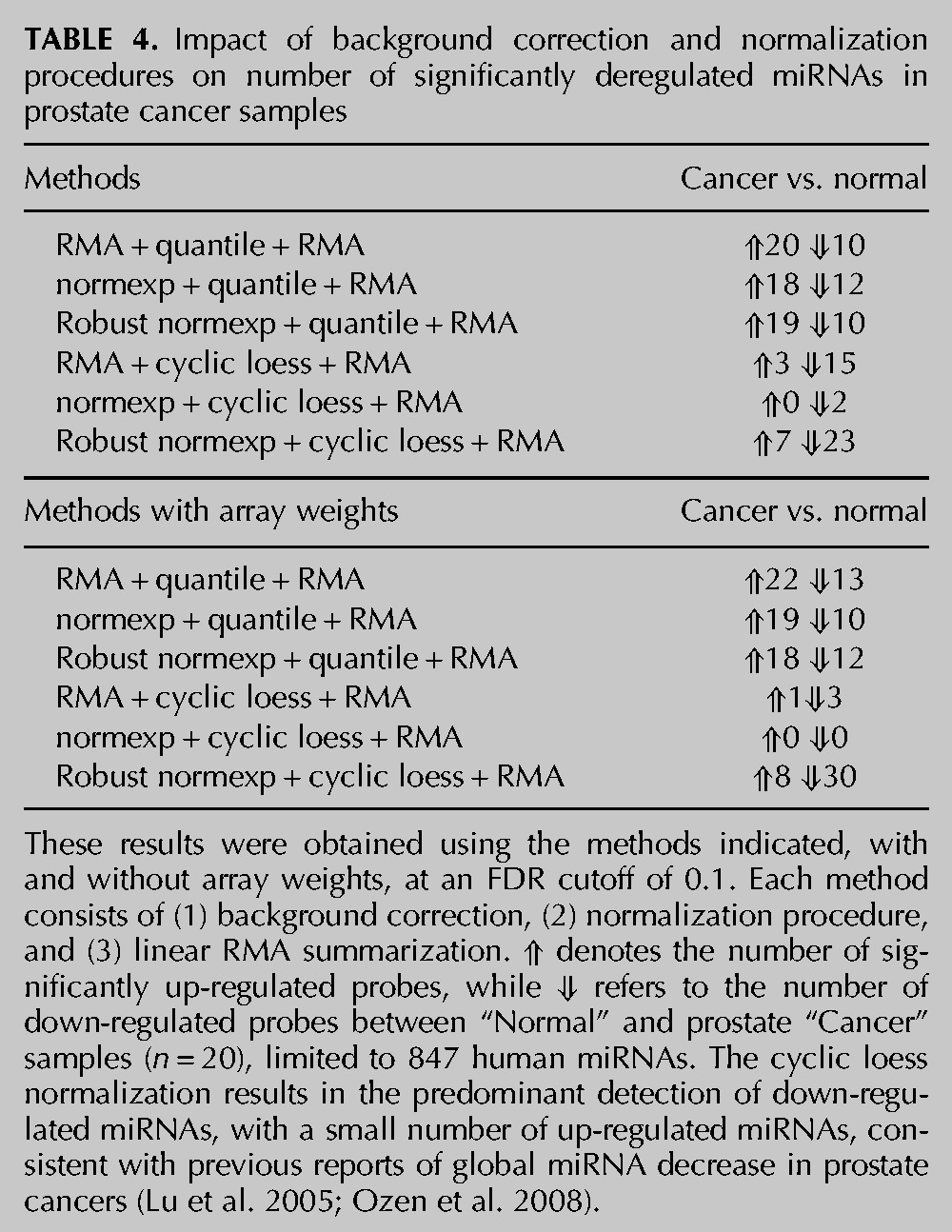
FIGURE 6.
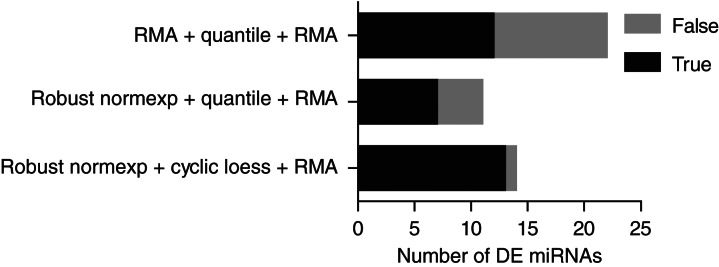
Comparison of different miRNA array normalization strategies and their consistency between miRNA arrays and RNA-seq in prostate cancer samples. Stacked bar graph showing the number of DE miRNAs in normal vs. prostate cancer miRNA microarrays (n = 20), with each normalization technique applied (see Supplemental Table 2 for details) at FDR cutoff of 0.1 with array weights. The analyses shown are restricted to 206 miRNAs detected by independent RNA-seq analyses of normal vs. prostate cancer samples. “True” DE miRNA refers to miRNAs that were DE in the same direction in the miRNA microarray and RNA-seq data. “False” DE miRNAs refer to miRNAs that were not validated to be up- or down-regulated in the RNA-seq analyses (see Supplemental Table 2). The precision (also called positive predictive value) of DE miRNA detection is shown as the ratio of the number of the “true” DE miRNAs to the number of the combined “true” and “false” DE (i.e., the black bar relative to the sum of black and gray bars).
DISCUSSION
As with whole-genome microarrays, miRNA microarray analyses can be strongly biased by hybridization, labeling, or batch-to-batch variations. Recent reports suggest that background correction and normalization procedures are beneficial for the identification of differentially regulated miRNAs (Hua et al. 2008; Rao et al. 2008; Pradervand et al. 2009; Risso et al. 2009; Meyer et al. 2010, 2012). However, all normalization procedures do not equate, and Risso et al. recently demonstrated that the choice of normalization procedure used could strongly impact on the overall identification of miRNAs as up- or down-regulated (Risso et al. 2009). The misidentification of deregulated miRNAs as up-regulated miRNAs is a critical issue in microRNA profiling studies, where miRNA profiles are used to classify tumors and bear prognostic value. Mutations resulting in global miRNA decrease are frequent across cancers and are associated with poorer outcomes (Karube et al. 2005; Merritt et al. 2008; Grelier et al. 2009).
Following their original description (Liu et al. 2004), glass-based microarray detection of miRNAs has rapidly become a very popular means of profiling miRNA expression in various samples, making up a vast proportion of the current literature. In addition to custom-made microarrays (where the probes are simply complementary to the miRNAs), several commercial platforms have been developed, with single-color miRNA microarrays being predominantly used (Meyer et al. 2010).
Interestingly, previous microarray profiling studies of samples with global miRNA decrease indicate a strong bias of microarrays in the identification of globally decreased miRNAs. Melo et al. recently reported monoallelic frameshift mutations impacting on XPO5 function in cancer cells. miRNA microarray analyses of such samples, while expected to reveal a global decrease in miRNAs, only identified 85 miRNAs that were significantly down-regulated out of about 300 that were expressed, through median normalization (Melo et al. 2010). Relying on the observations from Risso et al. (2009), we speculated that microarray analyses of samples with global decrease of miRNAs, as in those of Melo et al. (2010), could be strongly affected by the normalization method applied. This would be consistent with the fact that original analyses of large-scale cancer miRNA microarrays were performed with simple normalization procedures, such as median normalization (Volinia et al. 2006; Yanaihara et al. 2006). Median normalization assumes that there are few up- and down-regulated miRNAs among samples (with similar proportions of up- and down-regulated miRNAs) and, consequently, did not find the global miRNA decrease found with PCR-based technologies in cancer (Volinia et al. 2006; Yanaihara et al. 2006).
In this work, the use of an inducible deletion of miRNA biogenesis through Dicer1 deletion allowed us to generate samples with varying levels of decreased miRNAs to assess directly (1) the impact of normalization procedures on the identification of significantly deregulated miRNAs, and (2) the accuracy of these methods in a system where miRNAs are globally decreased. To our knowledge, this is the first description of a comparative study of miRNA normalization procedures on samples with truly defined miRNA expression. We opted for the commercial Affymetrix platform, which can conveniently be scanned using Affymetrix GeneChip scanners. We show that the use of the non-miRNA small RNA probes present on the Affymetrix arrays for the cyclic loess normalization procedure can help to improve substantially the identification of truly decreased miRNAs. Critically, we show that the better performance of cyclic loess is directly applicable to cancer samples with global miRNA decrease, where it strongly reduces the amount of false-positive up-regulated miRNAs while detecting a greater amount of decreased miRNAs. Our approach is, however, not limited to the Affymetrix platform and could be extended to other platforms, assuming that they have appropriate sets of control non-miRNA small RNA probes.
Quantile normalization has previously been proposed to be one of the most robust methods for the analysis of single-color miRNA microarrays (Rao et al. 2008; Pradervand et al. 2009; Meyer et al. 2010). This is in opposition to our findings, which clearly demonstrate the better performance of cyclic loess vs. quantile normalization in the accuracy and sensitivity of miRNA detection. This can, in part, be attributed to the fact that quantile normalization assumes stable intensities for most probes across microarrays, while our samples have a global decreased expression of all miRNAs. Conversely, loess normalization, when properly implemented, is able to tolerate 20%–30% of genes changing in one direction (Oshlack et al. 2007). However, the use of cyclic loess normalization is not restricted to the analyses of samples with unidirectional changes of miRNA expression. Rao et al. found an overall good performance for cyclic loess in their analyses of various tissues where miRNAs were both up- and down-regulated between samples (Rao et al. 2008). Our analyses of prostate cancer samples, where, although preferentially decreased (Ozen et al. 2008), some miRNAs are also up-regulated (Szczyrba et al. 2010; Wach et al. 2012), demonstrate that cyclic loess also performs well to detect “truly” up-regulated miRNAs with minimal false-positives. Note that cyclic loess, but not quantile normalization, allowed for the significant identification of miR-143 as being down-regulated in prostate cancer (Supplemental Table 2); this was independently validated by RT-qPCR and proposed to be a useful marker of prostate cancer (Wach et al. 2012). In contrast to our findings, Rao et al. found that cyclic loess performed slightly worse than quantile normalization in their studies (Rao et al. 2008). We note that the platform used by this group did not contain any non-miRNA small RNA probes, which we considered as “invariant” probes in our analysis. This further suggests an important role for such invariant probes in the ability of cyclic loess to outperform quantile normalization (as also indicated by the results shown in Table 2).
Our findings that cyclic loess normalization strongly reduces the misidentification of false up-regulated miRNAs reinforce the previous findings from Risso et al. and Meyer et al. that loess and loessM perform best (Risso et al. 2009; Meyer et al. 2012). While cyclic loess and loessM both address the normalization problems associated with the asymmetric modulation of a large proportion of miRNAs between microarrays, the two techniques, nonetheless, have important differences. Critically, loessM is currently restricted to two-color microarrays, which precluded its use for our analyses of the Affymetrix platform. Cyclic loess, which is used with single-color microarrays, uses pairs of microarray samples and allows the user to add varying weights to individual probes in order to calculate the normalizing constants. LoessM, on the other hand, does not pair microarrays and uses the data from the entire array to obtain median intensities used in the normalization. In the specific case of the Affymetrix platform, where about half of the probes are non-miRNA small RNAs and control RNAs and are, therefore, not expected to vary between samples, such an approach would likely introduce an important bias into the overall sensitivity of the analyses.
Our analyses point to an important contribution of non-miRNA small RNAs (snoRNAs) in the effect of cyclic loess normalization. To understand the contribution of these probes in cyclic loess normalization, it should be underlined that they represent a total of 10,090 probes (for 922 probe sets) on the Affymetrix microarray, which is about 2.5 times less than the total amount of miRNA probes (26,812). However, in our Dicer1-deficient cells, many of these non-miRNA snoRNAs cannot be detected due to species specificity of the probes (which are targeted to human snoRNAs, not mouse) or lack of expression in the cells. This suggests that only a small proportion of snoRNA probes are necessary for cyclic loess to improve sensitivity and sensibility. The non-miRNA snoRNAs used on the Affymetrix platform are families of small RNAs involved in the site-specific modifications of ribosomal RNAs, transfer RNAs, and spliceosomal RNAs. While snoRNA-driven specific nucleotide modifications of ribosomal RNAs are not essential to cell survival/division, they are thought to fine-tune the biological activities of ribosomal RNAs (Bachellerie et al. 2002). As housekeeping RNAs, their expression is expected to be stable overall across samples, including cancer vs. normal samples. Evidently, some of these snoRNAs will vary between treatments, but our findings with the cancer cohort samples indicate that the use of snoRNAs as “invariant” probes works well with cyclic loess in cancer samples with preferential global miRNA decrease.
As shown in Figure 4, the array weights can be useful to decrease the variation between replicates with the same biological significance. In the case of our MEF samples, array weights correction allows the incorporation of more divergent samples (day 2c and day 4a) and increased detection of decreased miRNAs (Table 1). However, this is a case-by-case issue, and we recommend performing analyses with and without array weights to see how this affects the repartition of samples with similar biological origin. With biological samples from cancer patients that have inherent variability, array weights should generally yield better results (as indicated in our prostate cancer analyses).
In conclusion, our analyses demonstrate that miRNA microarrays can be suitable for the identification of samples with globally decreased miRNAs, which are frequent across cancers. Our data show that the use of normexp background correction with cyclic loess normalization and array weights strongly reduces the number of false-positive up-regulated miRNAs in samples with globally decreased miRNAs for the single-color Affymetrix miRNA microarray platform. This approach also yielded a strong reduction in false-positive up-regulated miRNAs and the detection of the greatest amount of truly down-regulated miRNAs in the analysis of prostate cancer samples where miRNAs were preferentially down-regulated (Ozen et al. 2008; Szczyrba et al. 2010; Wach et al. 2012). Given their relatively low cost compared to other technologies such as RNA sequencing, miRNA microarrays remain a very popular method of characterizing miRNA profiles across samples. Nonetheless, as RNA-seq becomes more affordable, questions regarding miRNA normalization of RNA-seq data are becoming more important. A recent study comparing seven normalization procedures for the analysis of microRNA-seq data reported that loess and quantile performed best (Garmire and Subramaniam 2012). Given that small RNA-seq generates a high level of non-miRNA small RNA sequences, it will be interesting to define whether the use of cyclic loess relying on such non-miRNA sequences could improve microRNA-seq normalization compared to loess and quantile normalization (Garmire and Subramaniam 2012), as reported in this study.
MATERIALS AND METHODS
Cell culture
Dicerflox/flox × Cre/Esr1 MEFs used in the study have been reported previously (Gantier et al. 2011, 2012). MEFs were cultured in complete Dulbecco’s modified Eagle medium (DMEM) (Invitrogen Corporation) supplemented with 10% sterile fetal bovine serum (ICPBio Ltd.) and 1× antibiotic/antimycotic (referred to as complete DMEM). OHT (Sigma Aldrich) was resuspended in 0.5 mL of 100% ethanol (resulting in stock solution at 25 mM) kept at −80°C. Prior to cell treatment, the stock solution was first diluted to 2.5 mM in 100% ethanol before being diluted further to the final concentration of 500 nM in complete DMEM. The cells were incubated overnight with 500 nM OHT before being rinsed with fresh complete DMEM the next day (day 1).
miRNA microarray
Total RNA containing small RNAs was purified from cultured MEFs using the mirVana miRNA Isolation Kit (Applied Biosystems) and further processed by the Adelaide Microarray Centre, Adelaide, Australia. This study relies on the analyses of nine miRNA microarrays from RNA collected on days 2, 3, and 4 following Dicer1 deletion, with three independent biological samples per time point (see Fig. 1A). The RNA was labeled using the FlashTag Biotin RNA Labeling Kit (Genisphere LLC) and hybridized to GeneChip miRNA 1.0 microarrays (Affymetrix Inc.) as per the Genisphere manual. Briefly, 500 ng of RNA was poly-A tailed and a proprietary biotin-labeled dendramer molecule was joined to the 3′ end using DNA ligase. Labeled samples were hybridized to the arrays at 48°C for 16 h and then washed and stained with a Streptavidin-PE solution prior to imaging. Array images were scanned using Genepix. The Affymetrix miRNA 1.0 microarray contains perfect match probes (PM) only. There are 46,227 PM probes on the array, including 38,006 regular probes and 8221 background (called “BkGr” in the manufacturer annotation file). The regular probes comprise 6703 miRNA probe sets from different species and 922 non-miRNA small RNAs. Each probe set represents one miRNA/snoRNA. Among the 6703 miRNA probe sets, 609 are mouse (corresponding to 609 mouse miRNAs), while 847 are human. The BkGr probes are not specific to any miRNA and consist of 95 families of GC-binned negative controls. The annotation file was obtained from the manufacturer’s web site. The Dicer1 deletion miRNA microarray data have been submitted to GEO with accession number GSE45886. The prostate cancer miRNA microarray data were downloaded from GEO accession number GSE23022 and RNA-seq data from published supplemental data from Szczyrba et al. (2010).
Low-density miRNA arrays
TaqMan Array Rodent MicroRNA (A Cards v2.0, Applied Biosystems) were used for the detection of 335 murine miRNAs from one biological sample set, as previously reported (Gantier et al. 2011). Briefly, 900 ng of total RNA containing small RNAs was reverse-transcribed using the Megaplex RT Primers, Rodent Pool A (Applied Biosystems) with the TaqMan MicroRNA Reverse Transcription Kit, and each plate was run using the TaqMan Universal Master Mix II on the 7900 RT-qPCR system, according to the manufacturer’s instructions. Simultaneous analysis of the four different plates (one plate at days 2, 3, 4, and 5 following OHT treatment) was carried out using the RQ Manager software and using the RNAU6 probes as reference. We identified a list of 222 miRNAs that were significantly detected in our cells (with Cq—quantification cycle—inferior to 35) and are shown in Figure 1B (see Supplemental Table 1 for details). Two hundred twenty of these miRNAs were also present on the Affymetrix platform, and 209 were decreased with time in such a manner that Cq at day 2 ≤ Cq at day 4. In our analyses presented in Figure 5 and Table 3, we define as true “positives” all miRNAs that change significantly in the microarray in the same direction as in the qPCR data. Eleven of the 220 probes that are present in both the miRNA microarray and the qPCR array are up-regulated between day 4 and day 2 in the qPCR data (see Supplemental Table 1 for details). However, none of these probes are significantly up- or down-regulated between day 4 vs. day 2 in the microarray data at an FDR of 0.05 or 0.1. In other words, none of the miRNAs found to be up-regulated in the microarray were also up-regulated in the PCR data, and none of the down-regulated miRNAs in the microarray data were up-regulated in the PCR data. True “positives” are, therefore, limited to miRNAs that are down-regulated in both the microarray and the qPCR data, while true “negatives” are limited to miRNAs that are down in the PCR data and falsely up-regulated in the microarray.
Reverse transcription quantitative real-time PCR
For validation of global miRNA decrease following OHT treatment of the MEFs, individual miRNA TaqMan assays (Applied Biosystems) for the indicated miRNAs were used according to the manufacturer’s instructions, where 10 ng of total RNA was reverse-transcribed with pools of five miRNA specific reverse transcription primers (with the TaqMan MicroRNA Reverse Transcription Kit). miRNA levels were determined by RT-qPCR with the TaqMan Universal PCR Master Mix on a 7900 RT-qPCR system (Applied Biosystems), and fold-changes in expression were calculated by the 2−ΔΔCq method using snoRNA202 as a reference.
Microarray background correction and normalization procedures
All of the methods used in these analyses have previously been reported and are available as part of the R package Affy and limma (Gautier et al. 2004; Smyth 2005; R Development Core Team 2011), which are part of the Bioconductor project (http://www.bioconductor.org) (Gentleman et al. 2004). The version 2.15.1 of R was used. The raw CEL microarray files were read using the ReadAffy function in the Affy package. After background correction, normalization, and summation, the normalized data were fit to a linear model by using lmFit. The design matrix included the days after OHT treatment. An empirical Bayesian method was used to estimate the significance of differential expression of miRNAs (Smyth 2004). The comparisons between days after OHT treatment were made. To determine the differentially expressed miRNAs, we performed the following: We first obtained the nominal P value for each miRNA; we then applied the multiple testing adjustment using the Benjamini-Hochberg (BH) method to control the false discovery rate, allowing for getting adjusted P values for mouse miRNAs. We used a FDR cutoff. No log fold-change cutoff has been used to define differential expression.
RMA background correction
RMA, originally developed for the analysis of Affymetrix GeneChips, allows for the averaging of probes for the same miRNA target (four probes per miRNA on the Affymetrix GeneChip miRNA 1.0 microarrays) through the use of median polish. The default setup of the rma function in the Affy package includes RMA background correction, quantile normalization, and RMA summarization. For RMA background correction, we set normalization and summarization as FALSE. The expresso function was used to generate data for the MA plots.
Normexp-by-control background correction
Affymetrix GeneChip miRNA 1.0 microarrays include a set of 95 GC control probe families with varying probe length (17 to 25 nt long) and for each length, increasing numbers of GC content (for instance, ranging from 3 to 25 G/C for the 25-nt-long control GC probes). Each probe set contains many repeats over the microarray, as seen with the AFFX-BkGr-GC03 st family (25-nt-long probes with three GC), which has 25 different variants across the array. The total number of control probes represented by these 95 families is 8221, which is ∼17% of the total features present on the array. Previous studies of mRNA microarrays indicate that the use of negative control probes can provide a good estimate of the background noise (Ding et al. 2008; Shi et al. 2010a). We, therefore, relied on this set of GC control probes for background correction, as previously reported for mRNA microarrays (Shi et al. 2010b). To allow for the possibility that some negative control probes are subject to cross-hybridization with some miRNAs, we also used the option of robust estimation of the background mean and variance as previously published (Shi et al. 2010b). Nec stands for NormExp background Correction using control probes. The nec function in limma was used here. It returns a matrix of the same dimensions as the input, containing background-corrected intensities, on a raw scale.
Robust normexp-by-control background correction
The nec function with the robust option from the limma package was used, in which the robust estimators are used for the determination of the background mean and standard deviation.
Quantile normalization
Quantile normalization is an inter-array normalization procedure aimed at equalizing the distribution of probe intensities in a set of arrays (Bolstad et al. 2003). It assumes that the overall distribution of probe intensities is constant between arrays, which works very well for mRNA microarrays but is a strong assumption for miRNA microarrays. The rma function of the affy package was used (Gautier et al. 2004).
Cyclic loess normalization
Cyclic loess relies on an MA plot of the distribution of log2 intensity ratio (M) by the average log2 intensity (A) values and is applied to the intensities of probes from two arrays at a time, with the aim of reducing the divergence of the points from the M = 0 axis (Bolstad et al. 2003). This normalization method usually relies on normalization curves computed using ranked sets of invariant probes (Bolstad et al. 2003). In our analyses (with the exception of Table 2), this was achieved by using the set of non-miRNA probes (composed of small nucleolar RNAs, including small Cajal body-specific RNAs and C/D box and H/ACA box small RNAs) with a weight of 100 (representing 10,090 probes for 922 probe sets), while miRNA probes were attributed a weight of 0.001 (representing 26,812 probes for 6703 probe sets), and all other probes (GC control, spike in, hybridization control, 5.8S rRNA—totaling 9,325 probes) were given a weight of 1. The function normalizeCyclicloess in the limma package was used. It normalizes the columns of a matrix, cyclically applying loess normalization to normalize each pair of columns to each other (Ballman et al. 2004). Here, the default “pair” method was used (Ballman et al. 2004). Given that we were dealing with a total of nine microarrays, the loess normalization was applied to all distinct pairwise combinations (for a total of 36 combinations). This was repeated for several iterations.
DATA DEPOSITION
The Dicer1 deletion miRNA microarray data have been submitted to Gene Expression Ominbus (GEO) with accession number GSE45886.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Mark Van der Hoek (Adelaide Microarray Centre, Adelaide, Australia) for his help with RNA labeling and Affymetrix microarray processing, and Frances Cribbin for editorial assistance. This work was supported by grants from the Australian National Health and Medical Research Council (1022144 to M.P.G., 1036541 to D.W., 1006590 to B.R.G.W., 490970 to S.T., and 490036 to G.K.S.) and the Victorian Government’s Operational Infrastructure Support Program.
REFERENCES
- Aqeilan RI, Calin GA, Croce CM 2010. miR-15a and miR-16-1 in cancer: Discovery, function and future perspectives. Cell Death Differ 17: 215–220 [DOI] [PubMed] [Google Scholar]
- Bachellerie JP, Cavaille J, Huttenhofer A 2002. The expanding snoRNA world. Biochimie 84: 775–790 [DOI] [PubMed] [Google Scholar]
- Ballman KV, Grill DE, Oberg AL, Therneau TM 2004. Faster cyclic loess: Normalizing RNA arrays via linear models. Bioinformatics 20: 2778–2786 [DOI] [PubMed] [Google Scholar]
- Bolstad BM 2004. “Low level analysis of high-density oligonucleotide array data: Background, normalization and summarization.” Ph.D. dissertation, University of California, Berkeley, CA [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193 [DOI] [PubMed] [Google Scholar]
- Cheloufi S, Santos COD, Chong MMW, Hannon GJ 2010. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465: 584–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L-H, Xie Y, Park S, Xiao G, Story MD 2008. Enhanced identification and biological validation of differential gene expression via Illumina whole-genome expression arrays through the use of the model-based background correction methodology. Nucleic Acids Res 36: e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantier MP 2010. New perspectives in microRNA regulation of innate immunity. J Interferon Cytokine Res 30: 283–289 [DOI] [PubMed] [Google Scholar]
- Gantier MP, McCoy CE, Rusinova I, Saulep D, Wang D, Xu D, Irving AT, Behlke MA, Hertzog PJ, Mackay F, et al. 2011. Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res 39: 5692–5703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantier MP, Stunden HJ, McCoy CE, Behlke MA, Wang D, Kaparakis-Liaskos M, Sarvestani ST, Yang YH, Xu D, Corr SC, et al. 2012. A miR-19 regulon that controls NF-κB signaling. Nucleic Acids Res 40: 8048–8058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmire LX, Subramaniam S 2012. Evaluation of normalization methods in mammalian microRNA-Seq data. RNA 18: 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C, Ambros VR, Israel MA 2007. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res 67: 2456–2468 [DOI] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, Irizarry RA 2004. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20: 307–315 [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. 2004. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grelier G, Voirin N, Ay A-S, Cox DG, Chabaud S, Treilleux I, Leon-Goddard S, Rimokh R, Mikaelian I, Venoux C, et al. 2009. Prognostic value of Dicer expression in human breast cancers and association with the mesenchymal phenotype. Br J Cancer 101: 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S 2010. miRBase: microRNA sequences and annotation. Chapter 12. In Curr Protoc Bioinformatics, pp. 12.19.11–12.910 [DOI] [PubMed] [Google Scholar]
- Hua Y-J, Tu K, Tang Z-Y, Li Y-X, Xiao H-S 2008. Comparison of normalization methods with microRNA microarray. Genomics 92: 122–128 [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264 [DOI] [PubMed] [Google Scholar]
- Karube Y, Tanaka H, Osada H, Tomida S, Tatematsu Y, Yanagisawa K, Yatabe Y, Takamizawa J, Miyoshi S, Mitsudomi T, et al. 2005. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci 96: 111–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Loedige I, Filipowicz W 2010. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11: 597–610 [DOI] [PubMed] [Google Scholar]
- Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, Kirsch DG, Golub TR, Jacks T 2009. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev 23: 2700–2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenberger D, Cakir MV, Hoffmann S, Stadler PF 2013. Dicer-processed small RNAs: Rules and exceptions. J Exp Zool B Mol Dev Evol 320: 35–46 [DOI] [PubMed] [Google Scholar]
- Liu C-G, Calin GA, Meloon B, Gamliel N, Sevignani C, Ferracin M, Dumitru CD, Shimizu M, Zupo S, Dono M, et al. 2004. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc Natl Acad Sci 101: 9740–9744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. 2005. MicroRNA expression profiles classify human cancers. Nature 435: 834–838 [DOI] [PubMed] [Google Scholar]
- Melo SA, Esteller M 2011. Dysregulation of microRNAs in cancer: Playing with fire. FEBS Lett 585: 2087–2099 [DOI] [PubMed] [Google Scholar]
- Melo SA, Ropero S, Moutinho C, Aaltonen LA, Yamamoto H, Calin GA, Rossi S, Fernandez AF, Carneiro F, Oliveira C, et al. 2009. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet 41: 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Melo SA, Moutinho C, Ropero S, Calin GA, Rossi S, Spizzo R, Fernandez AF, Davalos V, Villanueva A, Montoya G, et al. 2010. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 18: 303–315 [DOI] [PubMed] [Google Scholar]
- Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R, Urbauer D, Pennacchio LA, Cheng J-F, Nick AM, et al. 2008. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med 359: 2641–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer SU, Pfaffl MW, Ulbrich SE 2010. Normalization strategies for microRNA profiling experiments: A ‘normal’ way to a hidden layer of complexity? Biotechnol Lett 32: 1777–1788 [DOI] [PubMed] [Google Scholar]
- Meyer SU, Kaiser S, Wagner C, Thirion C, Pfaffl MW 2012. Profound effect of profiling platform and normalization strategy on detection of differentially expressed microRNAs – a comparative study. PLoS One 7: e38946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Han Y-C, Betel D, Yao E, Squatrito M, Ogrodowski P, de Stanchina E, D’Andrea A, Sander C, Ventura A 2009. Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev 23: 2806–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshlack A, Emslie D, Corcoran LM, Smyth GK 2007. Normalization of boutique two-color microarrays with a high proportion of differentially expressed probes. Genome Biol 8: R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozen M, Creighton CJ, Ozdemir M, Ittmann M 2008. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene 27: 1788–1793 [DOI] [PubMed] [Google Scholar]
- Pradervand S, Weber J, Thomas J, Bueno M, Wirapati P, Lefort K, Dotto GP, Harshman K 2009. Impact of normalization on miRNA microarray expression profiling. RNA 15: 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team 2011. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- Rao Y, Lee Y, Jarjoura D, Ruppert AS, Liu C-G, Hsu JC, Hagan JP 2008. A comparison of normalization techniques for microRNA microarray data. Stat Appl Genet Mol Biol 7: Article22. [DOI] [PubMed] [Google Scholar]
- Risso D, Massa MS, Chiogna M, Romualdi C 2009. A modified LOESS normalization applied to microRNA arrays: A comparative evaluation. Bioinformatics 25: 2685–2691 [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Diyagama D, Neilson J, van Laar R, Dobrovic A, Holloway A, Smyth GK 2006. Empirical array quality weights in the analysis of microarray data. BMC Bioinformatics 7: 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Silver J, Oshlack A, Holmes M, Diyagama D, Holloway A, Smyth GK 2007. A comparison of background correction methods for two-colour microarrays. Bioinformatics 23: 2700–2707 [DOI] [PubMed] [Google Scholar]
- Shi W, de Graaf CA, Kinkel SA, Achtman AH, Baldwin T, Schofield L, Scott HS, Hilton DJ, Smyth GK 2010a. Estimating the proportion of microarray probes expressed in an RNA sample. Nucleic Acids Res 38: 2168–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Oshlack A, Smyth GK 2010b. Optimizing the noise versus bias trade-off for Illumina whole genome expression BeadChips. Nucleic Acids Res 38: e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver JD, Ritchie ME, Smyth GK 2009. Microarray background correction: Maximum likelihood estimation for the normal-exponential convolution. Biostatistics 10: 352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3. [DOI] [PubMed] [Google Scholar]
- Smyth GK 2005. Limma: Linear models for microarray data. In Bioinformatics and computational biology solutions using R and Bioconductor (ed. Gentleman R, et al. ), pp. 397–420 Springer, New York [Google Scholar]
- Smyth GK, Speed T 2003. Normalization of cDNA microarray data. Methods 31: 265–273 [DOI] [PubMed] [Google Scholar]
- Szczyrba J, Loprich E, Wach S, Jung V, Unteregger G, Barth S, Grobholz R, Wieland W, Stohr R, Hartmann A, et al. 2010. The microRNA profile of prostate carcinoma obtained by deep sequencing. Mol Cancer Res 8: 529–538 [DOI] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu C-G, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. 2006. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci 103: 2257–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach S, Nolte E, Szczyrba J, Stohr R, Hartmann A, Orntoft T, Dyrskjot L, Eltze E, Wieland W, Keck B, et al. 2012. MicroRNA profiles of prostate carcinoma detected by multiplatform microRNA screening. Int J Cancer 130: 611–621 [DOI] [PubMed] [Google Scholar]
- Wu Z, Aryee MJ 2010. Subset quantile normalization using negative control features. J Comput Biol 17: 1385–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, et al. 2006. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9: 189–198 [DOI] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP 2002. Normalization for cDNA microarray data: A robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang E, Liu H, Rotunno M, Koshiol J, Marincola FM, Landi MT, McShane LM 2010. Evaluation of normalization methods for two-channel microRNA microarrays. J Transl Med 8: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]