

Nano palladium coordination complexes, incorporating pincer ligands are reported to be highly efficient catalysts for C–C coupling reactions, giving excellent yields with low catalyst loading.1–8 A relatively new amino pincer palladium complex has been reported by Frech for C–C bond formation via the Heck, Sonogashira, Stille, Hiyama and Suzuki-Miyaura reactions.3–7 A myriad of palladium complexes exist to promote these C–C bond forming processes.9 However, a serious limitation to the use of these reactions for the synthesis of bioactive molecules stems from the lack of thermal stability and functional group tolerance of many palladium complexes as well as the requirement for relatively high catalyst loadings.4 Decomposition of catalysts at normal reaction temperatures (140–150°C) can result in highly contaminated products that require extensive purification, which markedly decreases the yields.8,10 Conventional catalysts also give modest results with heterocyclic substrates.11,12 In contrast, the Frech catalyst exhibits robust thermal stability due, in large part, to the P-Pd-P (PCP) moiety of the pincer ligand.5,13 This thermal stability, together with its inertness to oxygen and water, are unique qualities of the Frech catalyst and allow it to maintain high activity under a variety of reaction conditions.13 In the present work, we have evaluated the Frech catalyst and compared it with two conventional palladium catalysts for coupling highly-substituted, heterocyclic substrates in the final step of a synthesis of 2,4-diaminopyrimidine-based antibiotics, which have demonstrated activity against inhalation anthrax14–16 and multi-drug resistant staph.17 We now report results which validate the potential of this new catalyst in reactions involving multi-functional heterocyclic substrates.
Initially, an evaluation was made of the catalyst, base, and solvent required for the Heck reaction of 2,4-diamino-5-(5-iodo-3,4-dimethoxybenzyl)pyrimidine (1) with (±)-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (2a) to generate 3a (Scheme 1).14,15,18 Two conventional catalysts, (Ph3P)2PdCl2 and Pd(OAc)2,18 as well as the Frech complex, were examined and compared. The use of (Ph3P)2PdCl2 under standard conditions (round-bottomed flask, 1.25 mol% catalyst relative to substrates 1 and 2a, 1.10 equivalents of N-ethylpiperidine, DMF, argon atmosphere, 140–150°C, 18 h) gave a low yield (37%) of the coupled product 3a with a significant number of impurities. An improved return (42%) was realized in a sealed tube under the same conditions, but impurities still persisted. The use of Pd(OAc)2 (1.25 mol%) provided the products in similar yields (50–52%) under both standard and sealed tube conditions, but with only a slightly improved impurity profile. By comparison, the Frech catalyst afforded consistently high yields (80%) with far fewer contaminants at a loading of just 0.12 mol%. Moreover, the Frech catalyst allowed the reaction to be performed on a larger scale (see below).
Scheme 1.
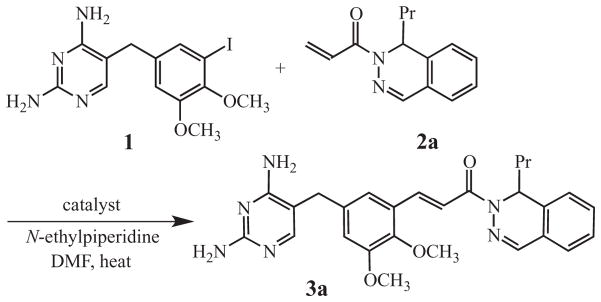
Preparation of 3a by Heck Coupling.
Heck reactions using pincer catalysts are often critically influenced by the solvent and base employed for the coupling process.19,20 To determine the optimum protocol, several solvents, including DMF, THF, and PhCH3, were studied. For the current application, DMF afforded the best results due to its superior solvating properties for the substrates and high boiling point. A series of bases, which included K2CO3, Cs2CO3, Et3N, DBU and N-ethylpiperidine, was also evaluated. In DMF, N-ethylpiperidine provided the highest yields of coupled products.
Reaction temperatures were also varied to optimize the conditions. Using DMF and N-ethylpiperidine, maximum conversions were realized at 140–150°C. Reactions at lower temperatures (110–120°C) were slow and gave low yields even after prolonged heating (36 h). At more elevated temperatures (≥160°C), complex mixtures were formed which hindered purification of the desired products. For catalyst comparison studies, reactions using (Ph3P)2PdCl2, Pd(OAc)2, and the Frech complex were run at 140–150°C for 16–20 h, although 3h–3j required only 8–12 h. Without exception, the Frech catalyst gave higher yields and cleaner products that were more easily purified.
Finally, catalytic loading for each catalyst was investigated. Our optimization studies indicated that 1.25 mol% of (Ph3P)2PdCl2 and Pd(OAc)2 was required to achieve complete conversions. Greater amounts of catalyst slightly decreased the yields and increased the number of impurities, while less catalyst resulted in incomplete reactions. In sharp contrast, the Frech complex afforded essentially complete conversions to products at a catalytic loading of only 0.12 mol%. For the class of compounds examined, isolated yields of products were highly reproducible with this quantity of catalyst.
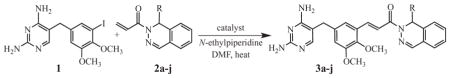
Under optimized conventional conditions, the reactions of 1.30 mmol each of 1 with 2a–j were carried out using 1.42 mmol of N-ethylpiperidine and 1.55 × 10−3 mmol (0.12 mol%) of the Frech catalyst in 4 mL of DMF under argon at 140–150°C for 16–20 h. The R groups [propyl (3a), isobutyl (3b), isobutenyl (3c), cyclohexyl (3d), phenyl (3e), 4-methylphenyl (3f), 4-fluorophenyl (3g), benzyl (3h), 4-methylbenzyl (3i) and 4-trifluoromethoxybenzyl (3j)] were carefully chosen to provide a range of agents with potential activity as antibiotics and also to ascertain the structural diversity tolerated by the catalyst. The results are summarized in Table 1. Products 3a–j were highly polar and retained water (from chromatography) or methanol (from recrystallizations) despite extensive efforts to remove them.21 Finally, though our study compared reactions run on a 1.30-mmol scale, the Frech catalyst (at a loading of 0.17 mol%) allowed us to run 20.0-mmol preparative scale reactions to generate lead compounds 3a and 3c in essentially undiminished yields of 78% and 74%, respectively.
Table 1.
Yields of 3a–j using Three Catalysts
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Product | R | Yield (%)
|
||||||
| Catalyst: | (Ph3P)2PdCl2
|
Pd(OAc)2
|
Frech
|
|||||
| Method: | A | B | A | B | C | D | ||
| 3a | n-C3H7 | 37 | 42 | 50 | 52 | 80 | 80 | |
| 3b | i-C4H9 | 30 | 65 | 67 | 67 | 82 | 81 | |
| 3c | i-C4H7 | 36 | 56 | 65 | 63 | 75 | 76 | |
| 3d | c-C6H11 | 32 | 47 | 54 | 57 | 82 | 82 | |
| 3e | C6H5 | 26 | 60 | 58 | 55 | 78 | 80 | |
| 3f | 4-CH3C6H4 | 38 | 67 | 66 | 64 | 76 | 74 | |
| 3g | 4-FC6H4 | 28 | 55 | 62 | 63 | 79 | 77 | |
| 3h | C6H5CH2 | 22 | 56 | 68 | 68 | 83 | 80 | |
| 3i | 4-CH3C6H4CH2 | 18 | 48 | 56 | 45 | 75 | 71 | |
| 3j | 4-CF3OC6H4CH2 | 10 | 25 | 38 | 40 | 74 | 77 | |
Method A: Round-bottomed flask, 1.30 mmol each of 1 and 2, 1.42 mmol of N-ethylpiperidine, 1.63 × 10−2 mmol of catalyst, 4 mL of DMF, 140°C, 18 h; Method B: Sealed tube, 1.30 mmol of 1 and 2, 1.42 mmol of N-ethylpiperidine, 1.63 × 10−2 mmol of catalyst, 4 mL of DMF, 140°C, 18 h; Method C: Round-bottomed flask, 1.30 mmol of 1 and 2, 1.42 mmol of N-ethylpiperidine, 1.55 × 10−3 mmol of catalyst, 4 mL of DMF, 140°C, 18 h; Method D: Sealed tube, 1.30 mmol of 1 and 2, 1.42 mmol of N-ethylpiperidine, 1.55 × 10−3 mmol of catalyst, 4 mL of DMF, 140°C, 18 h. All reactions were performed under argon.
Coupling of substrates incorporating such wide functional diversity–a diaminopyrimidine ring, two ethers, a tertiary amide, an imine and (in some cases) fluorine–is rare. The closest analogy to our work involved the use of the Frech catalyst to couple a variety of aryl halides to N,N-dimethylacrylamide.13 In this investigation, the reported transformations were assessed to be nearly quantitative by GC/MS analysis. Table 1 reports of products isolated in our current study. Although our optimized catalyst loading was 0.12 mol%, compared to 0.01 mol% for the acrylamide,13 this parameter would be expected to vary for different compounds. Nevertheless, the marked flexibility of the Frech catalyst to operate effectively on systems bearing such a large range of functional groups is remarkable and of great significance in organic synthesis.
In summary, we have used the Frech pincer catalyst to efficiently prepare a series of highly functionalized 2,4-diaminopyrimidine-based antibacterials for biological evaluation. The Frech catalyst proved superior to conventional palladium-based Heck catalysts, giving the desired products in higher yields and with fewer contaminants. The Frech catalyst also exhibited superior activity and thermal stability and reduced the required catalytic loading by a factor greater than ten compared to the other catalysts examined. Such remarkable utility, broad scope of action, and multi-functional group tolerance by the Frech catalyst mandates further exploration in organic synthesis.
Experimental Section
Commercial anhydrous N,N-dimethylformamide (DMF) was stored under dry argon and transferred by syringe into reactions where required. All other commercial reagents were used as received. Unless otherwise specified, all reactions were run under dry argon in oven-dried glassware. Reactions were monitored by thin layer chromatography on silica gel GF plates (Analtech No. 21521). Preparative separations were performed by column chromatography in quartz columns using silica gel (Davisil®, grade 62, 60–200 mesh) mixed with UV-active phosphor (Sorbent Technologies, No. UV-05). Band elution for all chromatographies was monitored using a hand-held UV lamp. Melting points were uncorrected. FT-IR spectra were run as thin films on NaCl disks. Both 1H-NMR and 13C-NMR spectra were measured in DMSO-d6 on a Varian GEMINI 300 instrument at 300 MHz (1H) and 75 MHz (13C) or on a Varian INOVA 400 instrument at 400 MHz and 100 MHz, respectively, and referenced to internal tetramethylsilane.
Representative Procedure for Heck Coupling using the Frech Catalyst to Prepare (±)-(E)-3-{5-[(2,4-Diamino-5-pyrimidinyl)methyl]-2,3-dimethoxyphenyl}-1-(1-propyl-2(1H)-phthalazinyl)-2-propen-1-one (3a)
Note: Methods A and B [for (Ph3P)2PdCl2 and Pd(OAc)2] differ from C and D [for the Frech catalyst] only in the mol% of catalyst used.
Conventional Conditions (Method C)
A solution of 500 mg (1.30 mmol) of 1,14,15 the corresponding phthalazine derivative (2a–j) (1.30 mmol),14,15 N-ethylpiperidine (161 mg, 0.195 mL, 1.42 mmol) and the Frech catalyst (1.00 mg, 1.55 × 10−3 mmol, 0.12 mol%) in 4 mL of anhydrous DMF was prepared in a 50-mL, round-bottomed flask equipped with magnetic stirring. The solution was heated at 140–150°C for a period of 18 h. During this time, the reaction mixture turned from brown to bright red in color. After cooling, the crude reaction mixture was transferred directly to a 30 cm × 2 cm silica gel column slurry packed in dichloromethane. Impurities were eluted using dichloromethane, and the final product was collected using 4% methanol in dichloromethane as the eluent. Evaporation of the solvent gave a pale yellow solid which was further purified using a 15-cm × 2-cm silica gel column, packed with 5% triethylamine-dichloromethane and eluted with 4% methanol in dichloromethane. The second chromatography removed yellow-colored impurities as well as several other minor contaminants. The products were recrystallized from methanol to give pure 3a–j. Using this procedure, 503 mg (80%) of 3a was isolated as a white powder, mp 121–124°C (shrinks to glass-like bead). The IR, 1H-NMR, and 13C-NMR data for 3a, as well as 3b–3j, matched those reported previously.14
Sealed Tube (Method D)
Sealed tube reactions were run using the same reactant, base, catalyst and solvent quantities given above. The reaction mixture was prepared in a 15-mL, screw-top Pyrex pressure vessel (Chemglass CG-1880–01 with O-ring CG-309-210), purged with argon for 1 min, sealed, and heated in an oil bath at 140–150°C for 18 h. This procedure also produced 3a in 80% yield.
Large-Scale Preparation of 3a
This procedure followed Method C above using 8.00 g (20.0 mmol) of 1, 5.20 g (22.0 mmol, 1.10 equiv) of 2, 2.58 g (3.12 mL, 22.0 mol, 1.10 equiv) of N-ethylpiperidine, and 22 mg of the Frech catalyst (3.41 × 10−2 mmol, 0.17 mol%) in 20 mL of DMF. The reaction was heated at 140–150°C for 18 h, and the product purified as above to give 7.86 g (78%) of 3a as a white powder.
Acknowledgments
We are very grateful to Dr. Ronaldo Mariez (Sigma-Aldrich) for a generous sample of the Frech catalyst used in this study. We gratefully acknowledge support of this work by the National Institutes of Allergy and Infectious Diseases [1-R01-AI090685-01] of the NIH/NIAID to WWB. Funding for the 300 MHz and 400 MHz NMR spectrometers of the Oklahoma Statewide Shared NMR Facility was provided by NSF (BIR-9512269), the Oklahoma State Regents for Higher Education, the W. M. Keck Foundation, and Conoco, Inc. The authors thank the OSU College of Arts and Sciences for funds to upgrade the Departmental FT-IR instruments.
References
- 1.van der Boom ME, Milstein D. Chem Rev. 2003;103:1759. doi: 10.1021/cr960118r. [DOI] [PubMed] [Google Scholar]
-
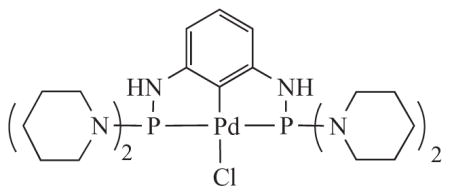
2.The Frech catalyst is [2,6-bis[(di-1-piperidinylphosphino)amino]phenyl]palladium(II) chloride.
- 3.Agarwal D, Schroder D, Frech CM. Organometallics. 2011;30:3579. [Google Scholar]
- 4.Bolliger JL, Frech CM. Chem Eur J. 2010;16:4075. doi: 10.1002/chem.200903309. [DOI] [PubMed] [Google Scholar]
- 5.Bolliger JL, Frech CM. Chimia. 2009;63:23. [Google Scholar]
- 6.Bolliger JL, Frech CM. Adv Synth Catal. 2009;351:891. [Google Scholar]
- 7.Bolliger JL, Blacque O, Frech CM. Angew Chem Int Ed. 2007;46:6514. doi: 10.1002/anie.200701804. [DOI] [PubMed] [Google Scholar]
- 8.Morales-Morales D. Rev Soc Quim Méx. 2004;48:338. [Google Scholar]; Chem Abstr. 2005;142:246825. [Google Scholar]
- 9.Narayanan R. Molecules. 2010;15:2124. doi: 10.3390/molecules15042124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tietze LF, Levy LM. In: the Mizoroki-Heck Reaction. Oestreich M, editor. Ch 8 Wiley-Chichester; 2009. [Google Scholar]
- 11.Li M, Hua R. Appl Organometal Cat. 2008;22:397. [Google Scholar]
- 12.Muller T, Bräse S. In: the Mizoroki-Heck Reaction. Oestreich M, editor. Ch 6 Wiley-Chichester; 2009. [Google Scholar]
- 13.Bolliger JL, Blacque O, Frech CM. Chem Eur J. 2008;14:7969. doi: 10.1002/chem.200800441. [DOI] [PubMed] [Google Scholar]
- 14.Nammalwar B, Bunce RA, Berlin KD, Bourne CR, Bourne PC, Barrow EW, Barrow WW. Eur J Med Chem. 2012;54:387. doi: 10.1016/j.ejmech.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourne CR, Bunce RA, Bourne PC, Berlin KD, Barrow EW, Barrow WS, Barrow WW. Antimicrob Agents Chemother. 2009;53:3065. doi: 10.1128/AAC.01666-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrow EW, Drier J, Reinelt S, Bourne PC, Barrow WW. Antimicrob Agents Chemother. 2007;51:4447. doi: 10.1128/AAC.00628-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bourne CR, Barrow EW, Bunce RA, Bourne PC, Berlin KD, Barrow WW. Antimicrob Agents Chemother. 2010;54:3825. doi: 10.1128/AAC.00361-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guerry P, Hubschwerlen C, Jolidon S, Specklin JL, Wyss PC. 9839328. World Patent WO. 1998; Chem Abstr. 1998;129:230736. [Google Scholar]
- 19.Beletskaya IP, Cheprakov AV. In: the Mizoroki-Heck Reaction. Oestreich M, editor. Ch 2 Wiley-Chichester; 2009. [Google Scholar]
- 20.Bonrath W, Létinois U, Netscher T, Schütz J. In: the Mizoroki-Heck Reaction. Oestreich M, editor. Ch 15 Wiley-Chichester; 2009. [Google Scholar]
- 21.X-ray data on related compounds show the 2,4-diaminopyrimidine ring H-bonded with water in the crystal structure: see Frey KM, Lombardo MN, Wright DL, Anderson AC. J Struc Biol. 2010;170:93. doi: 10.1016/j.jsb.2009.12.011.