

Abstract
Amyloid forming proteins Tau, alpha B crystallin, and amyloid P protein are all found in lesions of multiple sclerosis (MS). Our previous work established that amyloidogenic peptides from the small heat shock protein, alpha B crystallin(HspB5), and from amyloid β fibrils, characteristic of Alzheimer’s disease, were therapeutic in experimental autoimmune encephalomyelitis (EAE), reflecting aspects of the pathology of MS. To understand the molecular basis for the therapeutic effect, a set of amyloidogenic peptides composed of six amino acids, including those from tau, amyloid β A4, major prion protein (PrP), HspB5, amylin, serum amyloid P (SAP), and insulin B chain were shown to be anti-inflammatory, capable of reducing serological levels of IL-6, and attenuating paralysis in EAE. The chaperone function of the fibrils correlates with the therapeutic outcome. Fibrils composed of Tau 623–628 precipitated 49 plasma proteins, including apolipoprotein B-100, clusterin, transthyretin, and complement C3, supporting the hypothesis that the fibrils are active biological agents. Amyloid fibrils thus may provide benefit in MS and other neuroinflammatory disorders.
Keywords: Small heat shock proteins, chaperone, amyloid fibrils, inflammation, experimental autoimmune encephalomyelitis
INTRODUCTION
The accumulation of amyloid proteins in neurodegenerative diseases has long been associated with neuropathology, most notably in Alzheimer’s dementia. Nevertheless the biological functions of amyloid-forming proteins and the pathophysiological role of amyloid fibrils themselves are not well defined. The amyloid forming small heat shock protein, alpha B crystallin (HspB5)(1) was therapeutic in animal models of multiple sclerosis(2), stroke(3), cardiac and retinal ischemia-reperfusion injury(4, 5). We recently demonstrated that the Aβ 1–40 and 1–42 peptides from another amyloid protein, amyloid β A4, also are effective anti-inflammatory agents, ameliorating paralysisand reducing histopathological evidence inflammation in brain in EAE(6). Further, genetic deletion of HspB5(2) and amyloidβ A4(6) increased paralysis and inflammation in the EAE animal model of MS. This property of exacerbating EAE has been reported with genetic deletion of other amyloid forming proteins including major prion protein, PrP (7), serum amyloid P(8), and tau(9). Alpha B crystallin, tau, amyloid precursor protein, and serum amyloid P precursor protein are all found in MS lesions(10). Eisenberg and colleagues have established a common structural basis for the crossed β strands of amyloid fibrils, referred to as dry steric zippers, and have shown that peptides as short as hexamers are capable of forming this tertiary structure(11). Recent studies have established a correlation between the molecular chaperone activity of small heat shock proteins and their therapeutic function, with their mode of action arising from their capacity to bind proinflammatory proteins and reduce aggregation at the elevated temperatures within inflammatory foci(12). The reduction of proinflammatory stimulators would then have pleiotropic effects including reduction of pro-inflammatory cytokine production produced in the adaptive immune response. By mass spectral analysis a common set of approximately 70 ligands were precipitated by HspB5 from plasma(13). These proteins were distinguished from other precipitated molecules because they were enriched in the precipitate compared with their plasma concentrations, and they exhibited temperature dependent binding. Greater than 50% of these ligands were acute phase proteins, or members of the complement or coagulation cascades.
Additional structure activity correlations between chaperone activity and therapeutic function were established when linear peptide regions within HspB5 were examined(12). A single region, corresponding to residues 73–92 of HspB5 exhibited chaperone activity and was therapeutic, reducing paralysis and histological neuroinflammation in EAE. Only the peptide exhibiting chaperone activity was therapeutic, establishing a correlation between the two activities(12). Tanaka and colleagues demonstrated independently that only if 73–92, or a set of analogs, formed amyloid fibrils, did they have chaperone activity(14). This important correlation between the capacity to form amyloid fibrils and to act as a molecular chaperone, explained how a relatively short peptide could exhibit the equivalent biologic function as a fully folded protein. In this paper we have used minimal amyloidogenic hexapeptides to establish that the resultant simplified, and relatively homogeneous amyloid forming structures are effective therapeutics in neuroinflammation, with no apparent toxicity.
RESULTS
Amyloid Fibrils Composed of Hexapeptides Ameliorate EAE and Reduce Serum IL-6
The linkage between amyloid fibril formation, molecular chaperone function, and therapeutic activity in EAE led to the identification of two hexapeptides within or flanking residues 73–92 in HspB5 by applying the Rosetta-Profile algorithm(15, 16). In addition to the two HspB5 sequences, corresponding to residues 76–81, 89–94, ten other hexapeptides whose crystallographic solution has been determined, Tau 623–628, amyloid β A4 16–21, 27–32, 29–34, 35–40 and 37–42, major prion protein PrP 148–153, amylin 28–33, insulin B chain 11–16, and insulin A chain 12–17(11, 17), and one whose structure is unknown, serum amyloid P 213–218, were all tested for their ability to modulate the paralytic signs of EAE (Table 1).
Table 1. Hexameric peptides used in this study segregated by composition and their propensity toform fibrils(11, 17.
Those peptides whose crystal structure has been published are in italics with their denoted classes, with the hydrophobic amino acids highlighted in light grey, acidic residues and basic amino acids in dark grey. Those peptides used as therapeutics are listed in a bold font.
| anionic, requires low pH | nonionizable polar | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HspB5 76–81 | Ac | S | V | N | L | D | V | CONH2 | Major prion protein 148–153 | Ac | S | N | Q | N | N | F | CONH2 | class 2 | |
| Insulin B chain 11–16 | Ac | V | E | A | L | Y | L | CONH2 | class 7 | Apolipoprotein E 53–58 | Ac | S | S | Q | V | T | Q | CONH2 | |
| Insulin A chain 12–17 | Ac | L | Y | Q | L | E | N | CONHS | class 7 | Amylin 28–33 | Ac | S | S | T | N | V | G | CONH2 | class 1 |
| Ig Kappa chain 5–10 | Ac | S | V | S | S | S | Y | CONH2 | |||||||||||
| cationic, requires high pH | |||||||||||||||||||
| HspB5 89–94 | Ac | L | K | V | K | V | L | CONH2 | nonionizable hydrophobic | ||||||||||
| Amytoid beta A4 protein 27–32 | Ac | N | K | G | A | I | I | CONH2 | class 1 | ||||||||||
| Amyloid beta A4 protein 29–34 | Ac | G | A | I | I | G | L | CONH2 | class 6 | ||||||||||
| cationic, readily form at all pH | Amyloid beta A4 protein 35–40 | Ac | M | V | G | G | V | V | CONH2 | class 8 | |||||||||
| Amyloid beta A4 protein 37–42 | Ac | G | G | V | V | I | A | CONH2 | class 4 | ||||||||||
| Tau 623–628 | Ac | V | Q | I | V | Y | K | CONH2 | class 1 | Amylin 24–29 | Ac | G | A | I | L | S | S | CONH2 | |
| Serum amyloid P 213–218 | Ac | G | Y | V | I | I | K | CONH2 | |||||||||||
| Amyloid beta A4 protein 16–21 | Ac | K | L | V | F | F | A | CONH2 | class 7 | ||||||||||
Treating groups of ten mice at onset of hindlimb paralysis with daily injections of 1 µg of HspB5 76–81, 89–94, and Tau 623–628 resulted in statistically significant reductions of the paralytic signs, based on a standard clinical scoring system(18), compared with mice injected with PBS (Fig. 1A). As was the case for both treatment with small heat shock proteins and with residues 73–92 within these sHSPs, cessation of administration of the peptide resulted in most instances in a return ofgreater levels ofparalysis(12, 13). The importance of the linear sequence of HspB5 76–81 and Tau 623–628 was established by the demonstration that hexapeptides with equivalent amino acid content, but with the sequence shuffled did not modulate the disease (Fig. 1B and C).
Figure 1. Reduction of paralytic symptoms in mice with EAE by injection of amyloidogenic hexapeptides.

Groups of ten mice were injected daily with 1 µg of the listed peptides beginning at onset of hindlimb weakness. PBS or PBS containing 50% DMSO was injected in control littermates. (A) Hexapeptides corresponding to residues 76–81 and 89–94, and 623–628 of Tau effectively reduced the paralytic signs and the signs of clinical disease of EAE. (B,C) The shuffled analogs of HspB5 76–81 and Tau 623–628 were ineffective. (D) Hexamers corresponding to serum amyloid P 213–218, amyloid β A4 27–32, amylin 28–33 were therapeutic. (E) Two poorly soluble peptides, prion 148–153 and A-β A4 35–40 were therapeutic when administered in 50% DMSO. (F) Amyloid β A4 16–21, 29–34, and 37–42 were therapeutic when administered in 50% DMSO. (G) Administration of insulin B chain 11–16 did not statistically significantly reduce the signs of EAE. Bars represent the duration of the treatment. Values in graph represent mean +/− S.E.M. *p<0.05 for all peptides, ^p<0.05 for MVGGVV, +p<0.05 for A-β 29–34 by Mann Whitney U test.
To determine whether thetherapeutic effectiveness of amyloidogenic hexapeptides was a general property, three additional water soluble and six water insoluble peptides were tested. Peptides from various amyloid proteins, including serum amyloid P, amylin, and Aβ A4 all were effective therapeutics (Fig. 1D-F). Interestingly, so were four peptides that needed DMSO for solubilization, Aβ A4 29–34, 35–40, 37–42 and residues 148–153 of PrP, all of which reduced paralysis compared to animals receiving 50% DMSO in PBS as a control (Fig. 1E and F). The only peptide that did not exhibit statistically significant benefit was the amyloidogenic peptide from human insulin B chain, but even in this case the observed trend was a reduction in the neurological signs (Fig. 1G).
Histological analyses of the brain and spinal cords of treated and untreated mice confirmed the effects of the hexameric peptides were beneficial (Table 2). The number of inflammatory foci in the meninges and parenchyma of brains and spinal cords were quantified in mice treated with Aβ A4 hexapeptides or buffer control for 14 days and in mice after cessation of peptide treatment for 7 days. The signs, including worsening of paralysis, returned in mice following withdrawal of treatment, with a statistically significant increase in the clinical score as compared to treated mice. The reduction of signs with treatment corresponded to a significant decrease in the number of inflammatory foci in the meninges and parenchyma (Table 2).
Table 2. Histological analyses of inflammatory foci in brain and spinal cord confirm therapeutic benefit of hexapeptide administration.
Quantification of inflammatory foci in brains and spinal cords of mice with EAE treated with amyloid β hexapeptides or buffer control for 14 days and mice after cessation of treatment for 7 days. p values represent hexapeptide treatment compared to cessation of treatment.
| n* | c.s. | Meninges | Parenchyma | Total | Total Survival (%)* | |
|---|---|---|---|---|---|---|
| Control | 4 | 1.75 +/− 0.25 | 123.0 +/− 18.1 | 119.0 +/− 50.3 | 242.0 +/− 57.1 | 5/10 (50%) |
| Amyloid beta hexapeptides | 20 | 0.45 +/− 0.11 | 91.3 +/− 10.1 | 89.4 +/− 11.6 | 180.7 +/− 20.0 | 45/50 (90%) |
| Cessation of treatment | 15 | 2.40 +/− 0.21 | 125.3 +/− 11.3 | 155.4 +/− 12.8 | 280.7 +/− 21.6 | 45/50 (90%) |
| p values | 0.0001 | 0.0324 | 0.0006 | 0.002 | ||
Values represent meant +/− S.E.M. p values calculated using student t test; c.s., clinical score
Only a fraction of the total number of mice in the experiment were used for the histological analyses
In addition, no evidence of amyloid accumulation was found in pathological examination of major organs in mice treated with Aβ 37–42. Congo Red staining was absent from the central nervous system of mice treated with Aβ 37–42 who had histological evidence of EAE. These results were consistent with the inability of these peptides to catalyze seeding of amyloidogenic proteins or peptides in vivo.
To determine whether the hexapeptides modulated the plasma cytokine levels to the same extent as an intact small heat shock protein, groups of five mice were treated after the onset of hindlimb weakness with daily injections of either 1 µg of Tau 623–628 or 10 µg of HspB1 for two days and bled 12 hours later. Assaying the levels of 26 cytokines in the serum of untreated and treated mice revealed that administration of either HspB1 or the tau hexapeptide resulted in a large reduction of IL-6, with smaller reductions in IL-2, TGF-β, and IL-17 (Fig. 2).
Figure 2. Reduction of plasma cytokine levels in EAE mice after treatment with intact HspB1 or amyloid fibrils composed of residues 623–628 from human Tau.
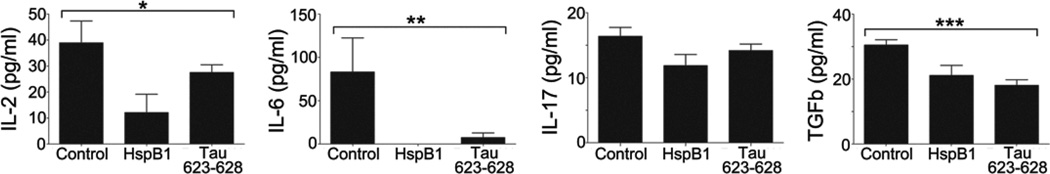
The concentration of IL-2, IL-6, IL-17,and TGFβ was measured from serum of mice treated for two days with 10 µg of HspB1 or 1 µg of tau 623–628 peptide (VQIVYK). Values in graph represent mean +/− S.E.M. *p<0.05, **p<0.04, and ***p<0.004 by one-way ANOVA.
Amyloid formation is pH dependent
To better understand the molecular behavior of the peptides and to optimize reproducibility of the biological experiments, the ability of each hexapeptide listed in Table 1 to form amyloid fibrils in aqueous buffers was examined. Surprisingly, when dissolved in PBS pH 7.4 only a fraction of the peptides formed amyloid fibrils as measured by staining with thioflavin T (ThT). Their differential propensity was used to help segregate them into functional sets (Table 1). The three peptides containing acidic residues, HspB5 76–81, insulin B chain 11–16, and insulin A chain 12–17 effectively formed amyloid fibrils only at pH 3–5, when the acidic residue would be protonated (Fig. 3A). Reciprocally, a second set of two peptides containing lysine, HspB5 89–94 and amyloid-βA4 27–32, bound ThT only at pH 10, when the lysine would not be protonated (Fig. 3B). In contrast, three peptides containing lysine formed amyloid fibrils in buffers from pH 3–10, Tau 623–628, amyloid-βA4 16–21, and serum amyloid P 213–218 (Fig. 3D). Two other sets were identified, both of which contained nonionizable amino acids. The first contained four polar sequences, the major prion protein, PrP 148–153, apolipoprotein E 53–58, amylin 28–33, and Ig kappa chain 5–10, which did not effectively bind ThT at any pH tested (Fig. 3C). The last set of peptides contained principally hydrophobic amino acids, three of which were from the amyloid-βA4 protein, residues 29–34, 35–40, and 37–42, and one from amylin, residues 24–29. These peptides had limited aqueous solubility, with the concentrated stock solutions being dissolved in 50% DMSO/PBS, which were then added to the solutions with ThT. Similar to the three peptides that formed amyloid fibrils at all pHs tested, Tau 623–628, serum amyloid P 213–218, and A-β A416–21, the four nonionizable hydrophobic peptides bound ThT at all pHs tested. As expected, the shuffled sequences for both Tau 623–628 and HspB5 76–81 were not stained by ThT at any pH, consistent with their inability to form fibrils.
Figure 3. Differential ability of the various hexamers to form amyloid fibrils and bind ThT allowed them to be segregated into functional sets.

Each of the hexamers (200 µg/50 µl) was added to 100 µl of 100 mM MES pH 7.4 and combined with 50 µl of 1M solution of at the appropriate pH and incubated at 37°C. ThT (10 µl of 10 µM solution) was added and the resultant fluorescence at 485nm from excitation at 440nm was measured using a fluorescent plate reader. The peptides can be segregated into an anionic set forming fibrils at pH 3,4, and 5 (A), a cationic group forming fibrils only at pH 10 (B), the three cationic sequences that form amyloid fibrils at all pHs measured (C), a nonionizable set of sequences weakly binding ThT at all pHs (D), and a hydrophobic set of sequences that bind ThT at all pHs measured (E).
In contrast with fibrils formed from intact proteins, those formed from the hexapeptide were rapidly reversible, associating or dissociating as the pH was modified. Consistent with a reversible system, insulin A 12–17 was an equally effective therapeutic when injected at pH 5.0 or 7.4 (fig. S1). This result and the failure of manyof the peptides to form amyloid fibrils at pH 7.4 in vitro were inconsistent with the high percentage of the peptides that were therapeutic in the MS animal model. To understand the behavior of the peptides and the importance of the resultant ThT positive structures, additional experiments were performed, examining whether any of the peptides were molecular chaperones, and if so, at what pH.
Amyloid formation correlates with chaperone function
To determine whether the structures that bind ThT correlated with molecular chaperone function, each of the peptides was assayed for their ability to inhibit insulin aggregation. Inhibition of aggregation has technical advantages over inhibition of fibril formation because both insulin and the inhibitory peptide fibrils bind ThT. In contrast, the peptides and their fibrils are both transparent at 360nm, the wavelength used to measure aggregation. Nevertheless, both Tau 623–628 and HspB5 are effective inhibitors of insulin fibril formation (fig. S2). Aggregation and subsequent amyloid fibril formation, can be divided into a rate determining nucleation step, followed by a relatively rapid fibrillation step, concluding with a final equilibration of fibrils dictated by the original concentration of the monomers(19, 20). Insulin aggregation is pH dependent(20), only allowing a dynamic range for the assay at pH between 5 and 8, which allows only a fraction of the pH range used in the ThT staining experiments to be analyzed.
When HspB5 76–81 and Tau 623–628 were assayed for inhibition of insulin aggregation, a clear distinction was apparent. The Tau peptide was a potent inhibitor at both pH 7.4 and pH 5, but the HspB5 peptide was only effective at pH 5 (Fig. 4A and B), which correlated with the pH dependence of ThT staining (Fig. 3A and D). The shuffled analogs of both peptides did not inhibit at either pH. The inhibition with the Tau peptide was titratable, allowingmeasurement of an IC50which in the case of the Tau peptide was approximately 50µg/ml (Fig. 4C). As previously documented the inhibition is dose dependent, and both inhibitors delay the appearance of the light scattering, and consequently inhibit the nucleation step of the process (Fig.4, A-C), thus limiting the amount of final aggregate or amyloid.
Figure 4. Only those peptides that form amyloid fibrils act as molecular chaperones and inhibit insulin aggregation.

(A) At pH 7.0, Tau 623–628 inhibits insulin aggregation, but a shuffled sequence does not. Neither HspB5 76–81 nor its shuffled sequence inhibit. (B) At pH 5.0, both Tau 623–628 and HspB5 76–81 inhibit, but neither shuffled analogs exhibit chaperone activity. (C) The inhibition of insulin aggregation by Tau 623–628 at pH 7 is dose dependent. (D) Anionic peptides insulin A chain 11–16 and insulin B chain 12–17 are molecular chaperones at pH 5, but not at pH 7.4. (E) The relative ability of three Ab peptides to inhibit insulin aggregation is proportional to their staining with ThT, consistent with their amyloid fibril content being responsible for the inhibition.
Assaying the insulin peptides also supported the conclusion that chaperone activity required ThT staining (Fig. 4D). Neither the peptide from the insulin A or B chain was able to inhibit at pH 7.4, but both were effective when assayed at pH 5.0. A third example that demonstrated that the formation of amyloid fibrils correlated with the molecular chaperone activity was the relative ability of three A-βA4 peptides to chaperone. To assay these nonionizable, hydrophobic peptides required modification of the assay to tolerate a small volume of DMSO, the buffer in which the peptides were dissolved. Addition of 10µl of DMSO resulted in a reduction, but not the elimination of aggregated insulin, allowing the three A-β A4 peptides to be assayed (Fig.4E). At pH 7.4 residues 35–40 bound greater amount of ThT, than residues 29–34, which bound more than residues 37–42 of A-β. A similar rank hierarchy was observed in their potency as molecular chaperones (Fig.4E.) Collectively, the results establish that the amyloid fibrils are molecular chaperones and not the individual peptides.
To investigate whether the Tau peptide inhibited in a similar or different manner as a small heat shock protein, the two types of inhibitors were added at different times after the aggregation was initiated. Very different effects were seen (fig. S3). When 150 µg of HspB5, a fully inhibitory dose when given at time 0, was added to a solution containing reduced insulin solution three minutes after the addition of dithiothreitol, there was a rapid increase in light scattering, which contrasted to the slight reduction in light scattering when an equivalent volume of buffer was added, due to the dilution of the solution (fig. S3A). In contrast, addition of the Tau fibrils, at a concentration that was inhibitory when added prior to the reducing agent, had little or no effect. The effects seen for the addition of the fibrils was equivalent to the dilution of the solution by adding buffer. A similar pattern was observed when either the sHsp or the Tau fibrils were added at 10 and 16 minutes; addition of the Hsp resulted in an increase in light scattering, albeit less of an increase at longer than earlier time points. The peptide fibrils had no apparent effect (fig. S4). Collectively, the results demonstrate the peptide fibrils, and the protein both can inhibit insulin aggregation/fibril formation, but do so by slightly different mechanisms.
Monomeric hexapeptides cannot inhibit insulin aggregation, and are not molecular chaperones, therefore the mechanisms underlying how the fibrils inhibit neuroinflammation, indicate that their fibrillar nature is essential in the process. Data in Supplemental Figure 3B and 4B demonstrate the peptide fibrils do not affect insulin fibrillization once it begins, whereas HspB5 can increase the light scattering after initiation of fibril formation, apparently by binding fibrils and increasing the size of the aggregates. This is consistent with previous experiments demonstrating that small heat shock proteins bind amyloid fibrils(21–23).
Amyloid fibrilsprecipitate specifically a set of plasma proteins
In contrast to the other peptides analyzed, none of the polar, nonionizable peptides bound ThT, or exhibited any molecular chaperone function, at any of the pHs examined (Fig. 3C). However, both the major prion peptide 148–153 and residues 28–33 of amylin were effective therapeutics, which demonstrates that the in vitro conditions used to analyze fibrillization do not reflect the in vivo milieu, in particular the absence of plasma proteins. Intact small heat shock proteins have been shown to bind a spectrum of proinflammatory mediators in plasma from patients with MS, rheumatoid arthritis and amyloidoses(13), which has been postulated to be basis of their immunosuppressive activity. Amyloid fibrils also are known to bind a spectrum of plasma proteins including apolipoproteins, A-I, A-IV, E(24), clusterin(25), and transthyretin(26). To determine what plasma proteins are bound by the amyloid fibrils, the Tau peptide was biotinylated and mixed with the unmodified hexapeptide to create a fibril that can be precipitated with streptavidin. Incubation of the two peptide mixtures with MS plasma, from three separate patients, with subsequent precipitation, elution, tryptic cleavage, and mass spectral analyses, allowed identification of 49 proteins whose relative concentration was enhanced compared to the set of proteins identified when the streptavidin resin alone was used (Fig. 5).
Figure 5. Compilation of proteins selectively precipitated by biotinylated fibrils composed of the hexamer, Tau 623–628.

The proteins are listed in descending concentration based on the difference between the amount precipitated by the biotinylated fibrils compared to streptavidin resin alone. The error bars represent the variations found from three separate MS plasma samples. The most prominent 49 proteins are shown in decreasing order in panel A-C. Components of HDL are shown in grey, others are in black histograms. Acute phase (*), complement (†), and coagulation proteins (‡) are demarcated. A Venn diagram displaying the number of common proteins precipitated by both amyloid fibrils composed of Tau 623–628 and HspB5 in panel D.
Reproducibility was high, both between triplicate injections of the same sample (technical triplicates) and between different patients’ plasma (biological replicates). Forty-one of the proteins (84%) were among those identified by precipitation with the small heat shock protein, HspB5(13). Among the precipitated proteins 29 are known components of HDL particles (59%), which emphasizes that not all the proteins precipitated will necessarily directly bind the amyloid fibril. Nevertheless, the Tau fibrils precipitated an extraordinarily high percentage of the proteins in the precipitate that were members of the acute phase response (19/49 39%), coagulation (13/49, 27%) or complement (11/49 23%) pathways, overall representing 33 of the 49 proteins (67%) precipitated (Fig 5D). Strong evidence that the set of proteins constituted biologically relevant ligands was the presence of five proteins known to bind amyloid fibrils, Apolipoproteins A-I, A-IV, E(24), clusterin(25), and transthyretin(26). Many of the molecules described here were identified in laser capture microdissected lesions from MS patients including Apolipoproteins A-I, A-II, B-100, and E, HspB5, tau, and amyloid precursor protein(10). In addition, many of the molecules identified here are known to modulate EAE. Earlier studies have shown that inhibition of angiotensin converting enzyme or angiotensin receptor, and inhibition of prothrombin limits the symptoms of EAE(10, 27). Interestingly, mice with genetic deletions of apolipoprotein E(28), tau(9), HspB5(2), and APP(6) all exhibit exacerbated EAE.
DISCUSSION
The role of amyloid fibrils in amyloidosis and a variety of neurodegenerative diseases has long been believed to be deleterious. The demonstrations that amyloidogenic hexapeptides are capable of reducing the paralysis and pathology in EAE reveal that some amyloid fibrils can be beneficial. This is consistent with the experimental data establishing that only those aggregates capable of forming pores in biological membranes are pathogenic(29–33). The therapeutic activity appears to be due to the unexpected capacity of the fibrils formed from the hexapeptides to inhibit the formation of amyloid fibrils of heterologous proteins and/or the binding of proinflammatory mediators in plasma. Reduction of protein amyloid fibrils could reduce inflammasome activity and stimulation of neutrophils(34–36). This was established by the inhibition of aggregation and amyloid fibril formation of insulin upon the addition of a reducing agent, an assay routinely used to assign molecular chaperone function to heat shock proteins(37, 38). The inhibitory activity of the fibrils distinguishes this mechanism from that proposed for peptide inhibitors of amyloid formation, many of which are believed to “cap” the growing fibril(39).
Fibrils composed of hexameric peptides represent therapeutic entities resembling, but distinct from, fibrils composed of fulllength amyloidogenic proteins and peptides. Support for this hypothesis is the observed inhibition of the aggregation of the B chain of insulin by fibrils composed of any of several hexameric peptides (Fig. 3). In no instance was there an increase in light scattering or fibril formation, which would be the case if the peptides were able to “seed” fibril formation. The structural basis for the inhibition, and inability to seed fibril formation, lies in the simplicity of the fibrils formed by hexameric peptides. The key feature is not only their short length, but also their limited secondary structure.
The structure of the two defined amyloid fibrils are composed of dry zippers, a point strongly argued by Eisenberg and colleagues(11), but are more complex than two associated homotypic hexamers. The unit cell, or minimal structural unit, of HET-s is a β-solenoid with a triangular hydrophobic core(40), whereas Aβ is a strand-turn-strand with a heterotypic zipper(41). Several groups have published models of the annular structures believed to be the toxic components of amyloid fibrils(1, 32, 42) and all require more complex secondary structure than a simple β-strand. Consequently, the hexameric peptides, or their fibrils, should not be able to “seed” heterotypic fibrils in vivo or in vitro, and should not be able to form toxic, annular structures capable of inserting into membranes because of their simplicity. Further experimental support of this idea is the recent work of Eisenberg and colleagues(1), who demonstrated that only an extended 12 amino acid peptide from HspB5, and not a hexamer, was able to form a strand-turn-strand and assemble into a cylindrin(1). Hexamers may thus be devoid of the pathogenic activities seen with larger domains of these notorious proteins.
The differential ability of the peptides to aggregate and form amyloid fibrils at physiological pH is consistent with the principal driving force for aggregation residing in the hydrophobic interactions of the side-chains at the zipper interface. Only if a charged amino acid is neutral within this interface will the fibril readily form. In the case of the two insulin peptides, which form “back to front” zippers (11, 17), the acidic residue is within the zipper interface, explaining why fibril formation occurs at, or near, the pKa of glutamate. Similarly, protonation of the lysine in Aβ A4 27–32 inhibits fibril formation, and presumably also in HspB5 89–94. In contrast, the lysine in the Tau peptide is opposite the zipper interface and an ionized amino group should not interfere with fibril formation, but rather should enhance the aqueous solubility of the resultant fibril. This also can explain the behavior of SAP 213–218 and Aβ A4 16–21. Consistent with the importance of hydrophobic interactions driving aggregation, the four hydrophobic, poorly aqueous soluble, peptides readily formed amyloid fibrils, while the polar, nonionizable did not.
The pH dependence of fibrillization was used to demonstrate that molecular chaperone activity correlated with the presence of amyloid fibrils in the case of each of the anionic peptides that required low pH for amyloid formation. A differential ability to form fibrils at physiological pH of three Aβpeptides confirmed that the amount of ThT staining was proportional to chaperone activity, consistent with previous studies using longer sHsp peptides(6, 12). A critical feature of the chaperone assays was that fibrils composed of the different peptides, all inhibited aggregation and amyloid formation of the B chain of insulin. The inhibition by a heterologous amyloid fibril, albeit one formed with a hexapeptide, was surprising. Based on much of the amyloid literature, the addition of a heterologous amyloid fibril, Tau, to a solution in which another amyloid is forming, insulin, should either have no effect, or catalyze the fibrillization by “seeding” by providing preexisting fibrils to circumvent the nucleation step(19, 43). Instead, each of the fibrils inhibited the nucleation of the insulin B chain. Addition of the fibrils to insulin after the initiation steps catalyzed by the addition of DTT did not have any inhibitory effects, indicating the heterologous fibrils are competitive only in the nucleation step and not in the elongation period.
In contrast, addition of intact small heat shock proteins after the nucleation step resulted in greater light scattering, consistent with the binding of the fibrils and increasing of the aggregate size upon addition. Neither the fibrillated peptide, nor the sHsp, reversed light scattering, indicating these reagents are not capable of reversing this process. A recent report has demonstrated that sHsps in concert with higher molecular weight ATP dependent chaperones in yeast reversed fibril formation(44). Both sHsps and the fibrils formed of hexameric peptides were equally potent therapeutics for EAE, whose continued administration was needed to limit signs.
The sHsp is expected to bind partially unfolded proteins and amyloid fibrils(21, 22), but such binding by fibrils composed of hexapeptides is unexpected. The precipitation of Apolipoprotein E(24) clusterin(25), and complement C3(45) is consistent with previous experiments demonstrating that Aβ fibrils bind these plasma proteins.That the relatively polar sequence of the Tau hexapeptide also binds these proteins indicates that the binding site is specific for fibrils and not just hydrophobic residues. These interactions with plasma proteins might represent sites where peptide concentration would be locally high and consequently increase fibrillization rates, explaining the discrepancy between the characteristic of some of the peptides to aggregate in vitro and in vivo. Alternatively, the precipitated proteins might represent a population of partially unfolded proteins with a propensity to form amyloid fibrils. Consequently, binding of these proteins by the hexapeptide containingamyloid fibrils and small heat shock proteins could reduce the local concentration of both proinflammatory proteins and precursors of amyloid fibrils. The reduction of the concentration of this population would reduce the activation of the innate immune response leading to a reduction in IL-6 and subsequent inflammatory processes involved in EAE.
Whenever peptides are used as therapeutics, their short biological half-life becomes an important impediment to their clinical development. However, the active agents in this study are the fibrils, whose molecular weight varies between 22 and 5 kDa based on mobility in gel filtration experiments, and not the monomeric peptides. Consequently, their circulation time would be expected to differ from the hexamer. Detailed kinetics have not been explored primarily because of the cost and difficulty of working with radiolabeled reagents. Nevertheless, the plots of the symptoms of the animals as a function of time (Fig. 1) provide data on the pharmacodynamics, with the rate of the reduction of symptoms and return of symptoms after cessation of treatment both being close to two days. Future studies will establish the biological half-life of these reagents more precisely.
Previous studies have shown that the acute phase protein SAA can induce the production of IL-6(46). Binding of SAA and other acute phase proteins in the plasma by the sHsps and amyloidogenic hexamers may lead to the reduced levels of IL-6, IL-2 and TGFβ in the serum of treated EAE mice. However, the mode of action appears to be more complex than solely the modulation of the pro-inflammatory cytokine IL-6. Previous studies have shown that anti-IL6 receptor monoclonal antibody can suppress the induction of EAE(47), but fails to suppress established EAE(48). The amyloidogenic hexapeptides are able to ameliorate the neurological signs of EAE even when given at the onset of hindlimb weakness, indicating that the peptides exert their effect beyond the reduction of the levels of serological IL-6.
Additional support for the therapeutic benefit of amyloid fibrils in EAE can be found from the increased succeptibility of mice with a single amyloidogenic protein knocked-out. The symptoms of EAE were exacerbated in mice lacking the expression of amyloid-β A4(6), the major prion protein(7), serum amyloid P(8), and tau(9) compared to wild type animals. Genetic deletion of molecules that bind amyloid including Apolipoprotein E is known also to exacerbate EAE(28). In other conditions where inflammation is seen, including stroke and focal brain trauma, reduction of expression of an amyloidogenic protein also exacerbated the disease. Genetic deletion of APP for brain trauma(49), also lead to worsening of neuropathology compared to wildtype. Taken together these ‘loss of function experiments’ showing worsened pathology and clinical signs in EAE, stroke and brain trauma, combined with the ‘gain of function’ experiments showing benefit in EAE, stroke and brain trauma, all support the notion that under many diverse neuropathologic conditions, amyloid fibrils appear to be beneficial. In these experiments the administration of the simplified amyloid fibrils composed of the hexapeptides adds further experimental support that fibrils might be active therapeutic agents and could represent a new class of drugs for treatment of neuroinflammation.
MATERIAL AND METHODS
Induction of EAE in mice by immunization with MOG and adjuvant and treatment with different hexamers
EAE was induced by procedures previously described(18). Briefly, EAE was induced in female C57BL/6J mice (Jackson Laboratories) at 9 weeks of age by subcutaneous immunization in the flank with an emulsion containing 200 µg myelin oligodendrocyte glycoprotein35–55 MOG35–55; MEVGWYRSPFSRVVHLYR NGK) in saline and an equal volume of complete Freund’s adjuvant containing 4 µg/ml mycobacterium tuberculosis H37RA (Disco Laboratories). All mice were administered 400 ng of pertussis toxin (List Biological) intraperitoneal at 0 and 48 h post-immunization. The neurological impairment was scored as follows: 0, no clinical disease; 1, tail weakness; 2, hindlimb weakness; 3, complete hindlimb paralysis; 4, hindlimb paralysis and some forelimb weakness; 5, moribund or dead. When animals exhibited level two symptoms they were injected in the peritoneum with either 1 µg of peptide, or PBS daily. All animal protocols were approved by institutional IACUC. Normal murine plasma was taken from age matched healthy C57BL/6J mice.
Peptide synthesis
Peptides were synthesized using solid phase techniques and commercially available Fmoc amino acids, resins, and reagents (PE Biosystems and Bachem) on an Applied Biosystems 433A peptide synthesizer as previously described (50). Purity of the peptides was shown to be greater than 90% using a PE Biosystems 700E HPLC and a reverse phase column (Alltech Altima).
Luminex assay of cytokine concentration
Sera from EAE mice were collected following one day of treatment with 10 µg HspB5 protein, 1 µg tau 623–628 (VQIVYK), or PBS. Cytokines were analyzed by the Stanford Human Immune Monitoring Core using multiplex-bead-analysis for the following mouse cytokines: eotaxin, G-CSF, GM-CSF, IFNγ, IL-10, IL-12p40, IL-12p70, IL-13, IL-17, IL-1α, IL-1β, IL-2. IL-3, IL-4, IL-5, IL-6, IL-23, IP-10, KC, MCP-1, MCP-3, MIP-1α, RANTES, TGFβ, TNFα, and VEGF.
Chaperone assays
The capacity of the proteins and peptides to inhibit DTT induced aggregation of the beta chain of insulin was assayed using procedures described previously by several authors(37, 51). Briefly, 150 µg of bovine insulin (Sigma) dissolved in 100mM NaCl, 20mM Tris, pH 7.4 with, or without, varying concentration of the peptides in a total volume of 380 µl and incubated at 37°C. DTT, 20 µl of a 100mM stock solution, was added at time zero, and the aggregation was measured by the increase in absorption at 360nm as a function of time over twenty minutes. Inhibition of amyloid fibril formation was monitored by the addition of ThT (10µl of 10µM solution) and the resultant fluorescence at 485nm from excitation at 440nm was measured using a Spectra ax 190 fluorescent microtiter plate reader.
Thioflavin T binding
The relative amount of amyloid present in each solution was measured by combining solutions of the hexamers (200µg, 50µl) with 100µl of 100mM MES pH 7.4 and 50µl of 1M solution of at the appropriate pH and incubated at 37°C black 96 well microtiter plate. ThT (10µl of 10µM solution) was added and the resultant fluorescence at 485nm from excitation at 440nm was measured using a Spectra ax 190 fluorescent microtiter plate reader.
Blood samples
Under the IRB protocol used in this study, the human blood samples were provided without further information other than they were collected from patients at the Stanford Multiple Sclerosis and Rheumatology Clinics from patients older than 18 years of age with each of the respective clinically defined indications.
Biotinylated Tau Amyloid-ligand precipitation from human plasma, reduction/alkylation, and trypsinization
BiotinylatedTau 623–628 was prepared by mixing Tau 623–628 with the analog with long chain biotin replacing the acetyl group on the amino terminus of the peptide at a ratio of 100:1. The peptide mixture was dissolved in physiological saline pH 7.4 at 1 mg/ml. The fibrillization of the resultant mixture was assayed by adding 50µl to 150µl of PBS with 10µl of 10µM ThT and the resultant fluorescence resulting from excitation at 440nm was measured at 485nm. Biotinylated fibrils ofTau 623–628, 50µg, were added to 600µl aliquots of fresh human plasma, diluted 1:1 with physiological saline and incubated at 37°C for one hour after which 50µl of streptavidin sepharose beads (Sigma) were added and the mixture were incubated an additional hour at 37°C. The resin was separated from the plasma by centrifugation, the plasma saved for later analysis while the resin was washed repeatedly with physiological saline pH 7.4. The biotinylated Tau 623–628 fibril and its ligands were eluted with 100µl water and heated to 90°C for two minutes. The eluted proteins were separated from the resin, precipitated with TCA, resuspended, reduced, alkylated, and trypsinized as described previously(13).
Liquid chromatography and mass spectrometry
Liquid chromatography was done using an Eksigent nano2D LC coupled to an in-house packed C18 analytical column as described previously(13). A Micro source was used for nana ESI at a flow of 750 nil/min. A one hour linear gradient from 2% mobile phase B (acetonitrile 0.1% formic acid) to 40% B was used. Data was acquired using a LTQ Orbitrap Volos mass spectrometer (Thermo Fischer), in which data acquisition was acquired in a data dependent fashion (MSMS on top 8 most intense precursor ions). The .RAW files were converted to mzXML format and searched on a Sorcerer (Sagan) processing system using SyQuest against the appropriate database (ipi Human or ipi Mouse). The search results were then uploaded into a Scaffold (Proteome Software) workstation for visualization, data filtering, and statistical analysis. Finally, the data was exported to Microsoft Xcel.
Statistical analyses
EAE clinical score and cytokine levels
EAE data are presented as means +/− S.E.M. and statistical significance was assessed by a Mann-Whitney U-test to detect differences between groups (each group n=10). The cytokine levels are presented as means +/− S.E.M. and statistical significance between groups (n=5) was assessed by one-way ANOVA. A p-value of 0.05 or lower was considered significant.
Mass Spectrometry Data Analysis
A global proteomic approach was used, interrogating the tryptic peptides (thereby protein assignment) as well as quantitatively measuring each of the samples using a label free spectral counting approach (52). The data was filtered to a false discovery rate (FDR) at the peptide level of 5% and at the protein level of 0.1%. The higher fidelity for protein assignment was the result of the constraint in which a minimum of 2 peptides was required to assign an individual protein. Statistical analysis of protein expression profiles of different sample sets was done using a Fishers exact test, and all p-values were appropriately reported (53). In addition to identifying each of the proteins precipitated, the software places a quantitative value (spectral count) on each peptide, and thereby protein assigned. This step allowed the albumin in the samples to be used as an internal standard, which enabled the direct comparison of the relative amounts of each of the client proteins between different samples. After normalizing the samples based on albumin content, the putative ligands could be placed in a rank hierarchy and compared with their concentration in plasma and with their concentration relative to different precipitates. The protein composition was consistent with variation seen principally in the constituents composing the lowest percentage composition (low spectral counts), while the quantitative values varied by less than 2% with only a few outliers.
Supplementary Material
Acknowledgements
We thank Jonathan Li for his experimental assistance.
Funding: M.P.K. was supported by a postdoctoral fellowship from the National Multiple Sclerosis Society (FG1859) and National Institutes of Health (T32 AI007290). We would like to acknowledge gratefully funding from the National Institutes of Health (R01NS55997 to L.S., UO1DK078123 to J.B.R., and 1R43AI0949 to J.B.R. and L.S.). This work was supported by the National MS Society (to J.B.R. and L.S.) and the Endriz Fund (to L.S.).
Footnotes
Author Contributions: M.P.K. established the therapeutic activity of the peptides and measured the modulation of the cytokines, J.B.R. provided the physical chemical framework for these studies, performed the molecular chaperone assays, the pH dependence of the fibril formation, and precipitated the plasma proteins using a biotinylated amyloid fibril, while C.A. performed the mass spectral analyses. R.A.S. quantified the inflammatory foci in the meninges and parenchyma of brains and spinal cords. L.S. oversaw all aspects of the research and wrote the paper with J.B.R. and M.P.K.
Competing Interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Laganowsky A, et al. Atomic view of a toxic amyloid small oligomer. Science. 2012 Mar 9;335:1228. doi: 10.1126/science.1213151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ousman SS, et al. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature. 2007 Jul 26;448:474. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- 3.Arac A, et al. Systemic augmentation of {alpha}B-crystallin provides therapeutic benefit twelve hours post-stroke onset via immune modulation. Proc Natl Acad Sci U S A. 2011 Aug 9;108:13287. doi: 10.1073/pnas.1107368108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pangratz-Fuehrer S, Kaur K, Ousman SS, Steinman L, Liao YJ. Functional rescue of experimental ischemic optic neuropathy with alphaB-crystallin. Eye (Lond) 2011 Jun;25:809. doi: 10.1038/eye.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Velotta JB, et al. alphaB-crystallin improves murine cardiac function and attenuates apoptosis in human endothelial cells exposed to ischemia-reperfusion. Ann Thorac Surg. 2011 Jun;91:1907. doi: 10.1016/j.athoracsur.2011.02.072. [DOI] [PubMed] [Google Scholar]
- 6.Grant JL, et al. Reversal of Paralysis and Reduced Inflammation from Peripheral Administration of beta-Amyloid in TH1 and TH17 Versions of Experimental Autoimmune Encephalomyelitis. Sci Transl Med. 2012 Aug 1;4:145ra105. doi: 10.1126/scitranslmed.3004145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gourdain P, Ballerini C, Nicot AB, Carnaud C. Exacerbation of experimental autoimmune encephalomyelitis in prion protein (PrPc)-null mice: evidence for a critical role of the central nervous system. J Neuroinflammation. 2012;9:25. doi: 10.1186/1742-2094-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ji Z, Ke ZJ, Geng JG. SAP suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. Immunol Cell Biol. 2012 Apr;90:388. doi: 10.1038/icb.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinger JG, et al. Mice devoid of Tau have increased susceptibility to neuronal damage in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol. 2012 May;71:422. doi: 10.1097/NEN.0b013e3182540d2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han MH, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008 Feb 28;451:1076. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- 11.Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012 Mar 16;148:1188. doi: 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurnellas MP, et al. Chaperone activity of small heat shock proteins underlies therapeutic efficacy in experimental autoimmune encephalomyelitis. J Biol Chem. 2012 Sep 6; doi: 10.1074/jbc.M112.371229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothbard JB, et al. Therapeutic effects of systemic administration of chaperone alphaB-crystallin associated with binding proinflammatory plasma proteins. J Biol Chem. 2012 Mar 23;287:9708. doi: 10.1074/jbc.M111.337691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka N, et al. Amyloid fibril formation and chaperone-like activity of peptides from alphaA-crystallin. Biochemistry. 2008 Mar 4;47:2961. doi: 10.1021/bi701823g. [DOI] [PubMed] [Google Scholar]
- 15.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci U S A. 2010 Feb 23;107:3487. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson MJ, et al. The 3D profile method for identifying fibril-forming segments of proteins. Proc Natl Acad Sci U S A. 2006 Mar 14;103:4074. doi: 10.1073/pnas.0511295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawaya MR, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007 May 24;447:453. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 18.Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006 Jul;60:12. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- 19.Jarrett JT, Lansbury PT., Jr Seeding "one-dimensional crystallization" of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993 Jun 18;73:1055. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 20.Nielsen L, et al. Effect of environmental factors on the kinetics of insulin fibril formation: elucidation of the molecular mechanism. Biochemistry. 2001 May 22;40:6036. doi: 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- 21.Shammas SL, et al. Binding of the molecular chaperone alphaB-crystallin to Abeta amyloid fibrils inhibits fibril elongation. Biophys J. 2011 Oct 5;101:1681. doi: 10.1016/j.bpj.2011.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waudby CA, et al. The interaction of alphaB-crystallin with mature alpha-synuclein amyloid fibrils inhibits their elongation. Biophys J. 2010 Mar 3;98:843. doi: 10.1016/j.bpj.2009.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xi D, Dong X, Deng W, Lai L. Dynamic behavior of small heat shock protein inhibition on amyloid fibrillization of a small peptide (SSTSAA) from RNase A. Biochem Biophys Res Commun. 2011 Dec 9;416:130. doi: 10.1016/j.bbrc.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 24.Strittmatter WJ, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993 Mar 1;90:1977. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghiso J, et al. The cerebrospinal-fluid soluble form of Alzheimer's amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem J. 1993 Jul 1;293(Pt 1):27. doi: 10.1042/bj2930027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Velayudhan L, et al. Plasma transthyretin as a candidate marker for Alzheimer's disease. J Alzheimers Dis. 2012;28:369. doi: 10.3233/JAD-2011-110611. [DOI] [PubMed] [Google Scholar]
- 27.Platten M, et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci U S A. 2009 Sep 1;106:14948. doi: 10.1073/pnas.0903958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karussis D, et al. Lack of apolipoprotein-E exacerbates experimental allergic encephalomyelitis. Mult Scler. 2003 Oct;9:476. doi: 10.1191/1352458503ms950oa. [DOI] [PubMed] [Google Scholar]
- 29.Kayed R, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003 Apr 18;300:486. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 30.Xue WF, et al. Fibril fragmentation enhances amyloid cytotoxicity. J Biol Chem. 2009 Dec 4;284:34272. doi: 10.1074/jbc.M109.049809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009 Jan;5:15. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 32.Jang H, et al. beta-Barrel topology of Alzheimer's beta-amyloid ion channels. J Mol Biol. 2010 Dec 17;404:917. doi: 10.1016/j.jmb.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quist A, et al. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci U S A. 2005 Jul 26;102:10427. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halle A, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008 Aug;9:857. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masters SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2010 Oct;11:897. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Azevedo EP, et al. Amyloid fibrils trigger the release of neutrophil extracellular traps (NETs), causing fibril fragmentation by NET-associated elastase. J Biol Chem. 2012 Aug 23; doi: 10.1074/jbc.M112.369942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong DP, Fink AL. Independent heterologous fibrillation of insulin and its B-chain peptide. Biochemistry. 2005 Dec 20;44:16701. doi: 10.1021/bi051658y. [DOI] [PubMed] [Google Scholar]
- 38.Sanger F. Fractionation of oxidized insulin. Biochem J. 1949;44:126. doi: 10.1042/bj0440126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sciarretta KL, Gordon DJ, Meredith SC. Peptide-based inhibitors of amyloid assembly. Methods Enzymol. 2006;413:273. doi: 10.1016/S0076-6879(06)13015-3. [DOI] [PubMed] [Google Scholar]
- 40.Wasmer C, et al. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008 Mar 14;319:1523. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 41.Luhrs T, et al. 3D structure of Alzheimer's amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005 Nov 29;102:17342. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stroud JC, Liu C, Teng PK, Eisenberg D. Toxic fibrillar oligomers of amyloid-beta have cross-beta structure. Proc Natl Acad Sci U S A. 2012 May 15;109:7717. doi: 10.1073/pnas.1203193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993 May 11;32:4693. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 44.Duennwald ML, Echeverria A, Shorter J. Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol. 2012 Jun;10:e1001346. doi: 10.1371/journal.pbio.1001346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu H, et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Abeta by microglia. Glia. 2012 May;60:993. doi: 10.1002/glia.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ather JL, et al. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol. 2011 Jul 1;187:64. doi: 10.4049/jimmunol.1100500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gijbels K, Brocke S, Abrams JS, Steinman L. Administration of neutralizing antibodies to interleukin-6 (IL-6) reduces experimental autoimmune encephalomyelitis and is associated with elevated levels of IL-6 bioactivity in central nervous system and circulation. Mol Med. 1995 Nov;1:795. [PMC free article] [PubMed] [Google Scholar]
- 48.Serada S, et al. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008 Jul 1;105:9041. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corrigan F, et al. sAPPalpha rescues deficits in amyloid precursor protein knockout mice following focal traumatic brain injury. J Neurochem. 2012 Jul;122:208. doi: 10.1111/j.1471-4159.2012.07761.x. [DOI] [PubMed] [Google Scholar]
- 50.Wender PA, et al. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000 Nov 21;97:13003. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong DP, Ahmad A, Fink AL. Fibrillation of human insulin A and B chains. Biochemistry. 2006 Aug 1;45:9342. doi: 10.1021/bi0604936. [DOI] [PubMed] [Google Scholar]
- 52.Bergeron JJ, Hallett M. Peptides you can count on. Nat Biotechnol. 2007 Jan;25:61. doi: 10.1038/nbt0107-61. [DOI] [PubMed] [Google Scholar]
- 53.Zhang B, et al. Detecting differential and correlated protein expression in label-free shotgun proteomics. J Proteome Res. 2006 Nov;5:2909. doi: 10.1021/pr0600273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.