

Abstract
Hydrocephalus is a common neurological disorder leading to expansion of the cerebral ventricles and is associated with significant morbidity and mortality. Most neonatal cases are of unknown etiology and are likely to display complex inheritance involving multiple genes and environmental factors. Identifying molecular mechanisms for neonatal hydrocephalus and developing non-invasive treatment modalities are high priorities. Here we employ a hydrocephalic mouse model of the human ciliopathy Bardet-Biedl Syndrome (BBS) and identify a role for neural progenitors in the pathogenesis of neonatal hydrocephalus. We found that hydrocephalus in this mouse model is caused by aberrant PDGFRα signaling, resulting in increased apoptosis and impaired proliferation of NG2+PDGFRα+ neural progenitors. Targeting this pathway with lithium treatment rescued NG2+PDGFRα+ progenitor cell proliferation in BBS mutant mice, reducing ventricular volume. Our findings demonstrate that neural progenitors are critical in the pathogenesis of neonatal hydrocephalus and we identify novel therapeutic targets for this common neurological disorder.
Keywords: hydrocephalus, Bardet-Biedl Syndrome, ciliopathy, neural progenitor cell, NG2, PDGFRα, AKT, GSK3β, lithium, apoptosis
Neonatal hydrocephalus is a common disorder affecting the human nervous system with an estimated incidence of 1 to 3 per 1,000 live births1–4 creating a healthcare burden of 2 billion dollars annually5,6. Hydrocephalus leads to the expansion of cerebral ventricles and is associated with significant morbidity and mortality7–9. Neonatal hydrocephalus remains an understudied disease despite being a common developmental anomaly10. Neonatal hydrocephalus has multiple causes including obstruction of cerebrospinal fluid (CSF) flow and CSF overproduction; however, a significant portion of neonatal hydrocephalus is idiopathic in nature9–16. Current therapies rely on invasive procedures associated with high failure and complication rates making the identification of molecular mechanisms underlying neonatal hydrocephalus a high priority for the medical community3,9,11,17,18.
Recently, mouse models with impaired cilia function have provided insight into mechanisms involved in hydrocephalus occurring in the absence of obstruction, a condition known as communicating hydrocephalus10,13,14,19,20. Mutations in genes that disrupt ependymal motile cilia structure and function hinder ependymal motile cilia beat frequency and CSF flow leading to the development of hydrocephalus13,14,19,20. Non-motile cilia known as primary cilia, extend from the surface of nearly all cell types. Primary cilia serve as sensory antennae facilitating many signaling pathways including Wnt21, sonic hedgehog (Shh)22,23, and platelet derived growth factor receptor alpha (PDGFRα)24 enabling cells to respond to developmental cues in several sites of neurogenesis in the central nervous system (CNS) including the periventricular regions25. These non-motile cilia are required for normal development of neural progenitor cells (NPCs)26,27.
Recent findings have demonstrated that ependymal motile cilia and CSF flow are required for normal development of NPCs suggesting an intimate link between the ventricular system and neural development28. The close proximity of NPCs to the periventricular regions suggests that these cells play a role in maintaining the integrity of the ventricular system25,29. However, a role for NPCs in the pathophysiology of hydrocephalus has not been studied. In this study we investigated whether abnormal signaling through primary cilia in NPCs may contribute to the genesis of neonatal hydrocephalus. To test this hypothesis, we utilize a mouse model of a genetically heterogeneous human disorder known as Bardet-Biedl syndrome (BBS) caused by mutations in one or more of 17 genes, seven of which (BBS 1,2,4,5,7,8 and 9) form a complex known as the BBSome30. The cardinal features of BBS include retinal degeneration, obesity and cognitive delay19. Some BBS patients have enlarged cerebral ventricles and BBS mouse models display communicating hydrocephalus19,31,32. Here we demonstrate that abnormal development of NPCs specifically expressing the chondroitin sulfate proteoglycan NG2 and PDGFRα leads to the development of neonatal ventriculomegaly in BBS mice. Our findings identify a novel mechanism underlying hydrocephalus and provide a therapeutic target for treatment.
RESULTS
BBS mutant mice develop neonatal hydrocephalus
We have previously shown that BBS mutant mice homozygous for the most common human BBS mutation (Bbs1M390R/M390R) develop ventricular dilatation similar to that reported in human BBS patients19,31,32 (Fig. 1a,b). Ventriculomegaly in Bbs1M390R/M390R mice is fully penetrant and is accompanied by neurological deficits, similar to patients with hydrocephalus3,9,19,31,32. We employ the term hydrocephalus in Bbs1M390R/M390R mice to describe the ventriculomegaly as previously reported in other mouse models4,9,10,14–16,20.
Figure 1.
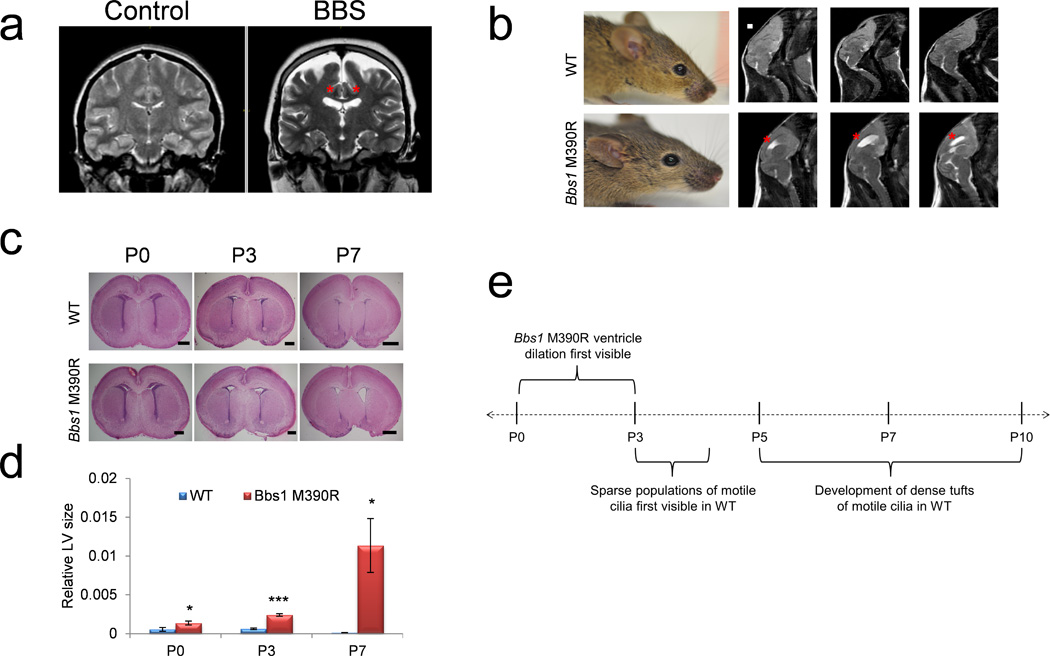
Hydrocephalus in BBS mutant mice occurs before motile cilia develop. (a) T2-weighted coronal MRIs of a Bardet-Biedl syndrome patient and age and sex matched control showing ventriculomegaly of the lateral ventricles (red asterisks). (b) Left, picture of 3 month old WT and Bbs1M390R/M390R mice showing a normal cranial vault in WT and Bbs1M390R/M390R. Right, T2-weighted sagittal MRIs showing hydrocephalus of the lateral ventricles (red asterisks) in a Bbs1M390R/M390R mouse. (c) Histology of WT and Bbs1M390R/M390R neonates showing perinatal onset of hydrocephalus in mutant pups and (d) the quantitations showing ventricular dilation at P3 and P7. (e) Timeline of the genesis of hydrocephalus in BBS mutant mice relative to motile cilia development showing that Bbs1M390R/M390R mice develop hydrocephalus prior to the development of motile cilia13,14. All error bars represent means ± s.e.m. *P<0.05, ***P<0.0005, results from unpaired t tests. All experiments utilized at least 3 mice per group and genotype. Scale bars equal 1 mm (b) and 500 µm (P0 and P3) and 1 mm (P7, c). E, embryonic day; LV, lateral ventricle; P, postnatal day.
We first examined the time of onset of hydrocephalus in Bbs1M390R/M390R brains by examining hematoxylin and eosin (H&E) stained sections. Dilation of the lateral ventricles begins at P0-P3 in Bbs1M390R/M390R mice (Fig. 1c,d). Importantly, this timing is prior to the maturation of ependymal motile cilia, which occurs from P5–P1010,13,14 suggesting that the onset of hydrocephalus in Bbs1M390R/M390R mice occurs independently of ependymal motile cilia function (Fig. 1e).
Increased apoptosis and reduced proliferation in the brains of Bbs1M390R/M390R mice
Next we studied whether known causes of hydrocephalus contribute to the phenotype observed in Bbs1M390R/M390R mice. Evan’s blue dye injection showed no evidence of obstructive hydrocephalus (Supplementary Fig. 1). We found no evidence of excess CSF production in Bbs1M390R/M390R mice based on normal choroid plexus ultrastructure and normal CSF ion concentrations (Supplementary Fig. 1).
Consequently, we sought to identify other potential mechanisms that may contribute to the early development of the communicating hydrocephalus in Bbs1M390R/M390R mice. Previous work has shown that these mice have a small corpus striatum19. Moreover, BBS patients have reduced white and gray matter in periventricular regions31,32. Therefore, we examined apoptosis and cell proliferation in the periventricular regions in Bbs1M390R/M390R mice to determine whether cell loss contributes to the pathophysiology of neonatal hydrocephalus. Immunofluorescent analysis using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), a marker of apoptotic cells and Bromodeoxy-Uridine (BrdU), a marker of proliferating cells, identified TUNEL+ cells adjacent to the lateral ventricles and BrdU+ cells in the subventricular zone (SVZ) in both wild-type (WT) and Bbs1M390R/M390R mice (Fig. 2a). Quantitation revealed that Bbs1M390R/M390R mice have a two-fold increase in apoptosis and a 50% reduction in cell proliferation in the periventricular regions at P3 and P7 relative to WT mice (Fig. 2b).
Figure 2.
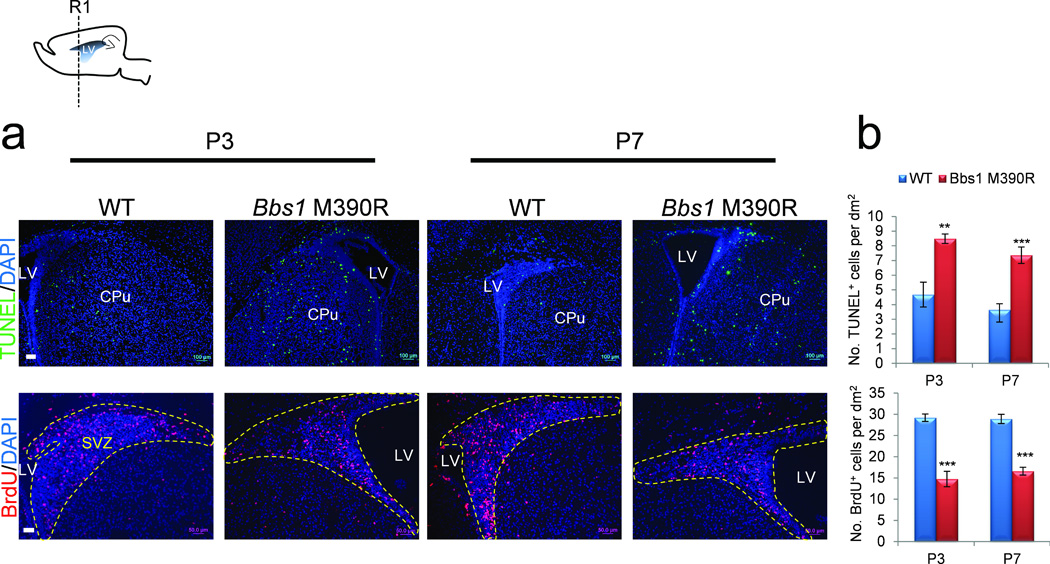
Increased apoptosis and reduced proliferation in the brains of Bbs1M390R/M390R mice. Top left, cartoon depicting the sagittal section of a mouse brain showing the region of subsequent analyses (R1). (a) Representative immunofluorescent images and (b) the quantitation of cells labeled with TUNEL (top row) or BrdU (bottom row) per area in P3 and P7 WT and Bbs1M390R/M390R brains in at least 3 mice per group and genotype (dotted yellow line outlines SVZ). All error bars represent means ± s.e.m. *P<0.05, **P<0.005, ***P<0.0005, results from unpaired t tests. Scale bars equal 100 µm (a, top) and 50 µm (bottom). CPu, caudate putamen; LV, lateral ventricle; NS, not significant; P, postnatal day; SVZ, subventricular zone.
Abnormal development of NG2+PDGFRα+ neural progenitor cells in Bbs1M390R/M390R mice
We next investigated the cell type responsible for the imbalance in apoptosis and cell proliferation by examining a number of markers of NPCs, neurons, and glia. Using immunofluorescence, we double stained coronal brain slices from WT and Bbs1M390R/M390R neonates with either TUNEL or BrdU and markers of these major cell types. We found no significant differences between WT and Bbs1M390R/M390R brains in the number of TUNEL labeled cells also staining positive for markers of developing (Nestin) and mature neurons (NeuN), astrocytes (GFAP) and oligodendrocytes (O4, refs. 33–35; Fig. 3a and Supplementary Fig. 2). Notably, we observed that nearly all TUNEL+ cells in both WT and Bbs1M390R/M390R mice expressed NG2 and PDGFRα, two markers of oligodendrocyte precursor cells (OLPs)36–40. Quantification revealed that a larger proportion (>two-fold) of TUNEL labeled cells are also NG2+ and PDGFRα+ in Bbs1M390R/M390R mice compared to WT (Fig. 3a). NG2 and PDGFRα have been previously shown to mark a particular class of OLPs expressed early in the lineage that are termed NG2+PDGFRα+ NPCs36–40. We also examined Olig2, a sonic hedgehog induced basic helix loop helix transcription factor expressed later in the oligodendrocytic lineage than NG2 and PDGFRα, which are both rapidly downregulated when differentiation to oligodendrocytes occurs36–39. We found no significant overlap between TUNEL+ and Olig2+ cells indicating that in both WT and Bbs1M390R/M390R mice more mature OLPs and oligodendrocytes are not undergoing apoptosis (Fig. 3a). Quantitation revealed no significant difference between WT and Bbs1M390R/M390R mice in the number of TUNEL+ cells also labeled with Olig2 (P=0.29, Fig. 3a)
Figure 3.
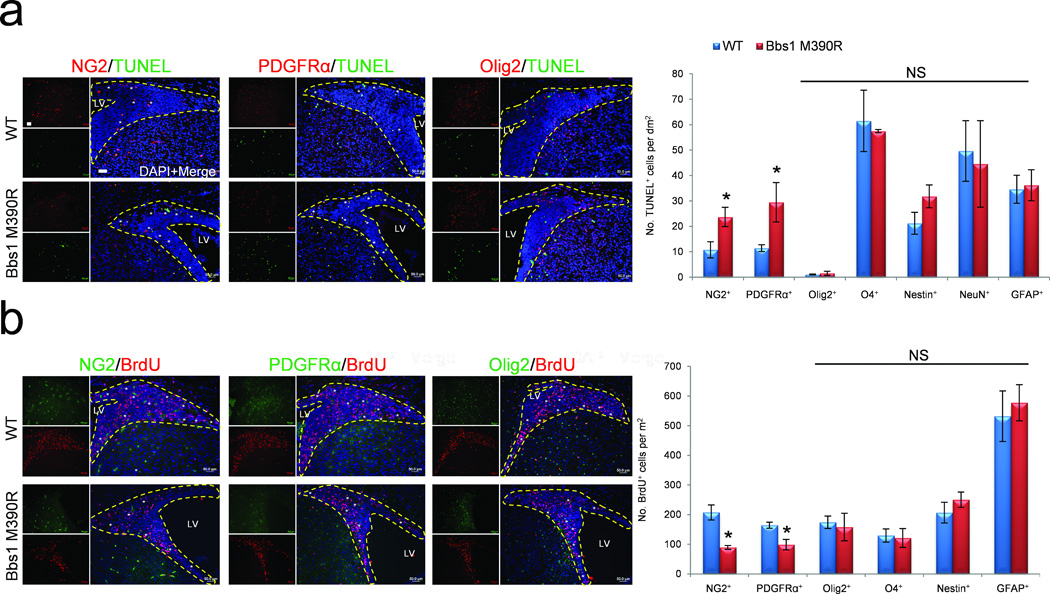
Abnormal development of NG2+PDGFRα+ neural progenitor cells in Bbs1M390R/M390R mice. (a,b) Representative immunofluorescent images showing TUNEL (a) and BrdU (b) labeled cells also expressing NG2, PDGFRα or Olig2 plus the quantitations (a,b, right). At least 3 mice per group and genotype were analyzed. All dotted yellow lines outline the SVZ. All error bars represent means ± s.e.m. *P<0.05, results from unpaired t tests. Scale bars equal 50 µm. CPu, caudate putamen; LV, lateral ventricle; NS, not significant; P, postnatal day; SVZ, subventricular zone.
Next we investigated the identity of the proliferating cells. We observed that NG2+, PDGFRα+ and Olig2+ OLPs constituted a large portion of BrdU+ cells in the SVZ of WT mice (Fig. 3b). However, in Bbs1M390R/M390R brains there was approximately 50% fewer BrdU labeled NG2+PDGFRα+ cells in the SVZ (Fig. 3b). WT and BBS mutant mice did not differ in the number of Olig2+ cells undergoing cell proliferation (BrdU+) (P=0.39, Fig. 3b). There was also no significant difference between WT and Bbs1M390R/M390R mice with respect to the other cell markers examined (Fig. 3b and Supplementary Fig. 2).
We then quantified the number of NG2+, PDGFRα+ and Olig2+ cells within the SVZ in order to determine the effect of impaired survival and proliferation in these precursor cells. We found significantly fewer of these cell populations in the SVZ of Bbs1M390R/M390R brains relative to WT (Supplementary Fig. 3).These results demonstrate that NG2+PDGFRα+ NPCs display increased apoptosis and reduced proliferation leading to reduced OLP populations in the brains of Bbs1M390R/M390R mice.
Conditional knockout of Bbs1 in PDGFRα+ cells leads to neonatal hydrocephalus
To confirm the involvement of NG2+PDGFRα+ NPCs in the genesis of neonatal hydrocephalus in BBS, we generated conditional knockout mice lacking Bbs1 in PDGFRα expressing NPCs (Bbs1loxP/loxP×PDGFRαCre; Bbs1CKO). Bbs1 mRNA was almost completely absent in cortex and significantly reduced in the hypothalamus of Bbs1CKO mice; moreover, Cre was expressed in NG2+, PDGFRα+ and Olig2+ NPCs, but not in the ependymal cells lining the ventricles (Supplementary Fig. 4a,b). These findings indicate that Bbs1 knockout in Bbs1CKO mice is specific to a particular class of periventricular NPCs in the cortex and hypothalamus. Bbs1CKO mice exhibit hydrocephalus with an onset at P3 in the absence of obstruction (Fig. 4a–d and Supplementary Fig. 4c,d). Moreover, neonatal hydrocephalus in Bbs1CKO mice was 100% penetrant. TUNEL and BrdU staining revealed a two-fold increase in apoptotic cells (Fig. 4e,f, top) and a 50% reduction in proliferating cells in Bbs1CKO mice relative to PDGFRαCre mice (controls) (Fig. 4g,h, top). We found that nearly all TUNEL+ and a majority of BrdU+ cells in the SVZ in PDGFRαCre and Bbs1CKO mice also stained positive for NG2 and PDGFRα, and to a lesser extent, Olig2 (Supplementary Fig. 5a,b). Quantitation revealed an approximately two-fold increase and 50% reduction in the number of TUNEL and BrdU labeled NG2+PDGFRα+ cells respectively in Bbs1CKO brains relative to control brains (Fig. 4f,h, bottom). No significant differences were found in the number of Olig2 labeled cells undergoing apoptosis (TUNEL+, P=0.32) or replication (BrdU+, P=0.22, Fig. 4f,h, bottom). These results demonstrate that the normal development of NG2+PDGFRα+ NPCs is disrupted following Bbs1 knockout in this specific cell type. Moreover, these results confirm the involvement of NG2+PDGFRα+ NPCs in the development of normal cerebral ventricles, disruption of which results in neonatal hydrocephalus.
Figure 4.
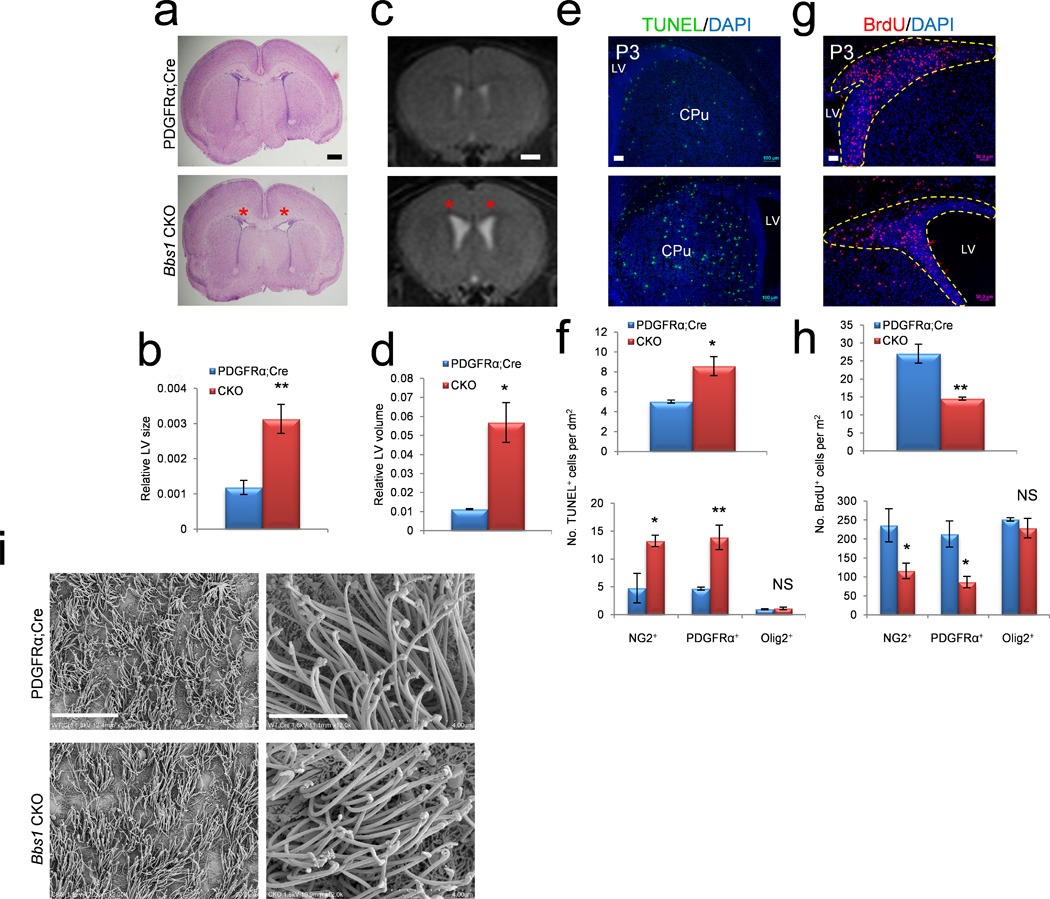
Conditional knockout of Bbs1 in NG2+PDGFRα+ progenitors causes neonatal hydrocephalus. (a,b) Representative histology of P3 PDGFRαCre (control) (n=3) and Bbs1CKO (n=3) pups and the quantitations showing dilated lateral ventricles of Bbs1CKO mice (red asterisks). (c,d) Representative T2-weighted MRIs of 3 month old PDGFRαCre (n=3) and Bbs1CKO (n=3) mice and the quantitations showing dilated ventricles in Bbs1CKO mice (red asterisks). (e–h) Representative immunofluorescent images and the quantitations of cells labeled with TUNEL (e,f) or BrdU (g,h) per area in P3 PDGFRαCre (n=3) and Bbs1CKO (n=3) brains (yellow dotted line outlines SVZ). (f,h, bottom) Quantifications of TUNEL (f, bottom) and BrdU (h, bottom) labeled cells that also express NG2, PDGFRα or Olig2 (at least 3 mice per group and genotype). (i) Scanning electron micrographs of the lateral wall of the lateral ventricles in PDGFRαCre and Bbs1CKO mice at low (left) and high (right) magnification. All error bars represent means ± s.e.m. *P<0.05, **P<0.005, ***P<0.0005, results from unpaired t tests. Scale bars equal 500 µm (a), 1 mm (c), 100 µm (e), 50 µm (g), 20 µm (left) and 4 µm (right, i). CPu, caudate putamen; CKO, conditional knockout; LV, lateral ventricle; NS, not significant; P, postnatal day; SVZ, subventricular zone.
Finally, we investigated whether dysfunctional motile cilia could contribute to the dilated ventricles observed in Bbs1CKO brains. We examined the ultrastructure of motile cilia in PDGFRαCre and Bbs1CKO brains at P14 and three months of age and found no abnormalities in the ultrastructure or number of tufts of motile cilia in the lateral ventricles of Bbs1CKO brains (Fig. 4i). This finding provides further evidence that hydrocephalus in BBS is caused by motile cilia-independent processes.
Bbs1 is required for PDGFRα signaling in NG2+PDGFRα+ neural progenitor cells
Next we examined cellular signaling pathways to assess the cause of impaired survival and proliferation of NG2+PDGFRα+ NPCs. We studied the PDGFRα signaling pathway because this pathway plays a major role in the survival and proliferation of NG2+PDGFRα+ NPCs36–40. We cultured primary OLPs from WT and Bbs1M390R/M390R neonates. Treatment of WT cultures with PDGFα, which specifically binds to PDGFRα, resulted in a large increase in the phosphorylation of PDGFRα and the two downstream effector proteins, AKT, master regulator of cell survival and proliferation and GSK3β, which regulates cell proliferation41–43 (Fig. 5a). The specificity of the response to PDGFα in WT cultures was shown by pretreatment with the PDGFR inhibitor, AG1296, which reduced the response to PDGFα. Bbs1M390R/M390R derived OLP cultures showed a blunted phosphorylation response to PDGFα stimulation (Fig. 5a).
Figure 5.
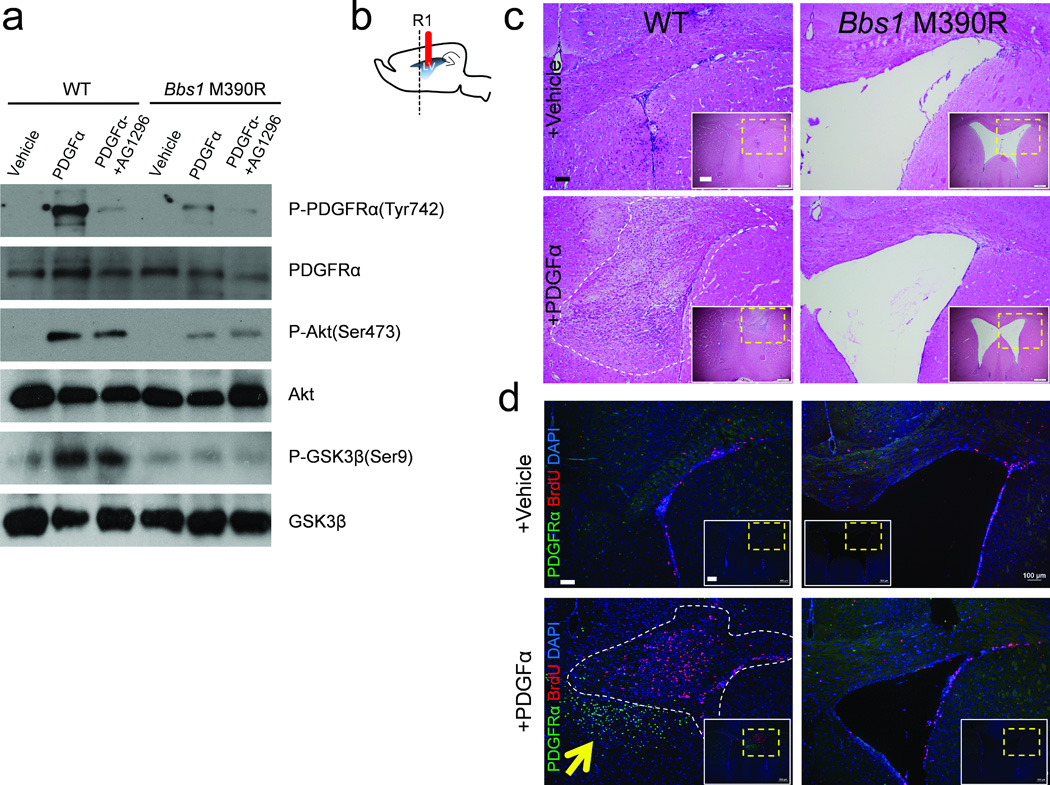
PDGFRα signaling is impaired in BBS. (a) PDGFα stimulation activates downstream signaling in primary oligodendrocytic precursor cells derived from WT but not Bbs1M390R/M390R brains. (b) Cartoon depicting the sagittal section of a mouse brain showing the cannula implantation site (solid red line) and the region of subsequent analyses (R1). (c) Representative histology of the ipsilateral brain hemisphere of vehicle infused WT (n=7) and Bbs1 KI (n=7); and PDGFα infused WT (n=7) and Bbs1M390R/M390R (n=7) mice (bottom). White panels show a low magnification image with the region of interest outlined by the yellow dotted line. (d) Representative immunofluorescent images of the ipsilateral infused hemisphere showing cells labeled with BrdU and PDGFRα in vehicle infused WT (n=3) and Bbs1M390R/M390R (n=3); and PDGFα infused WT (n=3) and Bbs1M390R/M390R (n=3) mice. The yellow arrow highlights the hyperplastic nodule consisting of PDGFRα+ cells and the white dotted line outlines the hyperplastic nodules observed in PDGFα infused WT mice. Scale bars equal 100 µm (larger image) and 500 µm (c, inset) and 100 µm (larger image) and 500 µm (d, inset).
In order to confirm these findings in vivo, we infused PDGFα into the lateral ventricles of control and mutant mice for six days. All infused WT mice developed atypical hyperplasias in either the medial or lateral wall of the ipsilateral ventricle while none of the treated Bbs1M390R/M390R mice showed this response (Fig. 5b,c). Furthermore, the hyperplastic nodules in WT infused brains contain a large proportion of small, round proliferating (BrdU+) cells (Fig. 5c,d). Immunostaining also identified a large increase in the population of PDGFRα+ NPCs in the medial wall of the PDGFα infused lateral ventricle in WT mice but not in Bbs1M390R/M390R mice (Fig. 5d). These findings demonstrate that Bbs1M390R/M390R NG2+PDGFRα+ NPCs do not respond to PDGFα and implicate a mechanism underlying the impaired survival and proliferation of NPCs in BBS. To study the role of BBS proteins in PDGFRα signaling we performed immunoprecipitation experiments. In vitro and in vivo experiments revealed that PDGFRα physically interacts with the BBSome (Supplementary Fig. 6a,b). These findings indicate a mechanism underlying the impaired PDGFRα signaling in BBS.
Lithium therapy rescues hydrocephalus through a GSK3β dependent mechanism
We attempted to modify the neonatal hydrocephalic phenotype in Bbs1M390R/M390R mice during the critical perinatal period by targeting defective PDGFRα signaling and OLP development. Lithium has been shown to enhance proliferation and promote survival of NPCs by stimulating phosphorylation of two downstream effectors in the PDGFRα pathway, AKT and GSK3β44–46. We treated Bbs1M390R/M390R heterozygous pregnant females, which had been previously mated to heterozygous male mice, with lithium chloride (LiCl) or an equimolar NaCl solution, administered in drinking water beginning at E14.5 (Fig. 6a). Histological analysis of P14 WT and Bbs1M390R/M390R brains revealed that lithium treatment in WT mice has no significant effect (NaCl WT vs. LiCl WT, P=0.17), whereas lithium treatment of Bbs1M390R/M390R mice (n=9) results in an approximately 50% reduction in the cross sectional area of the lateral ventricles relative to NaCl treated Bbs1M390R/M390R mice (n=5; Fig. 6a,b, top and c, left). Magnetic resonance imaging (MRI) at 3 months of age revealed that the ventricular volume of lithium treated WT mice does not differ from control treated WT mice (P=0.16, Fig. 6b, bottom and c, right). However, lithium treated Bbs1M390R/M390R mice show an approximately 50% reduction in ventricle volume relative to control treated Bbs1M390R/M390R mice (Fig. 6b, bottom and c, right). These effects of lithium were observed in all treated Bbs1M390R/M390R mice.
Figure 6.
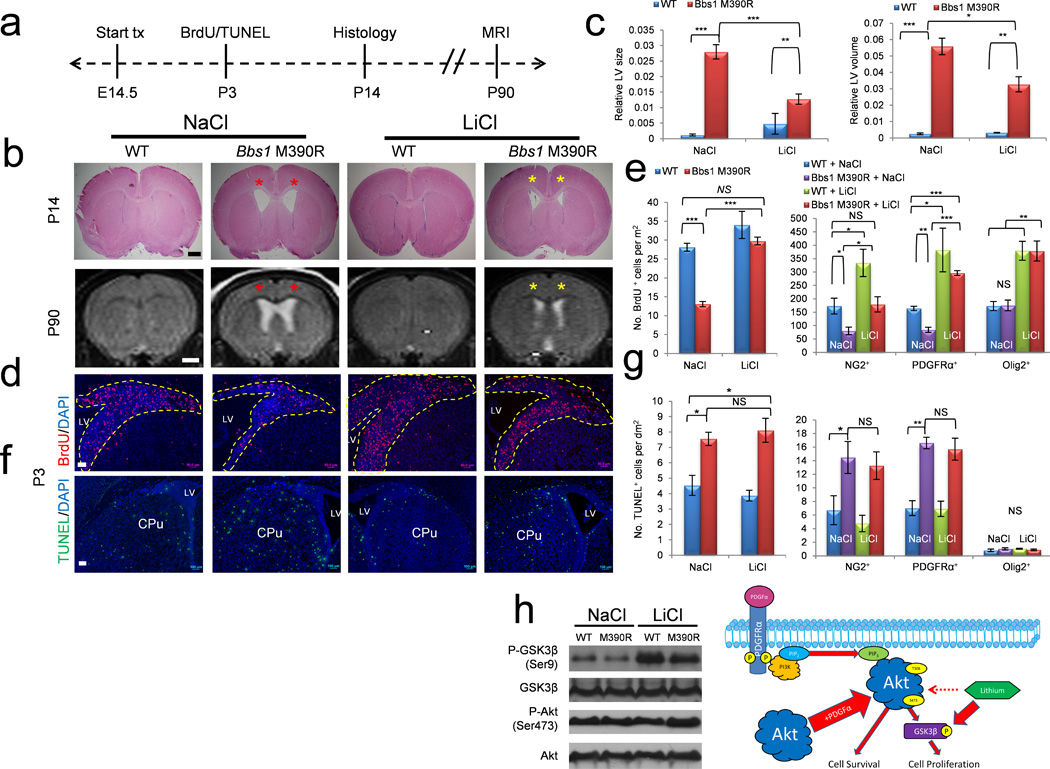
Lithium therapy rescues cell proliferation and hydrocephalus in Bbs1M390R/M390R mice. (a) Overview of the experimental timeframe. (b) Top row, representative histology plus quantitation (c, left) and representative MRI scans (bottom row) plus quantitation (c, right) of NaCl and LiCl treated WT and Bbs1M390R/M390R mice. Red asterisks highlight dilated lateral ventricles and yellow asterisks highlight reduced ventricle size following LiCl treatment in Bbs1M390R/M390R mice. (d,e) Representative immunofluorescent images of BrdU labeled cells in NaCl and LiCl treated mice (d) and quantitations (e, left). (e) Right, quantitation of BrdU labeled cells that also express NG2, PDGFRα or Olig2. (f,g) Representative immunofluorescent images (f) and quantitation (g, left) of TUNEL labeled cells in NaCl and LiCl treated mice. (g) Right, quantitation of TUNEL labeled cells expressing NG2, PDGFRα or Olig2. (h) Left, representative western blots of cortices from P3 NaCl and LiCl treated WT and Bbs1M390R/M390R mice. Right, drawing depicting the proposed mechanism of lithium’s effect on cell proliferation in treated WT and Bbs1M390R/M390R mice. Lithium increases phosphorylation of GSK3β leading to an increase in cell proliferation. We found no effect on the phosphorylation of AKT in contrast to previously reported results44. Error bars represent means ± s.e.m. *P<0.05, **P<0.005, ***P<0.0005, results from unpaired t tests. All experiments utilized ≥ 3 mice per group and genotype. Scale bars equal 500 µm (top, b) and 1 mm (bottom, b), 50 µm (d) and 100 µm (f). CPu, caudate putamen; E, embryonic day; LV, lateral ventricle; NS, not significant; P, postnatal day; SVZ, subventricular zone.
To study the mechanism underlying the lithium effect on neonatal hydrocephalus we examined cell proliferation and apoptosis in the periventricular regions of P3 mice using BrdU and TUNEL assays, respectively. Staining revealed that lithium has no significant effect on the number of proliferating or apoptotic cells in WT mice (NaCl WT vs. LiCl WT; BrdU, P=0.29; TUNEL, P=0.21; Fig. 6d–g). However, lithium treatment results in an approximately two-fold increase in proliferating cells in Bbs1M390R/M390R mice relative to NaCl treated Bbs1M390R/M390R (Fig. 6d,e, left). Importantly, there is no significant difference in the number of proliferating cells in NaCl treated WT and lithium treated Bbs1M390R/M390R mice indicating a complete rescue of the cell proliferation defect (P=0.16, Fig. 6d,e, left). There is also no significant difference in the number of apoptotic cells in NaCl and lithium treated Bbs1M390R/M390R mice indicating that the effect of lithium is specific to cell proliferation (P=0.29, Fig. 6f,g, left). Furthermore, lithium treatment specifically rescued NG2+PDGFRα+ NPC proliferation but not apoptosis in Bbs1M390R/M390R mice (NaCl vs. LiCl Bbs1M394R/M394R, NG2+, P=0.36; PDGFRα+, P=0.38) (Fig. 6e,g, right and Supplementary Figs. 7 and 8). Lithium also increased the number of proliferating Olig2+ cells in WT and Bbs1M390R/M390R mice by more than two-fold (Fig. 6e, right and Supplementary Fig. 7).
To determine the molecular mechanisms underlying the effects of lithium, we examined the phosphorylation of AKT and GSK3β. Western blot analysis revealed that lithium treatment increases phosphorylation of GSK3β yet has no effect on the phosphorylation of AKT (Fig 6h). These results indicate that defective NG2+PDGFRα+ NPC development leads to neonatal hydrocephalus in Bbs1M390R/M390R mice. Targeting the defective PDGFRα signaling pathway in these NPCs rescues the proliferation of these cells resulting in a partial rescue of hydrocephalus.
DISCUSSION
Hydrocephalus is a complex disorder involving both multiple genetic and environmental components3,4,9. Thus any single pathway or mechanism will not fully explain all of hydrocephalus. Previous studies implicate impaired ependymal motile cilia, CSF overproduction and cortical atrophy in the genesis of communicating hydrocephalus4,10,13–16,20,47. Here we demonstrate that NG2+PDGFRα+ NPCs play a key role in the pathogenesis of hydrocephalus. We found that impaired PDGFRα signaling in Bbs1M390R/M390R mice leads to increased apoptosis and reduced proliferation of NG2+PDGFRα+ NPCs resulting in hydrocephalus. We have also demonstrated that dysfunctional motile cilia are not the primary cause of neonatal hydrocephalus in BBS mouse models as evidenced by ventricular dilation occurring prior to the development of motile cilia and that ependymal cilia remain intact in mice lacking Bbs1 in PDGFRα+ cells. We have not excluded the possibility that motile cilia defects may contribute to the severity of the phenotype in older Bbs1M390R/M390R mice. These findings steer the study of hydrocephalus beyond the ciliopathy field as evidenced by the common theme of abnormal cellular signaling in other models of hydrocephalus14–16,48–51.
Although we found that NG2+PDGFRα+ cells exhibit impaired survival and proliferative capacities, we also found that Olig2+ cells appear normal despite both cell types existing within the oligodendrocytic lineage. This finding suggests that Bbs1 plays an essential role in the survival and proliferative capacities of NG2+PDGFRα+ cells but not Olig2+ cells. The abnormal survival and proliferative capacities and the reduced populations of NG2+PDGFRα+ NPCs in periventricular regions may explain recent observations that BBS patients and mouse models have reduced white and gray matter volume in periventricular subcortical structures19,31,32. The MRI findings in patients suggest a loss of cerebral tissue as a cause of ventriculomegaly31,32. Our data indicate that the underlying cause of reduced cerebral tissue and ventricular dilation in BBS patients is due to impaired survival and proliferation of NPCs rather than degeneration of mature neurons and glia.
The impaired PDGFRα signaling in BBS led us to explore the therapeutic potential of targeting this pathway to modify the hydrocephalic phenotype early in development. We targeted two downstream effector proteins in the PDGFRα signaling cascade, AKT and GSK3β, using lithium, a drug that stimulates phosphorylation of these proteins44–46,52. Phosphorylation of AKT and GSK3β has been shown to increase cell survival and cell proliferation41–43. We found that lithium treatment selectively rescues NG2+PDGFRα+ cell proliferation by stimulating phosphorylation of GSK3β leading to a reduction in the size of the dilated ventricles in Bbs1 mutant mice (Fig. 6h). These results are consistent with those of previous studies demonstrating that lithium stimulates NPC proliferation by increasing phosphorylation of GSK3β thereby suppressing its activity44–46,52. Although lithium rescues cell proliferation, it has no significant effect on cell death of NG2+PDGFRα+ NPCs. This finding may explain the partial rescue of the hydrocephalic phenotype. Rescue of both cell proliferation and apoptosis may result in a further reduction in ventricular size. To our knowledge, we are the first to target NPC development as a therapy to treat hydrocephalus in any model organism.
Finally, our results demonstrate that BBS proteins play a crucial role in PDGFRα signaling and NG2+PDGFRα+ NPC development in the CNS. The trafficking function of the BBSome, is disrupted in BBS mutant mice30. As a result, signaling proteins are mislocalized leading to an abnormal cellular response in BBS mutant mice23,30,53,54. Our findings suggest that the aberrant PDGFRα signaling observed in Bbs1M390R/M390R mice originates from the mistrafficking of PDGFRα based on our observations that PDGFRα interacts with primary components of the BBSome and hence, is likely a novel cargo protein of the BBSome. However, the exact mechanism underlying the PDGFRα signaling defects in BBS remains to be elucidated.
By targeting GSK3β we rescued the development of NG2+PDGFRα+ NPCs and hydrocephalus in Bbs1M390R/M390R mice. The strategy of targeting downstream effectors of signaling defects may be applicable to other BBS-associated phenotypes.
online Methods
Animals
We used male and female mice on pure 129/SvEv genetic background for Bbs1M390R/M390R mice and littermate controls. We generated PDGFRαCre (control) and Bbs1CKO mice by crossing Bbs1loxP/loxP (129/SvEv) × PDGFRαCre (C57BL/6NJ) (Jackson Laboratory, Bar Harbor, Maine). We used littermate and aged matched controls for all animal experiments. Animals were generated at the University of Iowa Carver College of Medicine and all experiments were performed in accordance with the Institute for Animal Care and Use Committee at the University of Iowa (Iowa City, Iowa).
Magnetic Resonance Imaging (MRI)
We obtained human T2-weighted MRI scans from K.M.K. and P.N. at the University of Iowa and Leslie Biesecker at the Clinical Research Center at the National Institutes of Health (NIH) in Bethesda, MD, USA. Informed consent was obtained for each human subject. Human subject research was approved by the by both the Institutional Review Board (IRB) of the National Human Genome Research Institute (NHGRI), at the National Institutes of Health (NIH), and by the IRB of the University of Iowa. For mice, we performed T2-weighted MRI scans while each mouse was under isoflurane induced anesthesia using a Varian Unity/Inova 4.7 T small-bore MRI system (Varian, Inc., Palo Alto, California) at the University of Iowa. There was an in-plane resolution of 0.13 ´ 0.25 mm2 and an approximate slice thickness of 0.6 mm acquired in the axial, sagittal, and coronal planes.
Histology, immunohistochemistry, BrdU and TUNEL assays
We sectioned and stained mouse brains fixed by intracardiac perfusion with 4% paraformaldehyde at 7 µm with hematoxylin and eosin. All mouse brains derived from fresh frozen tissue were cryosectioned (8 µm sections). For BrdU experiments, we injected mice intraperitoneally with 300mg/kg BrdU (Sigma, St. Louis, Missouri), exposed for four hours then subsequently sacrificed. We double stained cryosections with either an antibody to BrdU (1:200, Abcam) or Click-iT TUNEL assay (Life Technologies, Grand Island, New York) according to the manufacturer’s protocols and with the following antibodies: mouse Nestin (1:200; Abcam, Cambridge, Massachusetts), mouse O4 (1:200, Millipore), mouse NeuN (1:200, Millipore), rabbit GFAP (1:500, Abcam), rabbit NG2 (1:400, Millipore), rabbit PDGFRα (1:200, Santa Cruz Biotechnology Inc., Santa Cruz, California) and rabbit Olig2 (1:800, Abnova, Walnut, California). We blocked sections with 1% fetal calf serum (Life Technologies) plus 0.3% Triton X-100 (Sigma) in PBS for 1 hour at 25 °C, before primary antibody incubation at 4 °C overnight or at 25 °C for one hour. We used goat Alexa-Fluor secondary antibodies (Life Technologies) to image primary antigens and nuclei were counterstained with Vectashield containing DAPI (Vector Laboratories, Burlingame, California).
Cell Culture
We cultured oligodendrocyte precursor cells (OLPs) from dissociated cortices of P0 mice. We dissociated cortices in trypsin using a P1000 pipette after which we added DMEM (Gibco, Grand Island, New York) supplemented with 20% fetal calf serum (FCS, Gibco) and 1% pen-strep (PS, Gibco). We then plated cells in T25 culture flasks (Corning) and cultured these cells for 7 days at which point cells were shaken overnight in 37ºC at 250rpm to separate OLPs. At this point nearly all of the attached cells were astrocytes and the unattached cells were OLPs. Next we removed the media containing unattached OLPs and plated them on plastic culture dishes (Corning Inc., Corning, New York). We maintained all cells for an additional 2 days or until confluent using DMEM supplemented with 10% FCS and 1% PS. Prior to treatment, all cells were serum starved for 16 hours.
PDGFα treatment of cells and mice
For receptor activation analysis in vitro, we treated cells with 50ng/mL PDGFα (Cell Signaling Technologies, Danvers, Massachusetts) for 10 minutes following pretreatment with 8µM AG1296 (Cayman Chemical Company, Ann Arbor, Michigan) for 30 minutes. For in vivo experiments we infused PDGFα (80ng/day) in vehicle (1mg/mL BSA/PBS) or vehicle alone into the lateral ventricle of 3MO mice for 6 days using a miniosmotic pump per manufacturer’s instructions (Azlet).
Immunoprecipitation and western blotting
We performed in vitro immunoprecipitations as previously described23. For endogenous immunoprecipitation experiments, we isolated brain cortices from P3 mice, lysed tissues in RIPA buffer containing protease and phosphatase inhibitors (Roche Applied Science, Indianapolis, Indiana). and pre-cleared lysates with protein G beads (Thermo Fisher Scientific, Rockford, Illinois) overnight at 4 oC. We added a goat antibody to PDGFRα (1:100, R&D Systems, Minneapolis, Minnesota) and incubated overnight at 4 oC. We next incubated samples with protein G beads for 4 hours at 4 oC then washed briefly. We performed SDS-page and western blotting as described previously35 using 10 µg total protein per lane. We performed western blotting using the SuperSignal West Pico kit (Pierce Biotechnology, Rockford, Illinois) per manufacturer’s instructions. We blocked membranes for one hour in TBST+5% milk or BSA then incubated overnight at 4ºC with the following primary antibodies: mouse Flag (Sigma), mouse GFP (Santa Cruz), rabbit BBS230, rabbit BBS430, rabbit BBS8 (Sigma), rabbit BBS9 (Sigma), rabbit phospho-PDGFRα (Tyr720) (1:1000, Sigma), goat PDGFRα (1:5000) and the following antibodies from Cell Signaling Technologies: rabbit phospho-AKT (Ser473) (1:5000), rabbit AKT (1:1000),rabbit phospho-GSK3β (Ser9) (1:1000) and rabbit GSK3β (1:1000).
qRT-PCR experiments
We sacrificed mice by CO2 asphyxiation, and we excised four brain regions including cortex, hypothalamus, hippocampus and cerebellum. We extracted mRNA using Trizol reagent (Life Technologies). We performed Real-time RT-PCR to compare the mRNA levels of BBS1 between the control and BBS1 conditional knockout tissues. We used the following BBS1 primers: 5′-TTCTGCAGCTGGAGCTGAGTG-3′ (forward), 5′-TCAGTGCCTAGCACCAGACAG-3′ (reverse); RPL19 was used as an internal control.
Lithium therapy
We treated mice with lithium chloride (Sigma) in drinking water using a dose of 1.2 g/L starting from E14.5 (through pregnant and nursing dams). The drug was dissolved in double distilled water.
Electron microscopy
We obtained scanning electron micrographs of the lateral wall of the lateral ventricles dissected from mice using a Hitachi S-4800 scanning electron microscope (Hitachi, Pleasanton, California). We also obtained transmission electron micrographs of choroid plexi using a Gatan UltraScan 1000 (Pleasonton, California) 2kx2k CCD digital camera.
Dye injection and visualization of CSF flow
We anesthetized adult mice and injected Evans Blue dye into the lateral ventricles. The syringe was left in for 20 minutes to prevent CSF loss and to allow CSF to circulate. We then euthanized and cryopreserved mice at –20 oC. The next day, frozen heads were cut in the sagittal plane and imaged.
CSF collection and analysis
We anesthetized adult mice and inserted a glass micropipette into the cisterna magna for CSF collection. We froze all collected samples at –20 oC until analysis. We measured the chloride and sodium ion content using ion specific electrodes (Roche/Hitachi, Indianapolis, Indiana) in the Pathology Laboratory of the University of Iowa Hospitals and Clinics.
Image analysis and statistics
We cropped all images and adjusted the brightness/contrast using Adobe Photoshop CS5. For immunofluorescent images, we adjusted the brightness/contrast using Zen Light. We performed all cell quantifications, cross-sectional area and volumetric analyses using ImageJ (NIH). We performed statistical comparisons using unpaired t tests. Mean values ± standard error of the mean (s.e.m.) are reported.
Supplementary Material
Acknowledgements
We thank Leslie Biesecker for help obtaining the human MRI scans. We thank Kamal Rahmouni and Deng-Fu Guo for help with qRT-PCR and infusion experiments. We thank Khristofor Agassandian for help with dye injection and CSF collection. We thank Valerie Buffard and Lan Qian for their excellent technical assistance. We also appreciate valuable assistance from the University of Iowa Central Microscopy Research Facility. This work was supported in part by National Institutes of Health Grants R01EY110298 and R01EY017168 (to V.C.S.) and the Neurosurgery Research and Education Foundation to (to T.W.V.). C.S.C. is a National Science Foundation graduate research fellow and V.C.S. is an Investigator of the Howard Hughes Medical Institute.
This work has not been previously published in whole or in part or submitted elsewhere for review.
Footnotes
Author Contributions. C.S.C., T.W.V. and Q.Z. conceived of the project, designed, and performed experiments, coordinated collaborations, and wrote the manuscript. S.S. contributed to the experimental design and manuscript revisions. R.E.S. and M.C. performed TEM, CSF collection and dye injection experiments and revised the manuscript. T.O.M. coordinated microscopic experiments. K.M.K. and P.N. provided and analyzed human MRIs. D.R.T. performed magnetic resonance imaging for all mice. D.Y.N. and C.C.S. designed and developed the Bbs1 mouse model used in this experiment. K.B. coordinated mouse genotyping and mating. V.C.S. initiated the project, contributed ideas, analyzed and interpreted the results, and helped write the manuscript.
References
- 1.Bruni JE, Del Bigio MR, Clattenburg RE. Ependyma: Normal and pathological. A review of the literature. Brain Research Reviews. 1985;9:1–19. doi: 10.1016/0165-0173(85)90016-5. [DOI] [PubMed] [Google Scholar]
- 2.Del Bigio M. Ependymal cells: biology and pathology. Acta Neuropathologica. 2010;119:55–73. doi: 10.1007/s00401-009-0624-y. [DOI] [PubMed] [Google Scholar]
- 3.Williams MA, et al. Priorities for hydrocephalus research: report from a National Institutes of Health–sponsored workshop. Journal of Neurosurgery: Pediatrics. 2007;107:345–357. doi: 10.3171/PED-07/11/345. [DOI] [PubMed] [Google Scholar]
- 4.Vogel P, et al. Congenital Hydrocephalus in Genetically Engineered Mice. Veterinary Pathology Online. 2012;49:166–181. doi: 10.1177/0300985811415708. [DOI] [PubMed] [Google Scholar]
- 5.Simon TD, et al. Hospital care for children with hydrocephalus in the United States: utilization, charges, comorbidities, and deaths. Journal of Neurosurgery: Pediatrics. 2008;1:131–137. doi: 10.3171/PED/2008/1/2/131. [DOI] [PubMed] [Google Scholar]
- 6.Shannon CN, et al. The economic impact of ventriculoperitoneal shunt failure. Journal of Neurosurgery: Pediatrics. 2011;8:593–599. doi: 10.3171/2011.9.PEDS11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Camp G, et al. A duplication in the L1CAM gene associated with X-linked hydrocephalus. Nature genetics. 1993;4:421–425. doi: 10.1038/ng0893-421. [DOI] [PubMed] [Google Scholar]
- 8.Chi JH, Fullerton HJ, Gupta N. Time trends and demographics of deaths from congenital hydrocephalus in children in the United States: National Center for Health Statistics data, 1979 to 1998. Journal of neurosurgery. 2005;103:113–118. doi: 10.3171/ped.2005.103.2.0113. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Williams MA, Rigamonti D. Genetics of human hydrocephalus. Journal of neurology. 2006;253:1255–1266. doi: 10.1007/s00415-006-0245-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banizs B, et al. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development (Cambridge, England) 2005;132:5329–5339. doi: 10.1242/dev.02153. [DOI] [PubMed] [Google Scholar]
- 11.Patwardhan RV, Nanda A. Implanted ventricular shunts in the United States: the billion-dollar-a-year cost of hydrocephalus treatment. Neurosurgery. 2005;56:139–144. doi: 10.1227/01.neu.0000146206.40375.41. discussion 144-135. [DOI] [PubMed] [Google Scholar]
- 12.Vogel TW, Carter CS, Abode-Iyamah K, Zhang Q, Robinson S. The role of primary cilia in the pathophysiology of neural tube defects. Neurosurgical Focus. 2012;33:E2. doi: 10.3171/2012.6.FOCUS12222. [DOI] [PubMed] [Google Scholar]
- 13.Spassky N, et al. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:10–18. doi: 10.1523/JNEUROSCI.1108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tissir F, et al. Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nature neuroscience. 2010;13:700–707. doi: 10.1038/nn.2555. [DOI] [PubMed] [Google Scholar]
- 15.Talos F, et al. p73 is an essential regulator of neural stem cell maintenance in embryonal and adult CNS neurogenesis. Cell death and differentiation. 2010;17:1816–1829. doi: 10.1038/cdd.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang A, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 17.Drake JM. The surgical management of pediatric hydrocephalus. Neurosurgery. 2008;62(Suppl 2):633–640. doi: 10.1227/01.neu.0000316268.05338.5b. discussion 640-632. [DOI] [PubMed] [Google Scholar]
- 18.Drake JM, Kestle JR, Tuli S. CSF shunts 50 years on--past, present and future. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2000;16:800–804. doi: 10.1007/s003810000351. [DOI] [PubMed] [Google Scholar]
- 19.Davis RE, et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19422–19427. doi: 10.1073/pnas.0708571104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ibanez-Tallon I, et al. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Human molecular genetics. 2004;13:2133–2141. doi: 10.1093/hmg/ddh219. [DOI] [PubMed] [Google Scholar]
- 21.Lancaster MA, Schroth J, Gleeson JG. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nature cell biology. 2011;13:700–707. doi: 10.1038/ncb2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ocbina PJ, Eggenschwiler JT, Moskowitz I, Anderson KV. Complex interactions between genes controlling trafficking in primary cilia. Nature genetics. 2011;43:547–553. doi: 10.1038/ng.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Q, Seo S, Bugge K, Stone EM, Sheffield VC. BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes. Human molecular genetics. 2012;21:1945–1953. doi: 10.1093/hmg/dds004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider L, et al. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Current biology : CB. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annual review of neuroscience. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han YG, et al. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nature neuroscience. 2008;11:277–284. doi: 10.1038/nn2059. [DOI] [PubMed] [Google Scholar]
- 27.Breunig JJ, et al. Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13127–13132. doi: 10.1073/pnas.0804558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sawamoto K, et al. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science (New York, N.Y.) 2006;311:629–632. doi: 10.1126/science.1119133. [DOI] [PubMed] [Google Scholar]
- 29.Ihrie RA, Alvarez-Buylla A. Lake-front property: a unique germinal niche by the lateral ventricles of the adult brain. Neuron. 2011;70:674–686. doi: 10.1016/j.neuron.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nachury MV, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 31.Baker K, et al. Neocortical and hippocampal volume loss in a human ciliopathy: A quantitative MRI study in Bardet-Biedl syndrome. American journal of medical genetics. Part A. 2011;155A:1–8. doi: 10.1002/ajmg.a.33773. [DOI] [PubMed] [Google Scholar]
- 32.Keppler-Noreuil KM, et al. Brain tissue- and region-specific abnormalities on volumetric MRI scans in 21 patients with Bardet-Biedl syndrome (BBS) BMC medical genetics. 2011;12:101. doi: 10.1186/1471-2350-12-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Bohlen Und Halbach O. Immunohistological markers for staging neurogenesis in adult hippocampus. Cell and tissue research. 2007;329:409–420. doi: 10.1007/s00441-007-0432-4. [DOI] [PubMed] [Google Scholar]
- 34.Raponi E, et al. S100B expression defines a state in which GFAP-expressing cells lose their neural stem cell potential and acquire a more mature developmental stage. Glia. 2007;55:165–177. doi: 10.1002/glia.20445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson EL, et al. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006;51:187–199. doi: 10.1016/j.neuron.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 36.Rivers LE, et al. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nature neuroscience. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richardson WD, Young KM, Tripathi RB, McKenzie I. NG2-glia as multipotent neural stem cells: fact or fantasy? Neuron. 2011;70:661–673. doi: 10.1016/j.neuron.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tripathi RB, Rivers LE, Young KM, Jamen F, Richardson WD. NG2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:16383–16390. doi: 10.1523/JNEUROSCI.3411-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nature reviews. Neuroscience. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- 40.Kondo T, Raff M. Oligodendrocyte precursor cells reprogrammed to become multipotential CNS stem cells. Science (New York, N.Y.) 2000;289:1754–1757. doi: 10.1126/science.289.5485.1754. [DOI] [PubMed] [Google Scholar]
- 41.Datta SR, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 42.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nature reviews. Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 43.Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nature reviews. Molecular cell biology. 2001;2:760–768. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- 44.Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Su H, Chu TH, Wu W. Lithium enhances proliferation and neuronal differentiation of neural progenitor cells in vitro and after transplantation into the adult rat spinal cord. Experimental neurology. 2007;206:296–307. doi: 10.1016/j.expneurol.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 46.Li H, et al. Lithium-mediated long-term neuroprotection in neonatal rat hypoxia-ischemia is associated with antiinflammatory effects and enhanced proliferation and survival of neural stem/progenitor cells. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31:2106–2115. doi: 10.1038/jcbfm.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lechtreck KF, Delmotte P, Robinson ML, Sanderson MJ, Witman GB. Mutations in Hydin impair ciliary motility in mice. The Journal of cell biology. 2008;180:633–643. doi: 10.1083/jcb.200710162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goto J, Tezuka T, Nakazawa T, Sagara H, Yamamoto T. Loss of Fyn tyrosine kinase on the C57BL/6 genetic background causes hydrocephalus with defects in oligodendrocyte development. Molecular and cellular neurosciences. 2008;38:203–212. doi: 10.1016/j.mcn.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 49.Qin S, Liu M, Niu W, Zhang CL. Dysregulation of Kruppel-like factor 4 during brain development leads to hydrocephalus in mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:21117–21121. doi: 10.1073/pnas.1112351109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yung YC, et al. Lysophosphatidic acid signaling may initiate fetal hydrocephalus. Science translational medicine. 2011;3:99ra87. doi: 10.1126/scitranslmed.3002095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riviere JB, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nature genetics. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Azim K, Butt AM. GSK3beta negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia. 2011;59:540–553. doi: 10.1002/glia.21122. [DOI] [PubMed] [Google Scholar]
- 53.Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4242–4246. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seo S, et al. Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Human molecular genetics. 2009;18:1323–1331. doi: 10.1093/hmg/ddp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.