

Abstract
PURPOSE
To identify the chromosomal location of the gene involved in the pathogenesis of cavitary optic disc anomalies in a large pedigree with autosomal dominant inheritance of disease.
DESIGN
Linkage analysis of a pedigree affected with cavitary optic disc anomalies.
METHODS
Optic disc photographs were examined for the presence of cavitary optic disc anomalies. Sixteen affected family members and one obligate carrier were identified and studied with linkage analysis using both microarrays of single nucleotide polymorphisms (SNPs) and short tandem repeat polymorphism (STRP) markers.
RESULTS
Multipoint linkage analysis of SNP genotypes yielded a maximum nonparametric LOD score of 21.7 with markers located on chromosome 12q. Linkage was confirmed with 16 STRP markers in the 12q region. A maximum two point LOD score of 4.06 (Θ=0) was obtained with marker D12S1700. The disease interval defined by observed recombinants is 9.1 cM, which corresponds to 13.5 Mbp. Three candidate genes (GDF-11, NEUROD4, and WIF1) in the chromosome 12q locus were evaluated as possible disease-causing genes. No mutations were detected in the coding sequence of these genes.
DISCUSSION
The discovery of the chromosomal location of a gene responsible for cavitary optic disc anomalies is a key step in identifying the genetic basis of this condition and may ultimately provide important insight into the pathogenesis of more common optic nerve diseases such as normal tension glaucoma and primary open angle glaucoma.
INTRODUCTION
Excavation of the optic disc is the chief feature of some diseases of the optic nerve including adult-onset conditions which are progressive (the glaucomas) and congenital malformations of the optic nerve (optic pits, optic nerve coloboma, and morning glory disc anomaly) which are stationary. Optic pits, optic nerve coloboma, and morning glory disc anomaly have a similar appearance of the optic disc and are associated with a high frequency of serous retinal detachments. Consequently, these congenital conditions have been collectively referred to as cavitary optic disc anomalies.1, 2
Optic pits are focal depressions in the optic disc in which the normal tissue of the optic nerve is absent. Most optic pits are located in the temporal optic disc, however, they may also be seen centrally. Patients with optic pits may have excellent vision, however, schisis or splitting of the retina and serous retinal detachments of the macula frequently develop and are associated with severe vision loss. While the majority of optic pits are sporadic, autosomal dominant inheritance of this condition has been reported.3
Ocular colobomas are malformations due to defects in the closure of the optic cup fissure during embryogenesis, which may lead to structural defects in an inferonasal location involving the iris, ciliary body, lens, choroid, and optic nerve. If the optic nerve is involved in the coloboma, it is characterized by deep excavation of the optic disc and is frequently associated with serous retinal detachments of the macula and significant vision loss. Ocular colobomas are often part of “syndromes” which include other ophthalmic and systemic abnormalities. Many of these coloboma syndromes are hereditary and a subset of cases is caused by known genes4 including PAX6 (OMIM 607108),5 SHH (OMIM 600725),6 MAF, 7 and CHX10 (OMIM 142993).8 Although most cases involve multiple ocular structures, coloboma of the optic nerve may occur without involvement of any other ocular tissues. Isolated optic nerve coloboma is generally sporadic, however, familial cases have demonstrated autosomal dominant inheritance of this condition.9 No genes for isolated optic nerve coloboma have been identified.
The term “morning glory disc anomaly” was first used by Kindler,10 to describe a constellation of abnormal optic nerve features that have the appearance of a morning glory flower. The optic disc in this condition is enlarged and deeply excavated with an abnormal collection of centrally located glial tissue. Retinal vessels originating from the ciliary circulation radiate from the edge of the disc in an anomalous pattern. The optic disc has a funnel-shape and is surrounded by an elevated annulus of abnormal chorioretinal pigmentation. Morning glory disc anomaly is associated with an increased risk of retinal detachment and poor visual acuity. Most cases of morning glory disc anomaly are unilateral and sporadic, however a single bilateral case has been associated with a PAX6 mutation.5
While the majority of the different types of cavitary optic disc anomalies occur sporadically, rare cases have been reported in which optic pits, optic nerve colobomas, and morning glory disc anomaly have been observed within the same family.1, 2, 11 The gene associated with this heritable form of cavitary anomalies of the optic disc is unknown. In this study, a large family affected with a range of isolated cavitary anomalies of the optic disc was studied with genetic linkage analysis to identify the chromosomal location of the disease-causing gene.
METHODS
The study was approved by the University of Iowa’s Institutional Review Board and informed consent was obtained from study participants. Sixteen clinically affected family members had eye examinations (visual acuity, slit lamp examination, indirect ophthalmoscopy and retinal biomicroscopy) performed by one of the authors (RAH, MDM, or LMJ). Throughout the study, the clinicians remained masked to the genotypic data. Patients were included in the linkage analysis if: 1) they were found to have cavitary optic nerve abnormalities including optic pits, atypical optic nerve coloboma, or morning glory disc anomaly; or 2) they were obligate carriers, by virtue of having offspring with cavitary optic disc anomalies. Blood samples were obtained from all of the affected family members, obligate carriers, and spouses of affected patients with children. Seven to ten milliliters of blood were obtained from each patient in EDTA-containing glass tubes. DNA was prepared from the blood using a non-organic method.12
Pedigree members were first genotyped with short tandem repeat polymorphism (STRP) genetic markers flanking genes previously identified as ocular coloboma genes including PAX2 (OMIM 167409), PAX6, and SHH. Genotyping with STRP genetic markers was conducted using standard methods as previously described.13 A genome-wide scan was next performed with Affymetrix microarrays (GeneChip Human Mapping 10K Array Xba 2.0, Affymetrix, Santa Clara, CA) which interrogate 10,204 single nucleotide polymorphisms (SNPs). Sample processing and labeling were performed using the manufacturer’s instructions. The arrays were hybridized, washed, and scanned in the University of Iowa DNA core facility. Array images were processed with GeneChip DNA Analysis software.
Micro-array data were analyzed and multipoint non-parametric LOD scores were calculated using the Genespring GT software package (Agilent Technologies, Palo Alto, CA). Pairwise linkage analysis was performed with the MLINK and LODSCORE programs as implemented in the FASTLINK (v2.3) version14, 15 of the LINKAGE software package.16 Penetrance and disease gene frequency were set to 99% and 0.1% respectively. Multipoint linkage analysis was conducted using the VITESSE software package17 and parts of the LINKAGE software package (LINKMAP). The positions of 8 markers were obtained from the Marshfield map (ncbi.nlm.nih.gov/mapview/) and were held fixed while the disease locus was moved through the map in a stepwise fashion. Multipoint LOD scores were calculated using data from nearest 4 markers as the position of the disease locus was varied.
Due to variable expressivity of the disease, only affected patients and informative spouses were included in the linkage analysis. For the data given in figure 2, the allele frequencies were assumed to be equal for each marker. The true population allele frequencies for each marker could not be reliably estimated from the small number of spouses in the pedigree. In order to show that the assumption of the equal allele frequencies would not significantly affect our linkage results, we recalculated the LOD scores using allele frequencies for the "affected" allele of two of the most tightly linked markers (D7S1700 and D7S1702) ranging from 0.01 to 0.5. The Zmax for D7S1700 was 2.86 and the Zmax for D7S1702 was 2.09 when the “affected” allele frequency was arbitrarily set to 50%. In the 4 spouses that were studied, the actual frequencies of the "affected" alleles of D7S1700 and D7S1702 were much lower than 50%. In this small sample, the frequency of the “affected” of D7S1700 was 37.5% and the frequency of the “affected” allele of D7S1702 was 12.5%, suggesting that our use of equal allele frequencies for D7S1700 (14%) and D7S1702 (14%) were reasonable.
Figure 2.
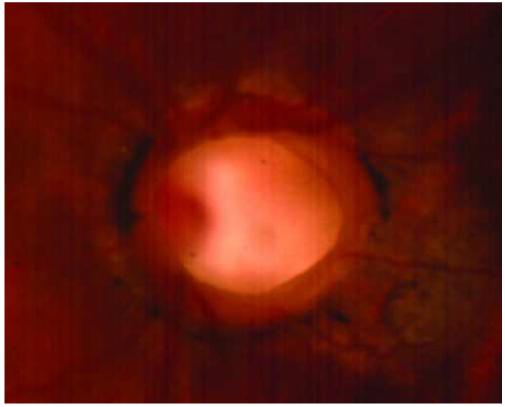
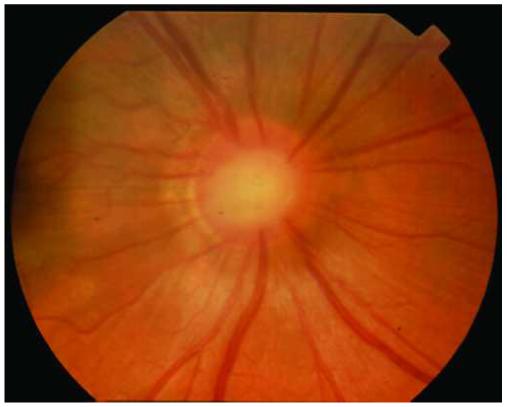
Appearance of cavitary optic disc anomalies. Affected members of the cavitary optic disc anomaly pedigree have abnormal optic discs with features of optic pits, optic nerve colobomas, and morning glory disc anomaly. A. The left optic disc of patient IV-11 is deeply excavated and exhibits features of an optic nerve coloboma and optic pit. B. The right optic disc of patient IV-8 is anomalous and has some characteristics that are similar to that of the morning glory disc anomaly including abnormal, radial vessels and a central tuft of glial tissue.
DNA samples from two affected family members and from two normal control subjects were tested for mutations in candidate genes (GDF11, OMIM 603936; NEUROD4; and WIF1, OMIM 605186) using bi-directional sequencing of PCR products that encompass the entire coding sequence. Sequencing was performed using dye-terminator chemistry on an ABI 3730 DNA sequencer (Applied Biosystems, Foster City, CA). PCR amplification was performed with a standard protocol18 using primer sequences that are available on request. Potential mutations were identified by comparing the DNA sequence of the affected family members and normal control subjects. Similarly, the DNA sequences of the affected family members were compared with NCBI reference sequences (GDF11, NM_005811; NEUROD4, NM_021191; WIF1, NM_007191). Identified sequence variations were evaluated as potential disease-causing mutations using standard criteria.19
RESULTS
The clinical features of the cavitary optic nerve anomalies in a large family (Figure 1) are described in detail in an accompanying report (Honkanen et al., accompanying manuscript). The optic disc phenotype observed in this family has features that lie within the spectrum of optic pits, optic nerve colobomas, and morning glory disc anomaly (Figure 2). Seventeen members of this pedigree have a cavitary optic nerve anomaly. DNA samples from 16 of these family members and 1 obligate carrier were subsequently studied with linkage analysis using a stepwise approach.
Figure 1.
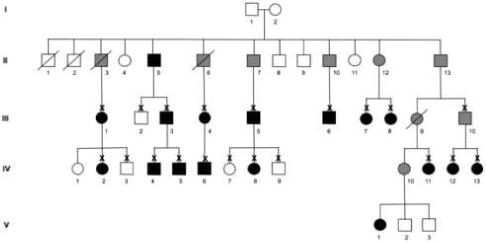
Pedigree affected with cavitary optic disc anomaly. Individuals found to be clinically affected with cavitary anomalies of the optic nerve including optic pits, optic nerve colobomas, or morning glory disc anomaly are represented by black symbols while unaffected individuals are depicted with open symbols. Family members who are obligate carriers are indicated with grey symbols. These family members were either unavailable for examination or they did not meet clinical criteria for having definite cavitary optic disc anomalies, however, they are obligate carriers by virtue of having offspring with cavitary optic disc anomalies. Individuals that are deceased are marked with a slash. Patients who were examined by the authors are indicated with an “X”.
After linkage to loci containing genes already associated with ocular coloboma was excluded (data not shown), a genome-wide scan for linkage was conducted by genotyping DNA samples from nine of the affected family members with microarrays of SNPs. Analysis of the SNP data identified a region of chromosome 12q with a maximum nonparametric multipoint LOD score of 21.7 with a p-value of 0.00097. All nine affected pedigree members were found to share an allele of each of the 49 consecutive SNPs in this locus. This 12q locus is 14.6 Mbp wide and is defined by the centromeric SNP, rs725029, and the telomeric SNP, rs722748.
The chromosome 12q linkage was confirmed by genotyping all 17 affected pedigree members with 16 STRP markers in this region (Figure 3). LOD scores over 3.0 were obtained from six STRP markers and a maximum LOD score of 4.06 (Θ=0) was obtained with marker D12S1700. The analysis of patients with recombination events near the linked interval is also shown in figure 3. These recombination events indicate that the disease-causing gene lies within the 9.1 cM (13.5 Mbp) interval between markers D12S1618 (centromeric) and D12S1702 (telomeric).
Figure 3.
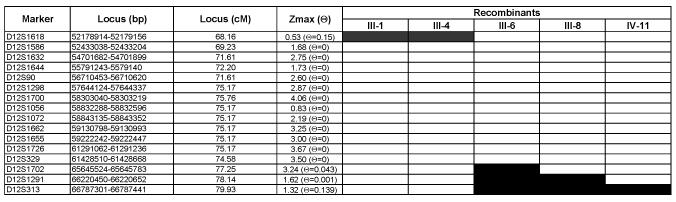
Cavitary optic disc anomalies linkage analysis: Two point linkage data and analysis of recombinant individuals. Sixteen genetic markers from the long arm of chromosome 12 are listed on the left of the figure with the most centromeric marker at the top. The physical position of the STRP markers is based on NCBI Build 36.1 of the human genome and the genetic position of the markers is based on the Marshfield map (http://www.ncbi.nlm.nih.gov/mapview/). The maximum LOD score (Zmax) is given for each marker as well as the recombination frequency at which the Zmax occurred. The patient designations correspond to those in Figure 1. A black box indicates that during the meiosis that gave rise to the individual (or that individual’s ancestor), an informative recombination event occurred between the marker and the disease gene. Parents were not available from any of the recombinant individuals. Consequently, it is not possible to identify those markers in which no recombinations occurred between the disease gene and the markers. The recombination events summarized in this figure suggest that the disease-causing mutations lie within the 9.1 cM (13.5 Mbp) interval bounded by D12S1618 and D12S1702.
Multipoint analysis was performed with the genotypic data from 8 STRP markers located within the 9.1 cM locus (D12S1618, D12S1586, D12S1632. DS12S1644, D12S329, D12S1726, D12S1700, and D12S1702). This analysis reveals a peak LOD score of 6.27 centered on marker D12S1726 (data not shown). The LOD-1 confidence interval20 is 5.1 cM.
The chromosome 12q locus, as defined by recombination events, contains 245 genes. Three of these genes (GDF11, NEUROD4, and WIF1) were considered good candidates for causing cavitary optic nerve defects based on their expression in the eye and presumed function in development and tissue differentiation. DNA from two affected family members was tested for mutations in the coding sequence of these three genes with bi-directional sequencing. No disease-causing mutations were discovered.
Discussion
Similarities in the optic disc appearance and clinical course associated with optic pits, optic nerve colobomas, and morning glory disc anomaly have been recognized suggesting that these conditions are part of a spectrum of disease (cavitary optic disc anomaly).2 Several large pedigrees demonstrating autosomal dominant cavitary optic disc anomalies have been reported.2, 11 We now report the genetic basis of the cavitary optic disc anomalies in one such pedigree. Our linkage study indicates that a single gene on chromosome 12 is capable of causing the cavitary optic disc anomalies in this pedigree. This disease-causing gene lies within a 9.1 cM (13.5 Mbp) locus that contains 245 known genes.
Three of the genes in the chromosome 12q locus (GDF11, NEUROD4, and WIF1) were considered promising candidates for causing cavitary optic disc anomalies due to their function and expression pattern. Gene differentiation factor 11 (GDF11) is a member of the TGF-β superfamily of genes and is expressed in the retina during retinal ganglion cell differentiation prior to closure of the optic fissure.21, 22 GDF11 regulates cell fate during retinal neurogenesis and controls the development of several retinal cell types, including retinal ganglion cells.22 Neurogenic differentiation factor-4 (NEUROD4) encodes a basic helix-loop-helix (bHLH) transcription factor that is expressed in the eye and has a role in the development and differentiation of the retina.23, 24 WNT inhibitory factor-1 (WIF1) encodes a secreted protein that is expressed in tissues of the eye and regulates development of the retina by binding proteins involved in the WNT signaling pathway.25, 26 Affected members of the cavitary optic disc anomaly pedigree were tested for disease-causing mutations in the coding sequences of these genes (GDF11, NEUROD4, and WIF1) with DNA sequencing. No disease-causing mutations were detected (data not shown).
Prior studies of pedigrees affected with POAG, primary congenital glaucoma and juvenile onset open angle glaucoma (JOAG) have identified potential glaucoma susceptibility loci on chromosome 1q24−25 (GLC1A),27 2cen-q13 (GLC1B),28 3q21−24 (GLC1C),29 8q23 (GLC1D),30 10p15-p14 (GLC1E),31 7q35-q36 (GLC1F),32 5q21-q22 (GLC1G),33 15q11-q13 (GLC1I),34 9q22 (GLC1J),35 20p12 (GLC1K),35 2p22-p21 (GLC3A),36 and 1p36 (GLC3B).37 A small fraction of glaucoma cases have been strongly linked to these loci. At present, however, the vast majority of glaucoma cases have not yet been associated with any genetic loci and the role of the chromosome 12q locus in these cases is not known.
The chromosome 12q gene responsible for cavitary optic disc anomalies has potent effects on the development of the optic nerve. A defect in this gene is capable of causing deep excavations of the optic disc during development. It is plausible that one set of mutations in this gene cause cavitary optic disc anomalies while another set of mutations might be associated with adult-onset forms of optic nerve disease with disc excavation, such as normal tension glaucoma (NTG) and primary open angle glaucoma (POAG). There is some support for this hypothesis. Two members of the cavitary optic disc anomalies pedigree in this report exhibited progressive cupping of the optic nerve head, which is a nearly pathognomonic feature of glaucoma (Honkanen et al., submitted and ref38) Also, the genes associated with other early-onset optic nerve conditions such as primary congenital glaucoma39 and dominant optic atrophy40 have been implicated in the pathophysiology of adult-onset glaucoma. Mutations in a gene that causes primary congenital glaucoma (cytochrome P450 1B1) have been shown to influence the phenotype of some POAG patients.41 Similarly, the OPA1 gene, which causes dominant optic atrophy (DOA), has also been investigated for a role in causing adult-onset glaucoma. While the classic feature of DOA is optic nerve head pallor, recent studies have shown that mutations in the OPA1 gene may be associated with optic nerve head changes similar to those seen in POAG.42 Also, association studies have suggested that the OPA1 gene might also be involved in POAG.43 In addition to clarifying the pathogenesis of cavitary optic disc anomalies, the identification of the disease-causing gene in our pedigree might also provide insight into the biologic processes of other more common optic nerve diseases such as NTG and POAG.
Acknowledgements
A. Funding / Support: This work was supported in part by grant 2R01EY010564−11A1 from the National Institutes of Health, Bethesda, MD and unrestricted grants from Research to Prevent Blindness, New York, N.Y. to the University of Iowa and Northwestern University. John H. Fingert was partially supported through a Career Development Award from Research to Prevent Blindness. Dr. Alward was supported by the Lew R. Wasserman Award of Research to Prevent Blindness.
B. Financial Disclosure: None of the authors have any financial disclosures relating to the research in this manuscript.
C. Contributions of the authors: Design and conduct of the study (JHF, SPS, VCS, EMS, WLMA); Collection, management, analysis, and interpretation of the data (JHF, RAH, SPS, LMA, MAE, DMD, LMJ, VCS, EMS, WLMA); Preparation, review, or approval of the manuscript (JHF, RAH, SPS, LMA, MAE, DMD, LMJ, VCS, EMS, WLMA)
Biography
John H. Fingert, M.D., Ph.D. is an Assistant Professor of Ophthalmology at the University of Iowa with research and clinical interests that focus on glaucoma and other inherited diseases of the optic nerve. Dr. Fingert received his medical and genetics degrees from the University of Iowa where he also completed an ophthalmology residency and fellowships in molecular ophthalmology and glaucoma.

Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Corbett JJ, Savino PJ, Schatz NJ, Orr LS. Cavitary developmental defects of the optic disc. Visual loss associated with optic pits and colobomas. Arch Neurol. 1980;37:210–213. doi: 10.1001/archneur.1980.00500530048006. [DOI] [PubMed] [Google Scholar]
- 2.Slusher MM, Weaver RG, Jr., Greven CM, Mundorf TK, Cashwell LF. The spectrum of cavitary optic disc anomalies in a family. Ophthalmology. 1989;96:342–347. doi: 10.1016/s0161-6420(89)32886-7. [DOI] [PubMed] [Google Scholar]
- 3.Stefko ST, Campochiaro P, Wang P, Li Y, Zhu D, Traboulsi EI. Dominant inheritance of optic pits. Am J Ophthalmol. 1997;124:112–113. doi: 10.1016/s0002-9394(14)71656-3. [DOI] [PubMed] [Google Scholar]
- 4.Gregory-Evans CY, Williams MJ, Halford S, Gregory-Evans K. Ocular coloboma: a reassessment in the age of molecular neuroscience. J Med Genet. 2004;41:881–891. doi: 10.1136/jmg.2004.025494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azuma N, Yamaguchi Y, Handa H, et al. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet. 2003;72:1565–570. doi: 10.1086/375555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schimmenti LA, de la Cruz J, Lewis RA, et al. Novel mutation in sonic hedgehog in nonsyndromic colobomatous microphthalmia. Am J Med Genet A. 2003;116:215–221. doi: 10.1002/ajmg.a.10884. [DOI] [PubMed] [Google Scholar]
- 7.Jamieson RV, Perveen R, Kerr B, et al. Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum Mol Genet. 2002;11:33–42. doi: 10.1093/hmg/11.1.33. [DOI] [PubMed] [Google Scholar]
- 8.Ferda Percin E, Ploder LA, Yu JJ, et al. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat Genet. 2000;25:397–401. doi: 10.1038/78071. [DOI] [PubMed] [Google Scholar]
- 9.Yamashita T, Kawano K, Ohba N. Autosomal dominantly inherited optic nerve coloboma. Ophthalmic Paediatr Genet. 1988;9:17–24. doi: 10.3109/13816818809031476. [DOI] [PubMed] [Google Scholar]
- 10.Kindler P. Morning glory syndrome: unusual congenital optic disk anomaly. Am J Ophthalmol. 1970;69:376–384. doi: 10.1016/0002-9394(70)92269-5. [DOI] [PubMed] [Google Scholar]
- 11.Savell J, Cook JR. Optic nerve colobomas of autosomal-dominant heredity. Arch Ophthalmol. 1976;94:395–400. doi: 10.1001/archopht.1976.03910030183002. [DOI] [PubMed] [Google Scholar]
- 12.Buffone GJ, Darlinton GJ. Isolation of DNA from Biological Specimens without Extraction with Phenol. Clinical Chemistry. 1985;31:164–165. [PubMed] [Google Scholar]
- 13.Heon E, Piguet B, Munier F, et al. Linkage of Autosomal Dominant Radial Drusen (Malattia Leventinese) to Chromosome 2p16−21. Arch Ophthalmol. 1996;114 doi: 10.1001/archopht.1996.01100130187014. [DOI] [PubMed] [Google Scholar]
- 14.Cottingham RW, Jr., Idury RM, Schaffer AA. Faster sequential genetic linkage computations. Am J Hum Genet. 1993;53:252–263. [PMC free article] [PubMed] [Google Scholar]
- 15.Schaffer AA, Gupta SK, Shriram K, Cottingham RW., Jr. Avoiding recomputation in linkage analysis. Hum Hered. 1994;44:225–237. doi: 10.1159/000154222. [DOI] [PubMed] [Google Scholar]
- 16.Lathrop GM, Lalouel JM. Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet. 1984;36:460–465. [PMC free article] [PubMed] [Google Scholar]
- 17.O'Connell JR, Weeks DE. The VITESSE Algorithm for Rapid Exact Multilocus Linkage Analysis via Genotype Set-Recoding and Fuzzy Inheritance. Nature Genetics. 1995;11:402–408. doi: 10.1038/ng1295-402. [DOI] [PubMed] [Google Scholar]
- 18.Fingert JH, Heon E, Liebmann JM, et al. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum Mol Genet. 1999;8:899–905. doi: 10.1093/hmg/8.5.899. [DOI] [PubMed] [Google Scholar]
- 19.Stone EM. Finding and interpreting genetic variations that are important to ophthalmologists. Trans Am Ophthalmol Soc. 2003;101:437–484. [PMC free article] [PubMed] [Google Scholar]
- 20.Conneally PM, Edwards JH, Kidd KK, et al. Report of the Committee on Methods of Linkage Analysis and Reporting. Cytogenet Cell Genet. 1985;40:356–359. doi: 10.1159/000132186. [DOI] [PubMed] [Google Scholar]
- 21.Nakashima M, Toyono T, Akamine A, Joyner A. Expression of growth/differentiation factor 11, a new member of the BMP/TGFbeta superfamily during mouse embryogenesis. Mech Dev. 1999;80:185–189. doi: 10.1016/s0925-4773(98)00205-6. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Wu HH, Lander AD, Lyons KM, Matzuk MM, Calof AL. GDF11 controls the timing of progenitor cell competence in developing retina. Science. 2005;308:1927–1930. doi: 10.1126/science.1110175. [DOI] [PubMed] [Google Scholar]
- 23.Inoue T, Hojo M, Bessho Y, Tano Y, Lee JE, Kageyama R. Math3 and NeuroD regulate amacrine cell fate specification in the retina. Development. 2002;129:831–842. doi: 10.1242/dev.129.4.831. [DOI] [PubMed] [Google Scholar]
- 24.Akagi T, Inoue T, Miyoshi G, et al. Requirement of multiple basic helix-loop-helix genes for retinal neuronal subtype specification. J Biol Chem. 2004;279:28492–28498. doi: 10.1074/jbc.M400871200. [DOI] [PubMed] [Google Scholar]
- 25.Hsieh JC, Kodjabachian L, Rebbert ML, et al. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature. 1999;398:431–436. doi: 10.1038/18899. [DOI] [PubMed] [Google Scholar]
- 26.Hunter DD, Zhang M, Ferguson JW, Koch M, Brunken WJ. The extracellular matrix component WIF-1 is expressed during, and can modulate, retinal development. Mol Cell Neurosci. 2004;27:477–488. doi: 10.1016/j.mcn.2004.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheffield VC, Stone EM, Alward WLM, et al. Genetic linkage of familial open angle glaucoma to chromosome 1q21-q31. Nature Genetics. 1993;4:47–50. doi: 10.1038/ng0593-47. [DOI] [PubMed] [Google Scholar]
- 28.Stoilova D, Child A, Trifan OC, Crick RP, Coakes RL, Sarfarazi M. Localization of a locus (GLC1B) for adult-onset primary open angle glaucoma to the 2cen-q13 region. Genomics. 1996;36:142–150. doi: 10.1006/geno.1996.0434. [DOI] [PubMed] [Google Scholar]
- 29.Wirtz MK, Samples JR, Kramer PL, et al. Mapping a gene for adult-onset primary open-angle glaucoma to chromosome 3q. Am J Hum Genet. 1997;60:296–304. [PMC free article] [PubMed] [Google Scholar]
- 30.Trifan OC, Traboulsi EI, Stoilova D, et al. A third locus (GLC1D) for adult-onset primary open-angle glaucoma maps to the 8q23 region. Am J Ophthalmol. 1998;126:17–28. doi: 10.1016/s0002-9394(98)00073-7. [DOI] [PubMed] [Google Scholar]
- 31.Sarfarazi M, Child A, Stoilova D, et al. Localization of the Fourth Locus (GLC1E) For Adult-Onset Primary Open- Angle Glaucoma to the 10p15-p14 Region. Am J Hum Genet. 1998;62:641–652. doi: 10.1086/301767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wirtz MK, Samples JR, Rust K, et al. GLC1F, a new primary open-angle glaucoma locus, maps to 7q35-q36. Arch Ophthalmol. 1999;117:237–241. doi: 10.1001/archopht.117.2.237. [DOI] [PubMed] [Google Scholar]
- 33.Monemi S, Spaeth G, Dasilva A, et al. Identification of a novel adult-onset primary open-angle glaucoma (POAG) gene on 5q22.1. Hum Mol Genet. 2005;14:725–733. doi: 10.1093/hmg/ddi068. [DOI] [PubMed] [Google Scholar]
- 34.Allingham RR, Wiggs JL, Hauser ER, et al. Early adult-onset POAG linked to 15q11−13 using ordered subset analysis. Invest Ophthalmol Vis Sci. 2005;46:2002–2005. doi: 10.1167/iovs.04-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiggs JL, Lynch S, Ynagi G, et al. A genomewide scan identifies novel early-onset primary open-angle glaucoma loci on 9q22 and 20p12. Am J Hum Genet. 2004;74:1314–1320. doi: 10.1086/421533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarfarazi M, Akarsu AN, Hossain A, et al. Assignment of a locus (GLC3A) for primary congenital glaucoma (Buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics. 1995;30:171–177. doi: 10.1006/geno.1995.9888. [DOI] [PubMed] [Google Scholar]
- 37.Akarsu AN, Turacli ME, Aktan SG, et al. A second locus (GLC3B) for primary congenital glaucoma (Buphthalmos) maps to the 1p36 region. Hum Mol Genet. 1996;5:1199–1203. doi: 10.1093/hmg/5.8.1199. [DOI] [PubMed] [Google Scholar]
- 38.Moore M, Salles D, Jampol LM. Progressive optic nerve cupping and neural rim decrease in a patient with bilateral autosomal dominant optic nerve colobomas. Am J Ophthalmol. 2000;129:517–520. doi: 10.1016/s0002-9394(99)00463-8. [DOI] [PubMed] [Google Scholar]
- 39.Stoilov I, Akarsu AN, Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum Mol Genet. 1997;6:641–647. doi: 10.1093/hmg/6.4.641. [DOI] [PubMed] [Google Scholar]
- 40.Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 41.Vincent AL, Billingsley G, Buys Y, et al. Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am J Hum Genet. 2002;70:448–460. doi: 10.1086/338709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Votruba M, Thiselton D, Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmol. 2003;87:48–53. doi: 10.1136/bjo.87.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aung T, Ocaka L, Ebenezer ND, et al. A major marker for normal tension glaucoma: association with polymorphisms in the OPA1 gene. Hum Genet. 2002;110:52–56. doi: 10.1007/s00439-001-0645-7. [DOI] [PubMed] [Google Scholar]