

Abstract
The family of fibroblast growth factors (FGFs) regulates a plethora of developmental processes, including brain patterning, branching morphogenesis and limb development. Several mitogenic, cytoprotective and angiogenic therapeutic applications of FGFs are already being explored, and the recent discovery of the crucial roles of the endocrine-acting FGF19 subfamily in bile acid, glucose and phosphate homeostasis has sparked renewed interest in the pharmacological potential of this family. This Review discusses traditional applications of recombinant FGFs and small-molecule FGF receptor kinase inhibitors in the treatment of cancer and cardiovascular disease and their emerging potential in the treatment of metabolic syndrome and hypophosphataemic diseases.
There are 18 mammalian fibroblast growth factors (FGF1–FGF10 and FGF16–FGF23) which are grouped into 6 subfamilies based on differences in sequence homology and phylogeny: FGF1 and FGF2; FGF3, FGF7, FGF10, FGF22; FGF4, FGF5 and FGF6; FGF8, FGF17 and FGF18; FGF9, FGF16 and FGF20; and FGF19, FGF21 and FGF23 (REF. 1). The numbered ‘FGFs’ that are unassigned to subfamilies — the FGF homologous factors (previously known as FGF11–FGF14) — have high sequence identity with the FGF family but do not activate FGF receptors (FGFRs) and are therefore not generally considered members of the FGF family2 (BOX 1); FGF15 is the mouse orthologue of human FGF19. FGFs are classically considered to be paracrine factors and are known for their roles in tissue patterning and organogenesis during embryogenesis: the first five subfamilies fall into this category. By contrast, the FGF19, FGF21 and FGF23 subfamily has recently been shown to function in an endocrine manner, dependent on the presence of klotho proteins in their target tissues, to regulate bile acid, cholesterol, glucose, vitamin D and phosphate homeostasis3–6.
Box 1. Fibroblast homologous factors.
Although fibroblast homologous factors (FHFs) have high sequence and structural homology with fibroblast growth factors (FGFs) and bind heparin with high affinity, they do not activate FGF receptors (FGFRs). The FHF core structure is similar to that of FGFs: they exhibit the same β-trefoil core that consists of 12 antiparallel β-strands. However, several key receptor-binding residues are divergent or occluded in FHFs. Val157, unique to FHFs, reduces binding to FGFRs by eliminating important hydrogen bonds with the D2–D3 linker of FGFR that are formed by asparagine, threonine or aspartate in FGFs2. Furthermore, the carboxyl terminus of FHF packs against the rest of the ligand in such a way as to preclude many FGFR binding residues from interacting278. Owing to the inability of FHFs to bind FGFRs, the inclusion of FHFs in the FGF family should be reconsidered. The principal targets of FHFs are the intracellular domains of voltage-gated sodium channels. FHF mutations in mouse models cause a range of neurological abnormalities and FHF mutations in humans are implicated in cerebellar ataxia263. Accordingly, FHFs are an intriguing area of research in their own right.
The involvement of FGF signalling in human disease is well documented. Deregulated FGF signalling can contribute to pathological conditions either through gain- or loss-of-function mutations in the ligands themselves — for example, FGF23 gain of function in autosomal dominant hypophosphataemic rickets7, FGF10 loss of function in lacrimo-auriculo-dento-digital syndrome (LADD syndrome)8, FGF3 loss of function in deafness9 and FGF8 loss of function in Kallmann syndrome10 — or through gain- or loss-of-function mutations in FGFRs, which contribute to many skeletal syndromes41, Kallmann syndrome36, LADD syndrome54 and cancer. Therapeutic approaches using exogenous FGFs, antibodies or small molecules are still relatively new, and many avenues of investigation remain open. Recombinant FGF7 is already in use for the treatment of chemoradiation-induced oral mucositis. Future application of the FGFs in renal disease, glucose and phosphate homeostasis, stem cell research, tissue repair and bioengineering, and angiogenesis is expected. Continued efforts to understand the structural biology of FGF–FGFR interactions will play a key part in driving the discovery of new therapies.
In this article, we briefly review current knowledge regarding FGF–FGFR signalling and then focus on the biology, pathology and recent developments regarding the pharmacological applications of each ligand.
The FGF–FGFR signalling system
FGFs
All FGFs, except those in subfamilies FGF1 and FGF2, and FGF9, FGF16 and FGF20, have signal peptides. The FGF9, FGF16 and FGF20 subfamily is nonetheless secreted through the traditional endoplasmic reticulum (ER)–Golgi secretory pathway11, whereas the FGF1 and FGF2 subfamily is secreted independently12. FGFs have a homologous core region that consists of 120–130 amino acids ordered into 12 antiparallel β-strands (β1–β12) flanked by divergent amino and carboxyl termini (FIG. 1a). In general, primary sequence variation of the N- and C-terminal tails of FGFs accounts for the different biology of the ligands13 (FIG. 1b). The heparan sulphate glycosaminoglycan (HSGAG) binding site (HBS) within the FGF core is composed of the β1–β2 loop and parts of the region spanning β10 and β12. For paracrine FGFs, the elements of the HBS form a contiguous, postively charged surface. By contrast, the HBS of the FGF19, FGF21 and FGF23 subfamily contains ridges formed by the β1–β2 loop and the β10–β12 region that sterically reduce HSGAG binding to the core backbone of the FGFs and lead to the endocrine nature of this subfamily14.
Figure 1. structural features of fibroblast growth factors (FgFs).

a | FGF1, showing its 12 antiparallel β-sheets and amino and carboxyl termini. b | The 18 FGFs, grouped according to subfamily. The sequence alignment in the region of the divergent N terminus proximal to the β-trefoil core is given. The β1 strand of FGF1 is provided to indicate the limit of the N terminus. c | FGF19 superimposed onto FGF2 from the FGF2–FGF receptor 1–heparin ternary structure (Protein Data Bank). FGF2 and FGF19 are rendered as ribbons and heparin is shown as sticks: oxygen (red), nitrogen (blue), carbon (beige), and sulphur (green) atoms are shown. The core regions of both ligands are coloured grey, and the heparin binding regions of FGF2 and FGF19 are coloured cyan and orange, respectively. Heparin from 1FQ9 clashes with the ridges in the heparin binding region of FGF19. To eliminate these clashes, heparin must translocate away from FGF19 but, in doing so, crucial contacts between heparin and the FGF19 backbone cannot be made. The weakened heparin binding observed in the FGF19 subfamily members is responsible for their endocrine behaviour.
FGFRs
The FGF ligands carry out their diverse functions by binding and activating the FGFR family of tyrosine kinase receptors in an HSGAG-dependent manner. There are four FGFR genes (FGFR1–FGFR4) that encode receptors consisting of three extracellular immunoglobulin domains (D1–D3), a single-pass transmembrane domain and a cytoplasmic tyrosine kinase domain13. A hallmark of FGFRs is the presence of an acidic, serine-rich sequence in the linker between D1 and D2, termed the acid box. The D2–D3 fragment of the FGFR ectodomain is necessary and sufficient for ligand binding and specificity, whereas the D1 domain and the acid box are proposed to have a role in receptor autoinhibition15 (FIG. 2a). Several FGFR isoforms exist, as exon skipping removes the D1 domain and/or acid box in FGFR1–FGFR3. Alternative splicing in the second half of the D3 domain of FGFR1–3 yields b (FGFR1b–3b) and c (FGFR1c–3c) isoforms that have distinct FGF binding specificities16 and are predominantly epithelial and mesenchymal, respectively. Each FGF binds to either epithelial or mesenchymal FGFRs, with the exception of FGF1, which activates both splice isoforms.
Figure 2. structural features of fibroblast growth factor receptors (FgFrs).

a | A schematic of the FGFR structure. b | The sequence alignment of the seven main FGFRs in the region of the βC′–βE loop, including the βC′ and βE strands. The vertical green bar divides the unspliced portion of the receptor at the left from the spliced portion that follows. c | A superimposition of the D3 domains of solved FGF–FGFR complex structures. FGF2–FGFR2c is shown in red, FGF8–FGFR2c is shown in purple, FGF1–FGFR1c is shown in blue, FGF1–FGFR2b is shown in green, FGF1–FGFR3c is shown in black, FGF1–FGFR2c is shown in brown, FGF2–FGFR1c is shown in pink, FGF3–FGFR2b is shown in grey and FGF10–FGFR2b is shown in cyan. The variation in the conformation of the βC′–βE loop between the structures as it interacts with divergent amino termini is evident. The plasticity of this loop is a major determinant of FGF–FGFR binding specificity.
After the binding of ligand and HSGAGs, FGFRs dimerize17–19, enabling the cytoplasmic kinase domains to transphosphorylate on A loop tyrosines to become activated (FIG. 3). A loop phosphorylation is followed by phosphorylation of tyrosines in the C tail, kinase insert and juxtamembrane regions20. The two main intracell-ular substrates of FGFR are phospholipase C (PLC) γ1 (also known as FRS1) and FGFR substrate 2 (also known as FRS2). Phosphorylation of an FGFR-invariant tyrosine (Y766 in FGFR1) at the C tail of FGFR creates a binding site for the SH2 domain of PLCγ and is required for PLCγ phosphorylation and activation. By contrast, FRS2 associates constitutively with the juxtamembrane region of the FGFR. Phosphorylation of FRS2 is essential for activation of the Ras–mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase–Akt signalling pathways21. FGFs are also known to function in the cytosol and nucleus of cells, both through endocytosis of activated FGF–FGFR complexes and through endogenous sources of ligand22.
Figure 3. Fibroblast growth factor receptor (FgFr) signalling.

Structurally unresolved regions are shown as grey lines. Amino-terminal and carboxy-terminal lobes of the kinase domain are coloured green and red, respectively. The two major intracellular targets, phospholipase (PLC)γ1 and FGFR substrate 2α (FRS2α), are shown. A loop, activation loop; GRB2, growth factor receptor bound 2; HS, heparan sulphate; IP3, inositol-1,4,5-trisphosphate; MAPK, mitogen-activated protein kinase; MAPKK, mitogen-activated protein kinase kinase; PH, pleckstrin homology domain; PIP2, phosphatidylinositol-4,5-bisphosphate; PKC, protein kinase C; PTB, phosphotyrosine binding domain; PTK, protein tyrosine kinase; SH, Src homology domain. Figure is modified, with permission, from REF. 13 (2005) Elsevier Science.
FGF–FGFR specificity
FGF–FGFR binding specificity is regulated both by primary sequence differences between the 18 FGFs and the 7 main FGFRs (FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3b, FGFR3c and FGFR4) and by temporal and spatial expression patterns of FGFs, FGFRs and HSGAGs. The alternative splice isoforms of FGFRs are generally tissue specific: the b isoform is usually expressed in epithelial tissue, whereas the c isoform is usually expressed in mesenchymal tissue23. Ligands are produced in either epithelial or mesenchymal tissue and generally activate receptors of the opposite tissue specificity: in normal physiology, a ligand produced in the epithelium will activate a mesenchymal receptor and vice versa. Several ligands, including FGF1 in particular, pose an exception to this general understanding by promiscuously binding to both b and c isoforms of certain FGFRs. Pathological states can result from a breakdown in binding specificity, as is common in cancers in which FGFs are overexpressed24. Structural studies of FGF1, FGF2, FGF8 and FGF10 with their cognate FGFRs show that sequence diversity at FGF N termini, variation in β1 strand length (FIG. 1b) and the alternatively spliced regions in D3 dictate their binding specificities (FIG. 2b, c).
The FGF–FGFR dimer
A functional FGF–FGFR unit consists of two 1:1:1 FGF–FGFR–HSGAG complexes juxtaposed in a symmetrical dimer18. Each ligand in the dimer binds both receptors, and the two receptors contact each other directly through a patch at the base of D2. Each ligand interacts with the D2 domain of a second receptor through a secondary receptor binding site, and mutation of ligand residues within this site reduces receptor dimerization and signalling without affecting ligand–receptor binding25.
HSGAG binding
HSGAG binds to a basic canyon formed on the membrane-distal end of the symmetric dimer to strengthen protein–protein contacts. HSGAG facilitates FGF–FGFR dimerization by simultaneously binding both FGF and FGFR, thereby promoting and stabilizing protein–protein contacts between ligand and receptor both within the 1:1 FGF–FGFR complex and between the two complexes in the 2:2 FGF–FGFR dimer. In addition to facilitating FGF–FGFR binding, HSGAGs stabilize FGFs against degradation, act as a storage reservoir for ligand and determine the radius of ligand diffusion26. Interestingly, the divergence in the morpho-genetic activities of FGF7 and FGF10 on branching organs appears to correlate with the differences in their HSGAG affinity and the HSGAG-dependent diffusion of these two ligands through the extra-cellular matrix (H. Makarenkova et al., unpublished observations).
Modulators of FGF signalling
FGF-binding protein (FGFBP) is a carrier protein27 that activates FGFs by releasing them from the extracellular matrix, where they are bound by HSGAGs28. FGFBP has been shown to increase FGF2-dependent proliferation of fibroblast cells29 and may have an important role in the development of some cancers30. Other activators of FGF signalling include fibronectin leucine-rich transmembrane protein 3 (FLRT3), which facilitates FGF8 activity through the MAPK pathway31.
The sprouty family of proteins play an important part in inhibiting receptor tyrosine kinase (RTK) signalling and were first discovered as inhibitors of FGFs in Drosophila melanogaster32. FGF signalling activates sprouty proteins, which can then in turn inhibit FGF stimulation of the MAPK pathway by interacting with GRB2 (growth factor receptor bound protein 2), SOS1 or RAF1 (REF. 33). MKP3 (MAPK phosphatase 3) is another general inhibitor of RTK signalling that also impinges on FGF activity by dephosphorylating extracellular signal-regulated kinase (ERK)34. SEF is a specific inhibitor of FGFs that can function at multiple points along the signalling pathway to attenuate signalling34.
FGFR pathophysiology and therapy
Germline gain-of-function mutations in FGFRs are responsible for various diseases, such as craniosynostosis, dwarfing syndromes and cancer. Most of the FGFR mutations are ligand independent, but a few — such as Ser252Trp and Pro253Arg in the ectodomain of FGFR2 — manifest only during ligand binding. These mutations cause Apert’s syndrome by enhancing ligand binding affinity and promoting the binding of inappropriate ligands35,278–280. Remarkably, many of the germline mutations that cause skeletal syndromes also contribute, through somatic mutations, to the development of cancer. Furthermore, mutations in FGFR1–FGFR3 often occur in homologous residues and account for multiple pathologies.
FGFR1
At least three genetic disorders can be attributed to mutations in FGFR1: Kallman’s syndrome36, osteoglophonic dyplasia and Pfeiffer’s syndrome37. Pathological FGFR1 signalling also occurs in various malignancies. Glioblastoma brain tumours exhibit FGFR1 kinase domain gain-of-function mutations38, and FGFR1 is abnormally activated in malignant prostate cells39. In 8p11 myeloproliferative syndrome (EMS), translocations fuse different proteins in frame with the FGFR1 kinase domain, causing constitutive dimerization of the kinase40.
FGFR2
Mutations in the kinase domain of FGFR2 have been identified in patients with various craniosynostosis syndromes, including Crouzon’s syndrome and Pfeiffer’s syndrome41. These mutations constitutively activate FGFRs by disengaging an autoinhibitory molecular brake at the hinge region of the kinase domain42. Many of these mutations that lead to skeletal deformity are also commonly observed in endometrial cancers43,44. Ectodomain FGFR2 mutations cause ligand-independent disulphide-mediated covalent receptor dimerization and activation in pathologies such as Crouzon’s syndrome45. Ligand-dependent gain-of-function ectodomain mutations in FGFR2c allow binding to FGFR2b-binding ligands46,278,280, which contributes to the development of Pfeiffer’s syndrome and Apert’s syndrome. The mutations involved in these syndromes also cause white matter pathologies, including callosal agenesis and ventriculomegaly47. Interestingly, through a dominant-negative effect, soluble FGFR2 can inhibit the osteoblastic differentiation typically observed in Apert’s syndrome48. Notably, single nucleotide polymorphisms (SNPs) in FGFR2c are associated with BRCA2 mutation-carrying breast cancers49,286.
FGFR3
Transmembrane mutations, such as Gly380Arg in FGFR3, promote non-covalent interactions between transmembrane helices and occur in nearly all cases of achrondroplasia, which is the most common genetic form of dwarfism50. Kinase-domain FGFR3 mutations increase catalytic activity independently of receptor dimerization51 by disengaging the molecular brake at the kinase hinge region4. A range of germline mutations affect three codons (Ile538, Asn540 and Lys650) in the FGFR3 kinase domain, yielding three dwarfing syndromes of varying clinical severity: hypochondroplasia, thanatophoric dysplasia type II and severe achondroplasia with developmental delay and acanthosis nigricans syndrome (SADDAN syndrome)52–53. Furthermore, overexpression and gain-of-function mutations in FGFR3 occur in multiple myeloma, an incurable B-cell malignancy55. Gain-of-function FGFR3 mutations are the most commonly observed mutations in bladder cancer56, and activating FGFR3 mutations are also observed in benign skin tumours57,58. Most of the FGFR3 mutations found in cancer are identical to the FGFR2 mutations involved in skeletal disorders. Kinase domain loss-of-function mutations also occur in FGFR2 and FGFR3 in LADD syndrome54 and in FGFR2 in melanoma281.
FGFR4
FGFR4 has potential value as a prognostic marker in cancer. Arg388 in FGFR4 is associated with increased aggressiveness of prostate cancer, and promotes metastasis by increasing cellular motility and invasiveness59. This same allele in FGFR4 is a predictor of poor clinical prognosis in head and neck squamous cell carcinoma60. In recurrent breast cancer, high FGFR4 expression correlates with low efficacy of tamoxifen treatment61.
Therapeutic potential of FGFRs
Direct inhibition of FGFRs may prove to be of clinical value. Sunitinib is a receptor tyrosine inhibitor that has received Food and Drug Administration (FDA) approval for indications in renal cell carcinoma and gastrointestinal stromal tumours, and, unlike imatinib mesylate (Gleevec; Novartis), it does have some activity against FGFRs62. SU5402, PD173074 and nordihydroguaiaretic acid are small-molecule FGFR inhibitors that have efficacy in multiple myeloma cell lines with deregulated FGFR3 expression63,64. Furthermore, PD173074 has the ability to induce cell cycle arrest in endometrial cancer cells with mutated FGFR2 (REF. 65). In addition to small-molecule inhibition, antibodies against FGFR3 have been shown to effectively cause apoptosis in mouse models of multiple myeloma and bladder cancer66,67. These instances are proof of principle that FGFR inhibition could be efficacious in the treatment of malignancy (TABLE 1). Mutation of Tyr766 in the PLCγ1 binding site of FGFR1 attenuates EMS68; therefore, interference with the FGFR–PLCγ1 interaction could prove to be a promising therapeutic strategy in the treatment of EMS. The use of PLCγ inhibitors alongside tyrosine kinase inhibitors could also slow the development of drug resistance to these tyrosine kinase inhibitors24.
Table 1.
Selected inhibitors of FGF signalling
| Drug | structure | Function | refs |
|---|---|---|---|
| Sunitinib |
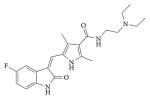
|
|
62 |
| Suramin |
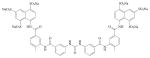
|
|
103–107 |
| Thalidomide |
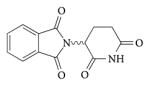
|
|
100–102 |
| SU5402 |
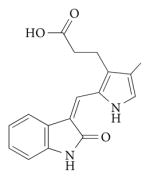
|
63–65 | |
| PD173074 |
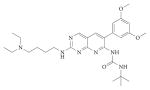
|
||
| NDGA |
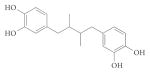
|
FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; NDGA, norhydroguaiaretic acid; RTK, receptor tyrosine kinase.
Paracrine FGF ligands
The paracrine FGF families are FGF1 and FGF2; FGF3, FGF7, FGF10 and FGF22; FGF4, FGF5 and FGF6; FGF8, FGF17 and FGF18; and FGF9, FGF16 and FGF20. Their high affinity for HSGAG causes them to act in a localized manner near the source of their expression (TABLE 2). Paracrine FGFs are being explored for their therapeutic potential in angiogenesis, cytoprotection and tissue repair (TABLE 3). For example, recombinant FGF7 is already used in the clinic to treat chemoradiation-induced mucositis; applications of recombinant FGF1, FGF2 and of FGF4 gene therapy to cardiovascular pathologies are being explored; and recombinant FGF18 is in the early stages of development for osteoarthritis treatment. Many paracrine FGFs are deregulated in cancers, and their overexpression stimulates proliferation and angiogenesis, which can contribute to cancer growth24.
Table 2.
The physiology of FGFs
| Fibroblast growth factor (FgF) | Phenotype of knockout mouse | Physiological role |
|---|---|---|
| FGF1 | Normal69 | Not established |
| FGF2 | Loss of vascular tone Slight loss of cortex neurons72–73 |
Not established |
| FGF3 | Inner ear agenesis in humans9 | Inner ear development9 |
| FGF4 | Embryonic lethal128 | Cardiac valve leaflet formation Limb development126–128 |
| FGF5 | Abnormally long hair129 | Hair growth cycle regulation129–131 |
| FGF6 | Defective muscle regeneration133 | Myogenesis132,133 |
| FGF7 | Matted hair Reduced nephron branching in kidney137,138 |
Branching morphogenesis138 |
| FGF8 | Embryonic lethal162 | Brain, eye, ear and limb development160,161 |
| FGF9 | Postnatal death Gender reversal Lung hypoplasia170 |
Gonadal development Organogenesis170,171 |
| FGF10 | Failed limb and lung development142 | Branching morphogenesis142 |
| FGF16 | Embryonic lethal172 | Heart development172 |
| FGF17 | Abnormal brain development163 | Cerebral and cerebellar development163 |
| FGF18 | Delayed long-bone ossification164,165 | Bone development164,165 |
| FGF19 | Increased bile acid pool189 | Bile acid homeostasis Lipolysis Gall bladder filling3,6,197–201 |
| FGF20 | No knockout model | Neurotrophic factor175 |
| FGF21 | No knockout model | Fasting response Glucose homeostasis Lipolysis and lipogenesis4,208–225 |
| FGF22 | No knockout model | Presynaptic neural organizer143 |
| FGF23 | Hyperphosphataemia Hypoglycaemia Immature sexual organs185,235 |
Phosphate homeostasis Vitamin D homeostasis226–261 |
Table 3.
Applications of FGFs and FGFRs
| Ligand or receptor | current/potential therapeutic application | refs |
|---|---|---|
| FGF1 | Recombinant FGF1 used with nerve grafts Treatment of peripheral ischaemia with FGF1 plasmids |
87,88, 90–92 |
| FGF2 | Use of thalidomide in prostate and renal cancer Implantation of FGF2-coated heparin beads post-MI Recombinant FGF2 modulates mood in mice |
97, 101–102, 122 |
| FGF4 | Potential gene therapy for stable angina in women | 136 |
| FGF5 | Potential of FGF5 inhibitors to aid hair growth | 129 |
| FGF7 | Treatment of mucositis (known as the drug palifermin) Recombinant FGF7 improves wound healing |
149, 157 |
| FGF18 | Recombinant FGF18 has an anabolic effect on cartilage | 167 |
| FGF19 | Potential of recombinant FGF19 in diabetes | 3,6,199,200 |
| FGF20 | Potential in Parkinson’s disease | 176 |
| FGF21 | Potential of recombinant FGF21 in diabetes | 4,208–225 |
| FGF23 | Use of anti-FGF23 antibodies in hypophosphataemia | 260,261 |
| FGFR1 | PLCγ inhibitors in the treatment of EMS and as an adjunct to TKIs | 24,68 |
| FGFR2 | Small-molecule inhibitors and anti-FGFR2 antibodies in endometrial cancer | 65 |
| FGFR3 | Small-molecule inhibitors and anti-FGFR3 antibodies in multiple myeloma | 63,64,66,67 |
| FGFR4 | Prognostic marker in prostate cancer and squamous cell carcinoma | 59,60 |
EMS, 8p11 myeloproliferative syndrome; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; MI, myocardial infarction; PLCγ, phospholipase Cγ; TKI, tyrosine kinase inhibitor.
The FGF1 subfamily
Biology
As both Fgf1−/− and Fgf2−/− mice are viable and fertile and Fgf1−/− mice are apparently completely normal69, the physiological roles of FGF1 and FGF2 are still unclear. However, it is likely that FGF1 and FGF2 play some physiological part in the maintenance of vascular tone, as administration of FGF1 and FGF2 lowers blood pressure in rats70 and can restore nitric oxide synthase activity in spontaneously hypertensive rats71. In addition, isolated vessels from Fgf2−/− mice have a reduced response to vaso-constrictors72. Although Fgf2−/− mice experience some hypotension owing to decreased smooth muscle contractility72, they are still able to regulate their blood pressure73.
The angiogenic properties of FGF2 are well known. Exogenous FGF2 stimulates migration and proliferation of endothelial cells in vivo74, has anti-apoptotic activity75 and encourages mitogenesis of smooth muscle cells and fibroblasts, which induces the development of large collateral vessels with adventitia76. However, as over-expression of Fgf2 does not lead to spontaneous vascular defects77, and normal vascularization is retained in double knockout Fgf2−/−;Fgf1−/− mice69, the physiological relevance of these effects is uncertain. Evidently, there is a high level of compensation among the growth factors mediating angiogenesis78.
Other possible physiological roles for FGF2 include inflammation, in which stress-induced activation of caspase 1 leads to release of FGF2 (REF. 79); and asthma, as FGF2 enables airway smooth muscle cells to proliferate in response to asthma triggers80.
Interestingly, FGF1 is a proliferative factor for human preadipocytes and may be important to the overall regulation of human adipogenesis81.
Pathophysiology
A possible role for FGF1 in humans is suggested by its increased levels in the pericardial fluid of patients with cardiac ischaemia82. Incubation of endothelial cells with FGF1 leads to microvascular branching83, and the ligand also has anti-apoptotic activity84, suggesting mechanisms through which it might function in vascular injury.
Therapeutic potential of FGF1
FGF1 has some therapeutic potential for cardiovascular disorders. Phase I trials have shown that intramyocardial injection of FGF1 during coronary artery bypass graft surgery improves collateral artery growth and capillary proliferation85. Beneficial effects of FGF1 on the peripheral circulation have also been shown. Injection of a plasmid that encodes FGF1 (NV1FGF) into the leg improved perfusion of end-stage lower-extremity ischaemia in a Phase I trial86 and led to a twofold reduction in the need for amputation in patients with critical limb ischaemia in a recent Phase II study87. Interestingly, distal blood and oxygen pressure were similar after injection of either NV1FGF or placebo88 and the mode of action of FGF1 might not have been primarily angiogenic. The TAMARIS (Therapeutic Angiogenesis for the Management of Arteriopathy in a Randomized International Study) Phase III trial is underway to evaluate NV1FGF and will further address the possibility of a systemic mechanism of FGF1 action.
FGF1 can repair nerve injuries. It enabled functional regeneration of transected spinal cords in rats89 and restored some motor function to paralyzed limbs in a 6-month-old boy with brachial plexus avulsion90. FGF1 administration has benefited patients with chronic transverse myelitis91, and the combination of sural nerve grafts with FGF1 treatment partly restored ambulation to a paraplegic92.
Therapeutic potential of FGF2 in cardiovascular disease
In an unblinded trial, a single bolus of FGF2 reduced the size of ischaemic regions in the myocardium, improved treadmill performance and reduced the frequency of angina93,94. However, in the FGF Initiating RevaScularization Trial (FIRST), FGF2 treatment conferred some benefit in the first few months, but these improvements were not sustained, whereas continued improvement was seen in the placebo group95,96. Using a different protocol, implanting heparin beads containing adsorbed FGF2 over ischaemic myocardium reduced the size of the ischaemic region and ameliorated the associated symptoms, with beneficial effects being retained for 3 years of follow-up97. This treatment method does require open-chest delivery, but it is one of the few examples of a sustained positive response among Phase I trials with FGF2.
FGF2 has also been examined for its efficacy in the peripheral circulation. Patients suffering from claudication who received intra-arterial FGF2 showed improved calf blood-flow compared with patients who received placebo98. However, in the TRAFFIC (Therapeutic Angiogenesis with Recombinant Fibroblast Growth Factor-2 for Intermittent Claudication) study, none of the immediate improvements, such as peak walking time, was ultimately statistically significant99 (BOX 2).
Box 2. Challenges to the treatment of cardiovascular disease with fibroblast growth factors.
The best method for administering growth factors for the purpose of angiogenic stimulation has been a matter of some discussion. The long-term presence and slow release of growth factors in the tissue is important for maintenance of new vasculature264, but the mean half-life of fibroblast growth factor 2 (FGF2) in the body is only about 7.6 hours. This half-life is extended when heparin is co-administered265,266. Protein engineering may prove useful, as the half-life of FGF1 in the presence of heparin can be increased by a single amino acid mutation267, and recent developments have shown that stabilizing mutations within the β-barrel can dramatically decrease the likelihood of protein unfolding268.
Only 3–5% of the dose is typically retained in the myocardium 150 minutes after intracoronary injection269,270. At 24 hours after intracoronary infusion, the myocardium no longer retains any portion of the dose94. The intravenous route is even less effective because of first-pass pulmonary metabolism of FGFs. Intramyocardial delivery of FGFs delivers the best dose of growth factor, as it allows targeting of ischaemic areas of the heart and has prolonged tissue retention — up to tenfold higher than that achieved by intracoronary injection271. However, intramyocardial injection may not be the most appropriate therapy if the goal is to cause growth of epicardial vessels. Adenoviral vectors may also not produce expression of FGFs for a sufficient length of time to achieve beneficial effects272.
It seems that a single intracoronary or intra-arterial injection of FGF, although helpful in animals, will be unlikely to affect clinical progress in patients78. Intramuscular or intramyocardial administration might yet be feasible, owing to a high retention of protein and its slow removal273. The potential for haemangioma formation274 or neovascularization of atherosclerotic plaques275 is a concern, however, for long-term safety. The advantages of protein therapy include precision in dosing, the ability to combine multiple proteins in a treatment and a well-characterized safety profile276. In summary, therapy with exogenous FGFs has not yet altered the course of cardiovascular disease in humans. Heparin derivatives are perhaps one alternative route of angiogenesis therapy277 and vascular endothelial growth factor A also holds promise, given its greater specificity for angiogenesis.
Therapeutic potential of FGF2 in cancer
Thalidomide is an inhibitor of FGF2-induced angiogenesis100, and Phase II trials have demonstrated its benefit in patients suffering from androgen-independent metastatic prostate cancer101 or renal cancer102. Suramin, a polysulphated naphylurea, interferes with FGF signalling by mimicking heparin, and is efficacious in bladder, kidney and prostate cancers103–107. Treatment of prostate cancer with suramin also enhances the activity of other chemotherapeutics, such as doxorubicin108 (TABLE 1), possibly by reducing the ability of FGF1 and FGF2 to enable broad-based resistance to anticancer drugs109. The high doses of suramin required for clinical efficacy produce substantial side effects, however, including coagulopathy110. Suramin is only one example of many heparinoids111. One of the most promising heparinoids is PI-88, a heparanase inhibitor that has been studied widely in recent clinical trials112.
Interferon-α (IFNα) and IFNβ can downregulate FGF2 in kidney, bladder and prostate human cell lines113; accordingly, administration of these interferons inhibits FGF2 expression and the growth of bladder carcinoma cells114. Some evidence suggests that inhibition of FGF2 signalling slows the growth of tumours by inhibiting vascularization115. However, FGF2 levels do not generally correlate with microvessel density in tumours116, indicating that the mechanism underlying the anti-tumour effects of interferons mediated through FGF2 may not be solely angiogenesis based. In 1995, the success of the ECOG (Eastern Cooperative Oncology Group) Trial 1684 led to the approval of IFNα for the treatment of patients with melanoma117. However, because of high toxicity, the use of IFNs in biochemotherapy regimens for metastatic melanoma is no longer recommended118. Gene silencing by antisense targeting of FGF2 and FGFR1 in models of human melanoma caused a dramatic reduction in the size of tumours115, but many challenges remain for the application of antisense technology in general119.
Therapeutic potential of FGF2 in other disorders
Interestingly, patients suffering from major depressive disorder have deregulated FGF transcript levels that are restored by serotonin reuptake inhibitors120. In rats, Fgf2 and Fgfr1 mRNA levels were downregulated in the hippocampi following social defeat121, and intra-cerebroventricular administration of FGF2 produced antidepressant-like effects122. These data suggest that manipulation of FGF signalling could yield benefits in the treatment of mood disorders.
FGF2 has also been studied in cartilage homeostasis123, recombinant FGF2 has been shown to have some efficacy in wound healing in patients suffering from ulcers124, and a recent Phase II study has suggested that recombinant FGF2 (trafermin) could aid in regenerating alveolar bone in patients with periodontitis125.
FGF4 subfamily
Biology
FGF4 has wide-ranging functions in development, including cardiac valve leaflet formation126 and limb development127. Fgf4 knockout mouse embryos experience post-implantation lethality owing to the necessity of FGF4 for trophoblast proliferation128. FGF5 negatively regulates a step of the hair follicle growth cycle. Fgf5 knockout mice exhibit abnormally long hair in the absence of any other defect129, and loss-of-function mutations in the Fgf5 gene account for hereditary variations in hair length in canines and felines130,131. FGF6 plays a part in myogenesis132, and Fgf6 knockout mice have defective muscle regeneration, with significantly increased fibrosis following a freeze–crush injury133.
Therapeutic potential
Whereas FGF2 has primarily been studied as preparations of recombinant protein, FGF4 has been administered by means of gene therapy. Alferminogene tadenovec (Ad5FGF4) is FGF4 encoded within replication-deficient human adenovirus serotype 5. Phase I and II clinical trials revealed improvements in treadmill exercise capacity, but Phase III trials were discontinued when a high placebo response was revealed134. The Phase III Angiogenic Gene Therapy trial (AGENT) demonstrated the safety of the therapeutic method, as flu-like symptoms or hepatic toxicity were rarely observed135. A review of all patients showed no significant benefit from the treatment, but a reanalysis revealed a gender-specific response that was traced in part to a reduced placebo response in women136. Cardium Therapeutics has initiated the AWARE (Angiogenesis in Women with Angina pectoris who are not candidates for Revascularization) Phase III trial to study the gender-specific response of Ad5FGF4 in women.
FGF7 subfamily
Biology
FGF7, also known as keratinocyte growth factor, is expressed specifically in mesenchyme. Fgf7−/− mice are viable and fertile, exhibiting only minor abnormalities, such as matted hair137, and about 30% fewer nephrons compared with controls138. FGF7 levels are increased by up to 150-fold in skin after cutaneous injury139, also being increased after bladder and kidney injury140,141.
Homozygous deletions in FGF3 were shown to cause hereditary deafness, leading to total inner ear agenesis in humans9. The specificity of FGF3 for this effect is impressive: this FGF3−/− human knockout showed no other symptoms apart from a few dental defects. Fgf10 (also known as Kgf2) knockout mice lack limbs and pulmonary structures282,283, in addition to exhibiting defects in all other branching organs. FGF22, along with FGF7 and FGF10, is a presynaptic organizer with roles in vesicle clustering and neurite branching143.
Pathophysiology
LADD syndrome, an autosomal-dominant disease characterized by hearing loss, dental anomalies, and lacrimal and salivary gland hypoplasia, is caused by FGF10 loss-of-function mutations8.
A correlation between disease and FGF7 subfamily expression is observed for several disorders. Overexpression of FGF7 correlates with inflammation in patients suffering from inflammatory bowel disease, suggesting that FGF7 may have a compensatory role144. FGF7 and FGF10 are also overexpressed in psoriatic skin145,146. FGF10 and FGF7 are thought to act as andro-medins (mediators of androgen action)147,148 and as such could have a role in the pathogenesis of prostate cancer by facilitating epithelial cell proliferation.
Therapeutic potential
Palifermin, an N-terminally truncated form of FGF7 with increased stability, is FDA approved for the treatment of chemoradiation-induced oral mucositis in patients undergoing bone marrow transplantation. When administered on 3 consecutive days before high-dose chemotherapy, as well as for 3 days following haematopoietic stem cell transplantation, palifermin reduced the median duration of mucositis from 9 to 6 days, and reduced the incidence of grade 4 mucositis from 62% to 20%. This corresponds with a significant improvement in the patients’ quality of life, as grade 4 mucositis is of such severity that oral feeding is impossible. Importantly, palifermin also reduced the patients’ use of opioid analgesics, which indicates that there was reduced pain. The adverse events associated with palifermin were mild and transient, and most were attributable to the underlying cancer or concurrent chemotherapeutic regimen149.
Palifermin acts mainly by increasing cell proliferation. Studies indicate that the increased epithelium thickness produced by a dose of palifermin can be maintained for up to 1 week150,151.
Other mechanisms of FGF7 action could include upregulation of NRF2, which activates genes that encode antioxidant enzymes152. Inflammatory cytokines are important to the pathogenesis of mucositis, and FGF7 may affect this aetiology both by reducing the ratio of T-helper type 1 to T helper type 2 cytokines153 and by reducing tumour necrosis factor-α and IFNγ levels through its induction of interleukin 13 (REF. 154).
New applications for palifermin are being investigated. Palifermin reduces the incidence of graft-versus-host disease and also improves immune function in animal models155. However, these findings have not yet been corroborated in clinical trials, perhaps because of the inclusion of methotrexate in the stem cell transplantation regimen, a compound that is cytotoxic to epithelial cells and could counteract the beneficial proliferative effects of palifermin156. Treatment of injured epithelia with FGF7 results in an improved wound-healing response157, suggesting the potential use of FGF7 for tissue engineering.
Human Genome Science explored recombinant FGF10 (repifermin) as a treatment option for ulcerative colitis and mucositis, but its development was terminated in 2004 after it failed in several clinical trials158,159.
FGF8 subfamily
Biology
FGF8 is involved in brain, limb, ear and eye development160 and, along with FGF17, is crucial for forebrain patterning161. Fgf8−/− mice do not undergo gas-trulation162, Fgf17−/− mice exhibit abnormalities in the development of cerebral and cerebellar structures163, and Fgf18−/− mice have decreased expression of osteogenic markers and delayed long-bone ossification164,165.
Pathophysiology
Loss-of-function mutations in FGF8 that affect its binding to FGFR1c or cause degradation of FGF8 lead to Kallmann’s syndrome, a developmental disorder characterized by anosmia and hypogonadism10.
Therapeutic potential
FGF18 is currently under investigation by Merck Serono for the treatment of osteoarthritis, which is a disease involving degeneration of cartilaginous tissue. FGF18 has an anabolic effect on cartilage: a single intravenous injection of FGF18 leads to increased deposition of cartilage in the ribs, trachea, spine and joints166. In a rat model of osteoarthritis, intra-articular injection of FGF18 increased cartilage formation167. Merck Serono is now following up this preclinical data with Phase I clinical trials to study the effects of FGF18 on osteoarthritis progression in humans.
Monoclonal antibodies against FGF8 have shown some efficacy in mouse models of breast cancer and prostate cancer168,169.
FGF9 subfamily
Biology
Fgf9 knockout mice demonstrate male-to-female sex reversal and lung hypoplasia that quickly leads to postnatal death170. Importantly, the FGF9 subfamily, which signals from epithelium to mesenchyme, functions in a reciprocal way to the FGF7 subfamily, which signals from mesenchyme to epithelium. FGF9 stimulates mesenchymal proliferation, and mesenchyme produces ligands of the FGF3, FGF7, FGF10 and FGF22 subfamily. Accordingly, knocking out FGF9 disrupts the mesenchymal–epithelial signalling loop that helps regulate these FGFRb-binding ligands. Reduced mesen-chymal proliferation in turn leads to a reduced production of FGF3, FGF7, FGF10 and FGF22 subfamily ligands, which is the proximate cause of lung hypoplasia171. Fgf16 knockout mice exhibit significant cardiac defects172.
Pathophysiology
SNPs in FGF20 have recently been associated with Parkinson’s disease173, in which they have been shown to increase FGF20 translation in vivo, leading to increased expression of α-synuclein, one of the causative agents of this disease 174.
Therapeutic potential
The potential therapeutic application of FGF20 in Parkinson’s disease is beginning to be explored. FGF20 is a neurotrophic factor for rat midbrain dopaminergic neurons175, and monkey stem cells differentiated in vitro into dopaminergic neurons after treatment with exogenous FGF20 and FGF2 have been transplanted into a primate Parkinson’s disease model, which alleviated some symptoms176. Thus, despite the negative role of FGF20 in Parkinson’s disease aetiology in vivo, the ligand shows some promise in stem cell biology in vitro.
Under the name velafermin, FGF20 was investigated by Curagen for the purpose of alleviating oral mucositis. Although Phase I clinical trials were promising177, the project was terminated in October 2007 when Phase II trials failed to meet therapeutic targets.
Endocrine FGF ligands
The endocrine ligands, FGF19, FGF21 and FGF23, currently have the greatest promise for pharmacological development among the FGFs (FIG. 4; TABLE 3). The decreased HSGAG binding of the endocrine FGFs leads to increased diffusion of these FGFs from their source, but it also reduces the ability of HSGAGs to promote the binding of these FGFs to their receptors. In order to signal, the endocrine FGFs depend on the presence of α-klotho or β-klotho (encoded by Kl and Klb, respectively) in their respective target tissues. The klotho proteins bind both the endocrine FGFs and their cognate FGFRs to increase ligand–receptor affinity186,187,191–196.
Figure 4. The physiology of fibroblast growth factor 19 (FgF19), FgF21 and FgF23.

a | Bile acids activate the FXR receptor in the intestine, leading to expression of FGF19 in the ileum. FGF19 circulates to the liver, where it acts through FGF receptor 4 (FGFR4) to inhibit bile acid synthesis and lipogenesis. b | FGF21 mediates the fasting response and is regulated by peroxisome proliferator-activated receptor-α (PPARα) and PPARγ in liver and adipose tissue, respectively. The biology of FGF21 in model systems and humans is still being elucidated, but among its many functions are increasing glucose uptake in adipose tissue, improving β-cell function, inhibiting glucagon secretion, increasing ketogenesis and regulating lipolysis and lipogenesis in a complex manner. FGF21 is expressed in liver, adipose and pancreatic tissue. It acts primarily on adipose tissue. The effects of FGF21 on liver function are probably accomplished through indirect mechanisms as it does not signal through FGFR4. c | FGF23 production is upregulated in bone in response to high serum phosphate and vitamin D levels. FGF23 then circulates to the parathyroid gland, intestine and kidney. In the intestine, FGF23 downregulates 1α-hydroxylase so as to reduce the levels of activated vitamin D, thereby inhibiting absorption of phosphate from the diet. The repression of parathyroid hormone (PTH) by FGF23 also helps to downregulate 1α-hydroxylase. In the kidney, FGF23 inhibits Na+–phosphate ion co-transport and thus increases excretion of phosphate. CYP7A1, cytochrome P450 7A1; SCD1, stearoyl CoA desaturase 1.
α-Klotho was first discovered when mice that lacked the gene aged prematurely178. Overexpression of Kl can extend the lifespan of mice179. The extracellular domain of the α-klotho protein is secreted into the blood and cerebrospinal fluid, where it acts as a humoral factor180,181. In particular, α-klotho regulates Ca2+ metabolism by binding the Na+–K+-ATPase182 and by acting as a β-glucuronidase to hydrolyse the extracellular sugar residues of the TRPV5 ion channel, thereby trapping the channel on the cell membrane183.
Abnormalities of phosphate metabolism and bone mineral density in Kl−/− mice were similar to those observed in Fgf23 knockout mice184,185. This phenotypical similarity led to the discovery that FGF23 requires α-klotho to activate FGFRs186,187. Similar reasoning identified the necessity of β-klotho for FGF19 signalling: both Klb−/− and Fgf15−/− (the orthologue of human FGF19) mice have increased expression of the liver-specific gene CYP7A1 (cytochrome P450 7A1) and increased bile acid pools188,189. A similar phenotype is also seen in Fgfr4−/− mice190, which lack a principal receptor for FGF19 and the principal liver FGFR. In vitro studies have confirmed that FGF19 requires β-klotho for signalling191–193. Some overexpression studies also suggested that FGF19 might bind α-klotho, but this may only occur in non-physiological conditions193. FGF21 is also a β-klotho-dependent ligand191,193–196.
FGF19
Biology
FGF19 transcripts are found in brain, cartilage, skin, retina, kidney, gall bladder and small intestine197,198. Expression occurs primarily in the ileum from which the ligand circulates to the liver and carries out its main functions189. Interest in FGF19 was stimulated after decreased adiposity, increased energy expenditure, reduced liver triglycerides, increased fatty acid oxidation, reduced glucose levels and improved insulin sensitivity were observed in Fgf19 transgenic mice6. Moreover, these mice did not become obese or diabetic on a high-fat diet. These metabolic effects were not mediated through insulin-like growth factor 1, growth hormone, the thyroid hormone triiodothyronine or leptin, none of which was increased in the transgenic mice6. Metabolic rate was similarly increased in mice given recombinant FGF19, thereby confirming the genetic data. FGF19 treatment was also able to prevent or reverse diabetes in mice that were made obese by ablation of brown adipose tissue or genetic knockdown of leptin3.
FGF19 mediates its physiological effects in the liver through the regulation of transcription3. FGF19 gene expression is directly induced by the farnesoid X receptor, a nuclear receptor that recognizes bile acids. In turn, FGF19 inhibits CYP7A1, the enzyme that catalyses the rate-limiting step in bile acid synthesis199. Studies in humans have shown that serum FGF19 levels vary diurnally, with rises in serum FGF19 of up to 250% occurring 1–2 hours after a post-prandial increase in bile acids200. FGF19 also downregulates acetyl CoA carboxylase 2 (ACC2), which converts acetyl CoA to malonyl CoA, a repressor of carnitine palmitoyl transferase 1 (CPT1)-initiated fatty acid oxidation. By reducing ACC2 activity, FGF19 increases fatty acid oxidation. FGF19 also down-regulates the lipogenic enzyme stearoyl CoA desaturase 1 (SCD1)3,199. FGF19 additionally regulates gall bladder filling in part by a cAMP-dependent relaxation of gall bladder smooth muscle201.
FGFR4 is the predominant receptor by which FGF19 mediates its liver-specific effects. Experiments in Fgfr4−/− mice showed that FGF15 (the mouse orthologue of FGF19) was unable to repress CYP7A1 activity189, and Fgfr4−/− mice have a phenotype that is indicative of reduced FGF19 activity, such as increased bile acid pools190. FGF19 has been believed to be specific for FGFR4 since 3T3 fibroblast cell lines, which lack FGFR4, were found to be unresponsive to FGF19 (REF. 198). This is supported by more recent in silico modelling of the interaction of FGF19 with FGFRs202 and pull-down experiments in the presence of β-klotho192,193. However, it is unlikely that FGF19 is entirely specific for FGFR4. Overexpression studies in HEK293 and 3T3-L1 cells show that FGF19 can bind and activate other FGFRs in the presence of β-klotho191,194. Most importantly, FGF19 can also cause an increase in gall bladder volume in Fgfr4−/− mice, indicating that FGFRs other than FGFR4 can mediate the effects of FGF19 in gall bladder201. Although the overlapping phenotypes of FGF19- and FGF21-overexpressing mice suggest that FGF19, like FGF21, might act on adipose tissue that predominantly expresses FGFR1, FGF19 only weakly activates cells in excised white adipose tissue191.
Pathophysiology
Deregulated FGF19 signalling or FGF19 mutant proteins have not yet been associated with human metabolic disease, and plasma FGF19 levels in patients with anorexia nervosa are the same as in controls203.
Therapeutic potential
One major concern for the potential translation of FGF19 to the clinic is the evidence that Fgf19 transgenic mice develop hepatocellular carcinomas with age204. Nonetheless, it might be possible to find a therapeutic window in which FGF19 is efficacious but not tumorigenic.
Interestingly, Genentech has shown that anti-FGF19 monoclonal antibodies inhibit growth of colon tumour xenografts in vivo and prevent hepatocellular tumours in FGF19 transgenic mice205. Some of this tumour growth inhibition is mediated by downregulating β-catenin signalling206. This further confirms a role for FGF19 in oncogenesis and suggests that its mitogenicity could be controlled pharmacologically.
The need for experimental data on FGF19 action in primates has been noted in another review270, and target identification through FGF19 administration to mice lacking different metabolic enzymes should be useful207.
If FGF19 does eventually prove to be safe for use in humans, it might represent an important therapeutic option in the treatment of type 2 diabetes and its associated disorders.
FGF21
Biology
FGF21 is expressed in liver and thymus208, adipose tissue209 and islet β-cells in the pancreas210. Its expression can be induced in skeletal muscle in response to Akt activation211. The role of FGF21 in metabolic regulation was first discovered in association with its adipocyte-specific ability to cause glucose uptake, which is accomplished in part by upregulating transcription of the glucose transporter GLUT1 (REF. 4). Daily injections of FGF21 for 7 days in various murine models of diabetes (ob–ob mice, db–db mice, and Zucker diabetic fatty rats) reduced the levels of plasma glucose, triglycerides, gluca-gon and insulin4. Administration of FGF21 to ob–ob mice for 2 weeks reduced body weight by 20% and ameliorated hyperglycaemia212, and similar results were found in mice with diet-induced obesity213. Likewise, Fgf21 transgenic mice exhibited improved insulin sensitivity and glucose clearance, lower fasting glucose levels, lower glucagon levels, reduced weight, leaner hepatic tissue, increased retention of brown adipose tissue and smaller adipocytes, relative to controls4. The Fgf21 transgenic mice consumed twice as much food as did control mice, but were nonetheless resistant to diet-induced obesity. In fact, Fgf21 transgenic mice are markedly smaller than their control littermates, which may be a consequence of the suppression of STAT5 (signal transducer and activator of transcription 5) — a mediator of growth hormone signalling — by FGF21 (REF. 214) Adenoviral knockdown of FGF21 in mice leads to fatty liver, lipaemia, reduced levels of serum ketones and increased cholesterol levels215. These results are consistent with data from experiments with rhesus monkeys, in which levels of fasting glucose, triglycerides, glucagon and insulin were reduced after FGF21 administration216. A small reduction in weight and improved lipoprotein profiles were also observed. In particular, high density lipoprotein c levels were 80% higher than control levels after 6 weeks of FGF21 administration.
Another action of FGF21 is the preservation of β-cell function. Although FGF21 has no effect on normal rat pancreatic islets, it does increase insulin secretion from diabetic islets and protects β-cells from apoptosis by activating the ERK1–ERK2 and Akt pathways, respectively. Under conditions of glucolipotoxicity or inflammation, FGF21 reduces caspase 3 and caspase 7 activity, probably by Akt-induced phosphorylation of BCL2-antagonist of cell death (BAD), a suppressor of apoptosis210. The anti-apoptotic effects of FGF21 are probably also exerted by reducing glucose and triglyceride levels, creating a less toxic environment for β-cells210. β-Cell mass was preserved in db–db mice after 8 weeks of FGF21 administration, and treated animals had 280% more β-cells per histological section than untreated animals210. Under diabetic conditions, insulin biosynthesis becomes important for the insulin response, and FGF21 supports this function by preserving β-cells.
A role for FGF21 in the fasting response became apparent when it was observed that FGF21 expression was induced in mice by starvation or a ketogenic diet215,217 and that FGF21 is also induced by fasting and suppressed by refeeding in rats218. In fact, microarray data show that FGF21 is the most markedly upregulated gene in ketotic mice215.
The molecular mechanisms by which FGF21 mediates the fasting response is still being elucidated. It is known that peroxisome proliferator-activated receptor-α (PPARα) regulates FGF21 activity215,217. PPARα is a nuclear receptor that responds to fatty acid metabolites, mediates the starvation response and upregulates genes that are involved in fatty acid transport and oxidation219. PPARα directly induces FGF21 mRNA transcription in mouse liver and human hepatocytes, and chromatin immuno-precipitation experiments show that PPARα binds to the Fgf21 promoter in mouse liver tissue217. PPARα is not solely responsible for inducing FGF21 expression, however, as FGF21 expression can be induced by a ketogenic diet even in Ppara−/− mice215. FGF21 also reduces physical activity and induces torpor in fasting mice, indicating that FGF21 may also be involved in a neurological response to fasting217.
Several lines of evidence suggest that FGF21 is involved in lipolytic processes. Adenovirus-mediated short hairpin RNA knockdown of FGF21 in mice down-regulated genes that are involved in β-oxidation as well as triglyceride accumulation215. Although the PPARα target genes were not upregulated in Fgf21 transgenic mice, increased numbers of mRNA transcripts of lipases were observed in the liver. Two mediators of ketogenesis — CPT1a (carnitine palmitoyltransferase 1a) and HMGCS2 (hydroxymethylglutaryl-CoA synthetase 2) — were also post-transcriptionally upregulated217. Interestingly, as there was less adrenaline in the urine of transgenic mice compared with controls217, the lipolytic activities of FGF21 are not mediated through catecholamines.
In tension with these results is the observation that FGF21 is also a target of PPARγ, which is a key regulator of adipogenesis220. Moreover, PPARγ agonists act in synergy with FGF21 to promote glucose transport and triglyceride formation221. Some experiments have shown that FGF21 does not upregulate lipolytic genes in adipocytes222, which contradicts data from earlier experiments217. Furthermore, FGF21 significantly attenuated noradrenaline-stimulated lipolysis in human adipocytes in vitro222. These observations are consistent with the fact that Fgf21 knockdown in mice leads to lipaemia215, and perhaps that some of the pro-lipolytic changes seen in transgenic mice are adaptative217. It has also been proposed that the anti-lipolytic effect of FGF21 contributes to its role in insulin sensitization222.
Some coherence may be brought to the conflicting data by the fact that PPARγ agonists upregulate FGF21 expression in adipose tissue but not in liver, whereas PPARα agonists upregulate FGF21 expression in liver, but not in adipose, tissue223. Furthermore, administration of FGF21 to ob–ob mice for 2 weeks dramatically suppressed liver lipogenic genes, such as stearoyl CoA desaturase 1 (Scd1), and at the same time upregulated the expression of Scd1 and other lipogenic genes such as acetyl CoA carboxylase 2 (Acc2) in white adipose tissue212. Several lipases and PPARγ co-activator 1α (PGC1α), a regulator of oxidative metabolism, were also upregulated in white adipose tissue. This led to the hypothesis that FGF21 leads to a futile cycling of lipogenesis and lipolysis in white adipose tissue212.
The potential for tissue-specific activity of FGF21 raises the question of its receptor specificity. FGFR1 and FGFR4 are the principal FGFRs in white adipose tissue and liver, respectively191. FGF21 can bind FGFR4 in in vitro overexpression experiments195, but cannot activate H4IIE hepatocytes that express β-klotho191. This suggests that the action of FGF21 on liver may be indirect, as already suggested by its repression of STAT5 levels214. FGFR1 from adipose tissue is therefore probably the main receptor for FGF21.
Studies in humans have further clarified the profile of FGF21 biology. Although a ketotic diet induces FGF21 expression in mice215, it does not do so in humans, in whom ketogenesis is independent of FGF21 and FGF21 levels only become increased after prolonged fasting for 7 days224. Earlier induction of FGF21 expression may occur in liver and adipose tissue during fasting, but these tissues were not specifically examined. In humans, FGF21 levels varied 250-fold among 76 healthy individuals and did not correlate with serum triglycerides, glucose, body mass index, age or gender. There was no diurnal variation and FGF21 was unrelated to bile acid synthesis. FGF21 expression is induced by PPARα agonists in humans224. Interestingly, although acute fasting increases FGF21 levels, FGF21 levels are significantly reduced in individuals suffering from chronic malnourishment as a result of anorexia nervosa203.
Pathophysiology
As for FGF19, deregulated FGF21 has not been shown to be a causative factor in human metabolic disorders. In human cross-sectional studies, a positive association of serum FGF21 with adiposity, insulin resistance, and adverse lipid profiles has been observed209, although the correlation with insulin resistance is abolished when controlling for body mass index. Fasting FGF21 levels are also increased in individuals with type 2 diabetes225, which may indicate that resistance develops to FGF21 or may represent a compensatory increase in FGF21.
Therapeutic potential
FGF21 is currently of great therapeutic promise as, unlike FGF19, it has an excellent safety profile. FGF21 did not show significant mitogenic potential in cell lines and Fgf21 transgenic mice did not demonstrate any tissue hyperplasia until they were 10 months of age4. Furthermore, FGF21 administration does not lead to either hypoglycaemia or oedema, which are two common side effects of current diabetes therapies4,212,213,216.
The biological profile of FGF21 gives this ligand the potential to address the causative factors of type 2 diabetes. The progressive loss of β-cells through β-cell apoptosis and hyperglycaemia caused by inappropriately increased glucagon levels that are unrepressed following feeding are among the aetiologies of type 2 diabetes. FGF21 has been shown to increase β-cell survival210 and inhibit glucagon secretion4,216. The ability of FGF21 to normalize glucose levels and facilitate insulin sensitization is well attested and reproducible4,212,216.
The pharmacology of FGF21 remains to be fully elucidated but it seems that, by initiating a wide range of cellular responses, FGF21 has a pharmacodynamic action that long outlasts the presence of the ligand4. Interestingly, the ability of FGF21 to ameliorate hyper-glycaemia was apparent at doses of 0.1 mg per kg per day that achieved steady-state levels of about 7.4 ng per ml in mice, whereas the effect of FGF21 on weight loss was increased by higher doses212.
FGF23
Biology
FGF23 was initially shown to be preferentially expressed in the ventrolateral thalamic nucleus226. FGF23 was also identified as a gene that is mutated in patients with hypophosphataemic rickets7. Since then, studies have revealed that FGF23 is a key humoral regulator of phosphate homeostasis.
FGF23 is most highly expressed in bone227,228, from which it circulates through the blood to regulate vitamin D and phosphate metabolism in kidney. Renal phosphate reabsorption is suppressed in Fgf23-overexpressing mice229,230, owing to downregulation of type IIa and IIc sodium–phosphate co-transport on the apical surface of renal proximal tubular epithelial cells231–233. FGF23 also downregulates enzymes that metabolize vitamin D, leading to reduced levels of available active 1,25-dihydroxy-vitamin D. Because 1,25-dihydroxyvitamin D enhances intestinal phosphate absorption, this effect of FGF23 also leads to reduced phosphate levels231.
FGF23 also acts on the parathyroid gland to inhibit parathyroid hormone (PTH) secretion234. PTH increases the uptake of phosphate from bone and upregulates 1α-hydroxylase, leading to increased vitamin D activation and enhanced phosphate reabsorption in the intestine. Notably, FGF23 can still normalize serum phosphorous levels in thyroparathyroidectomized rats232.
Fgf23−/− mice suffer from hyperphosphataemia, increased 1,25-dihydroxyvitamin D levels, hypoglycaemia, atrophy of the thymus, immature reproductive organs and increased serum triglycerides185,235. Hyperphosphataemia and soft tissue calcification in Fgf23−/− mice are ameliorated by additionally knocking down the genes encoding 1α-hydroxylase or the vitamin D receptor236–238. This indicates that an increase in 1,25-dihydroxyvitamin D levels is responsible for the hyperphosphataemia and calcification seen in FGF23-deficient mice239. Indeed, FGF23 has been shown to lower 1α-hydroxylase levels by a vitamin D receptor-independent mechanism240. High vitamin D levels lead to tissue atrophy through apoptosis, and so FGF23 can prevent vitamin D-induced apoptosis by suppressing 1α-hydroxylase241.
Pathophysiology
Mutations in FGF23 are implicated in a wide range of disorders. Autosomal dominant hypophos-phataemic rickets is caused by mutations in a subtilisin-like proprotein convertase cleavage site in FGF23 that render the protein less susceptible to degradation, thereby increasing the biological activity of FGF23 and leading to hypophosphataemia7. X-linked hypophosphataemic rickets is caused by inactivating mutations of PHEX, a gene that encodes a metalloprotease of the M13 family242. By an unknown mechanism, this leads to increased FGF23 levels in many patients with this disease243,244.
FGF23 levels are increased tenfold above controls in patients with tumour-induced osteomalacia, a tumour-associated syndrome of renal phosphate wasting243,244. These data, combined with the observation that FGF23 serum concentrations decrease after tumour removal245,246, show that FGF23 is important to the pathology of phosphate wasting in TIO. Circulating FGF23 levels are also increased and correlate with disease burden in patients with fibrous dysplasia, a disorder in which normal bone is replaced by fibro-osseous tissue227.
Reduced FGF23 signalling also causes pathology. Familial tumoural calcinosis (FTC) is a disorder marked by hyperphosphataemia in which individuals develop calcified masses, often within the joints247. Even though Fgf23−/− mice do not develop calcified masses185, several mutations in the Fgf23 gene have been shown to contribute to hyperphosphataemic tumoral calcinosis248–250. These missense mutations destabilize the tertiary structure of the FGF23 protein and increase its susceptibility to degradation, such that full-length species of FGF23 occur at low concentrations in affected patients284. Mutations in α-klotho have also been implicated in FTC251,285; in such cases, insensitivity to circulating FGF23 causes FTC, rather than the increased processing of FGF23 that results from FGF23 mutations. Further defects that cause FTC include loss-of-function mutations in the glycosyltrans-ferase GALNT3. Although FGF23 is O-glycosylated252 and GALNT3 selectively directs O-glycosylation at Thr178 of FGF23 (REF. 253), FGF23 probably does not contribute to FTC caused by GALNT3 mutations, as FGF23 is increased in these patients, probably in compensation for their hyperphosphataemia254.
FGF23 is increased in patients with renal failure by 100–1,000-fold, partly owing to decreased renal clearance but also suggesting that it might have a compensatory role in this disease255,256. However, whether the increased FGF23 levels in chronic kidney disease are beneficial or harmful is still a matter of debate257. In any case, FGF23 levels do correlate strongly with disease outcome. Increased levels of serum FGF23 at the beginning of dialysis treatment predict a significant increase in 1 year mortality in patients with chronic kidney disease258. FGF23 serum levels are also predictive of the development of secondary hyperparathyroidism259.
Therapeutic potential
Given its involvement in the pathogenesis of human disease, FGF23 holds promise as a therapeutic target, and a range of studies have confirmed its potential. Administration of neutralizing antibodies that target FGF23 normalized phosphate and vitamin D concentrations in the serum of mice with hypophosphataemia260, which points to the possible application of FGF23 neutralizing antibodies to the treatment of hypophosphataemic disorders. Neutralizing antibodies against N- and C-terminal regions of FGF23 have also proved successful at increasing serum phosphate and activated vitamin D levels in mice261. Another potential avenue of therapy could be the use of C-terminal peptides of FGF23. FGF23 binds to klotho through its C-terminal region, and these peptides could therefore abrogate binding and eliminate FGF–FGFR klotho-dependent signalling14. This possibility is currently being investigated in our laboratory (R. Goetz et al., unpublished observations). The contribution of FGF23 to chronic kidney disease is unclear, but this ligand may also have pharmacological significance in this context.
Concluding remarks
The mitogenic and cytoprotective properties of FGF7 are already being put to advantageous use in the clinic. Other FGFs, including FGF1, FGF2 and FGF4, have been tested in clinical trials and may eventually be used to treat cardiovascular disease. FGF18 is in the beginning stages of development for the treatment of osteoarthritis, and FGF5 inhibitors may find a niche in the treatment of some forms of non-autoimmune alopecia. The precise role of FGFs in mood disorders requires considerably more investigation262, but it is possible that some therapeutic application will arise in this field, especially as so many of the FGFs are involved in brain patterning and neurological development.
Therapies that target RTKs are already common. It remains to be seen whether FGFR-specific inhibitors will have an impact on the treatment of cancer. The recent work that revealed the ability of small-molecule FGFR inhibitors to cause cell death in cancer cells is at least a proof of principle65. Currently, the development of inhibitors of the FGFR–PLCγ interaction looks promising because their concomitant use with RTK inhibitors may slow the onset of drug resistance.
Of the endocrine ligands, FGF21 currently holds the most potential as a drug target, owing to its beneficial impact in animal models of diabetes and its lack of toxicity. Although recombinant FGF19 also improves aspects of the metabolic syndrome in mouse models, its ability to initiate tumour growth in transgenic mice as well as its expression in human tumours is a significant cause for concern. Further investigation of its side-effect profile is vital.
The involvement of FGF23 in disease makes it a particularly attractive therapeutic target. Antibodies against FGF23 or peptide analogues of the FGF23 C terminus should ultimately prove useful in combating human hypophosphataemic diseases. Increased levels of FGF23 in chronic kidney disease is a subject of intense study; further applications of the ligand to the treatment of renal disorders may yet be found.
FGF-based therapies are still relatively new to the clinic and the broad biology of this family of growth factors has yet to be fully exploited in the treatment of human disease. Many new developments, both in further elucidation of FGF biology and in their pharmacological application, are expected in the future.
Acknowledgments
This work was supported by the US National Institutes of Health (NIH) grant R01-DE13686 (to M.M.) and by NIH/ NIGMS training grant T32-GM066704-05 (to A.B.).
Glossary
- Autosomal dominant hypophosphataemic rickets
A hereditary disorder of phosphate wasting characterized by rickets, lower extremity deformities and osteomalacia
- Lacrimo-auriculo-dento-digital syndrome (LADD)
A syndrome characterized by abnormalities of the digits and teeth, low-set ears and aplasia of the lacrimal and salivary glands. Mutations in FGFR2 and FGF10 are known to cause LADD
- Kallmann syndrome
This syndrome results from a deficiency of gonadotropin-releasing hormone, which leads to hypogonadism. Mutations in FGFR1c and FGF8 are known to cause Kallmann syndrome
- Oral mucositis
This condition results from injury to the epithelium of the oral cavity and can vary widely in severity. In the worst cases, oral mucositis can lead to ulceration, infection and the need for assisted feeding
- Heparan sulphate glycosaminoglycan (HSGAG)
HSGAGs are long chains of repeating disaccharide units that can be variably sulphated or acetylated, allowing for considerable structural diversity. HSGAGs are located in the extracellular matrix at the surface of every cell, where they modulate the activity of a wide range of growth factors and morphogens
- Exon skipping
A specific type of alternative splicing in which an exon is entirely skipped
- Alternative splicing
This process increases protein diversity by dividing up the primary RNA gene transcript, excluding certain exons, and then reconnecting the transcript. These alternative ribonucleotide sequences are then translated, giving a variety of protein isoforms
- Craniosynostosis
This condition results from the premature closure of sutures of a developing skull before the completion of brain growth. The brain continues to grow in areas of the skull where sutures have not closed, leading to a malformed cranium
- Apert’s syndrome
One of the most common craniosynostosis syndromes that exhibits severe syndactyly (digit fusion) of the hands and feet. Apert’s syndrome is often associated with visceral abnormalities of the cardiovascular, respiratory and urogenital systems
- Osteoglophonic dysplasia
A bone disorder presenting with dwarfism, vertebral fragility, craniosynostosis and failure to thrive. The term osteoglophonic refers to the ‘hollowed out’ appearance of the metaphyses in X-rays, which are the growth zones of long bones
- Pfeiffer’s syndrome
A craniosynostosis disorder that can also present with polydactyly
- Glioblastoma
An aggressive tumour derived from glial cells that exhibits high levels of neovascularization
- Myeloproliferative syndrome
A progressive disease that can transform into acute leukaemia. Also known as stem cell leukaemia or lymphoma syndrome, it often presents with a T-cell lymphoblastic lymphoma and eosinophilia
- Crouzon’s syndrome
A craniosynostosis syndrome presenting with a beaked nose and bulging, excessively separated eyes (exopthalmos and hypertelorism, respectively)
- Callosal agenesis
An absence of the corpus callosum, the tissue that connects the two hemispheres of the brain
- Ventriculomegaly
A condition associated with enlarged lateral ventricles in the brain. Ventriculomegaly can have many causes, one of which is callosal agenesis
- Hypochrondroplasia
A mild dwarfism syndrome generally presenting with nearly normal cranial and facial characteristics
- Thanatophoric dysplasia type II
A lethal neonatal skeletal dysplasia associated with a severe cloverleaf-shaped skull deformity
- Severe achondroplasia with developmental delay and acanthosis nigricans syndrome
This dwarfism syndrome is accompanied by substantial neurological disorders and acanthosis nigricans, which involves a hyperpigmentation of the skin
- Nitric oxide
Among its many functions, this small molecule relaxes the smooth muscle surrounding blood vessels
- Brachial plexus
The bundle of nerves located in the axilla (armpit) that descends into the upper limb to provide sensation and motor control
- Chronic transverse myelitis
Inflammation across the width of one segment of the spinal cord that can lead to destruction of myelin and neurological impairment
- Heparin
A highly sulphated heparan sulphate glycosaminoglycan (HSGAG). Although it does not act physiologically on FGF–FGFR signalling, it can substitute for other HSGAGs in experimental studies
- Trophoblast
These cells form the outer layer of the developing embryo and are responsible for its implantation into the endometrium
- Osteomalacia
Demineralization of the bones often associated with a lack of vitamin D
- Secondary hyperparathyroidism
This condition is marked by excessive secretion of parathyroid hormone as a result of low serum calcium levels. It is often seen in patients suffering from kidney disease
Footnotes
DATABASES
UniProtKB: http://www.uniprot.org
FGF1 | FGF2 | FGF4 | FGF7 | FGF8 | FGF9 | FGF19 | FGF21 | FGF23 | FGFR1 | FGFR2 | FGFR3 | FGFR4
FURTHER INFORMATION
Moosa Mohammdi’s homepage:
http://saturn.med.nyu.edu/~mohammad/
rcsB Protein Data Bank web site:
http://www.rcsb.org/pdb/home/home.do
ALL Links Are AcTive in The onLine PDF
Contributor Information
Andrew Beenken, Email: Andrew.Beenken@med.nyu.edu.
Moosa Mohammadi, Email: Moosa.Mohammadi@nyumc.org.
References
- 1.Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Olsen SK, et al. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J Biol Chem. 2003;278:34226–34236. doi: 10.1074/jbc.M303183200. [DOI] [PubMed] [Google Scholar]
- 3.Fu L, et al. Fibroblast growth factor19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology. 2004;145:2594–2603. doi: 10.1210/en.2003-1671. [DOI] [PubMed] [Google Scholar]
- 4.Kharitonenkov A, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115:1627–1635. doi: 10.1172/JCI23606. The first paper to describe the metabolic profile of FGF21 in mice and rats. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol. 2007;194:1–10. doi: 10.1677/JOE-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomlinson E, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143:1741–1747. doi: 10.1210/endo.143.5.8850. Initiated interest in FGF19 as a metabolic regulator by detailing the phenotype of FGF19 transgenic mice. [DOI] [PubMed] [Google Scholar]
- 7.White KE, et al. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nature Genet. 2000;26:345–348. doi: 10.1038/81664. This study showed that an FGF23 mutation caused ADHR, which began to unravel the physiology of FGF23. [DOI] [PubMed] [Google Scholar]
- 8.Milunsky JM, Zhao G, Maher TA, Colby R, Everman DB. LADD syndrome is caused by FGF10 mutations. Clin Genet. 2006;69:349–354. doi: 10.1111/j.1399-0004.2006.00597.x. [DOI] [PubMed] [Google Scholar]
- 9.Tekin M, et al. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am J Hum Genet. 2007;80:338–344. doi: 10.1086/510920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falardeau J, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118:2822–2831. doi: 10.1172/JCI34538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Revest JM, DeMoerlooze L, Dickson C. Fibroblast growth factor 9 secretion is mediated by a non-cleaved amino-terminal signal sequence. J Biol Chem. 2000;275:8083–8090. doi: 10.1074/jbc.275.11.8083. [DOI] [PubMed] [Google Scholar]
- 12.Nickel W. Unconventional secretory routes: direct protein export across the plasma membrane of mammalian cells. Traffic. 2005;6:607–614. doi: 10.1111/j.1600-0854.2005.00302.x. [DOI] [PubMed] [Google Scholar]
- 13.Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16:107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 14.Goetz R, et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27:3417–3428. doi: 10.1128/MCB.02249-06. Elucidates the structural rationale for the reduced binding of the FGF19 subfamily to heparan sulphate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang F, Kan M, Yan G, Xu J, McKeehan WL. Alternately spliced NH2-terminal immunoglobulin-like loop I in the ectodomain of the fibroblast growth factor (FGF) receptor 1 lowers affinity for both heparin and FGF-1. J Biol Chem. 1995;270:10231–10235. doi: 10.1074/jbc.270.17.10231. [DOI] [PubMed] [Google Scholar]
- 16.Johnson DE, Lu J, Chen H, Werner S, Williams LT. The human fibroblast growth factor receptor genes: a common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol Cell Biol. 1991;11:4627–4634. doi: 10.1128/mcb.11.9.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ornitz DM, et al. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol Cell Biol. 1992;12:240–247. doi: 10.1128/mcb.12.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlessinger J, et al. Crystal structure of a ternary FGF–FGFR–heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 19.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- 20.Mohammadi M, et al. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol Cell Biol. 1996;16:977–989. doi: 10.1128/mcb.16.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16:233–247. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 22.Wiedlocha A, Sorensen V. Signaling, internalization, and intracellular activity of fibroblast growth factor. Curr Top Microbiol Immunol. 2004;286:45–79. doi: 10.1007/978-3-540-69494-6_3. [DOI] [PubMed] [Google Scholar]
- 23.Orr-Urtreger A, et al. Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2) Dev Biol. 1993;158:475–486. doi: 10.1006/dbio.1993.1205. [DOI] [PubMed] [Google Scholar]
- 24.Grose R, Dickson C. Fibroblast growth factor signaling in tumorigenesis. Cytokine Growth Factor Rev. 2005;16:179–186. doi: 10.1016/j.cytogfr.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Ibrahimi OA, et al. Analysis of mutations in fibroblast growth factor (FGF) and a pathogenic mutation in FGF receptor (FGFR) provides direct evidence for the symmetric two-end model for FGFR dimerization. Mol Cell Biol. 2005;25:671–684. doi: 10.1128/MCB.25.2.671-684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hacker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nature Rev Mol Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 27.Wu DQ, Kan MK, Sato GH, Okamoto T, Sato JD. Characterization and molecular cloning of a putative binding protein for heparin-binding growth factors. J Biol Chem. 1991;266:16778–16785. [PubMed] [Google Scholar]
- 28.Aigner A, et al. An FGF-binding protein (FGF-BP) exerts its biological function by parallel paracrine stimulation of tumor cell and endothelial cell proliferation through FGF-2 release. Int J Cancer. 2001;92:510–517. doi: 10.1002/1097-0215(20010515)92:4<510::aid-ijc1227>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 29.Tassi E, et al. Enhancement of fibroblast growth factor (FGF) activity by an FGF-binding protein. J Biol Chem. 2001;276:40247–40253. doi: 10.1074/jbc.M104933200. [DOI] [PubMed] [Google Scholar]
- 30.Abuharbeid S, Czubayko F, Aigner A. The fibroblast growth factor-binding protein FGF-BP. Int J Biochem Cell Biol. 2006;38:1463–1468. doi: 10.1016/j.biocel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 31.Bottcher RT, Pollet N, Delius H, Niehrs C. The transmembrane protein XFLRT3 forms a complex with FGF receptors and promotes FGF signalling. Nature Cell Biol. 2004;6:38–44. doi: 10.1038/ncb1082. [DOI] [PubMed] [Google Scholar]
- 32.Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow M. A sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell. 1998;92:253–263. doi: 10.1016/s0092-8674(00)80919-8. [DOI] [PubMed] [Google Scholar]
- 33.Cabrita MA, Christofori G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis. 2008;11:53–62. doi: 10.1007/s10456-008-9089-1. [DOI] [PubMed] [Google Scholar]
- 34.Tsang M, Dawid IB. Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci STKE. 2004;228:pe17. doi: 10.1126/stke.2282004pe17. [DOI] [PubMed] [Google Scholar]
- 35.Ibrahimi OA, et al. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc Natl Acad Sci USA. 2001;98:7182–7187. doi: 10.1073/pnas.121183798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dode C, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nature Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 37.Muenke M, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nature Genet. 1994;8:269–274. doi: 10.1038/ng1194-269. [DOI] [PubMed] [Google Scholar]
- 38.Rand V, et al. Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci USA. 2005;102:14344–14349. doi: 10.1073/pnas.0507200102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res. 1999;5:1063–1071. [PubMed] [Google Scholar]
- 40.Cross NC, Reiter A. Fibroblast growth factor receptor and platelet-derived growth factor receptor abnormalities in eosinophilic myeloproliferative disorders. Acta Haematol. 2008;119:199–206. doi: 10.1159/000140631. [DOI] [PubMed] [Google Scholar]
- 41.Kan SH, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002;70:472–486. doi: 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen H, et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27:717–730. doi: 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dutt A, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci USA. 2008;105:8713–8717. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pollock PM, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene. 2007;26:7158–7162. doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neilson KM, Friesel RE. Constitutive activation of fibroblast growth factor receptor-2 by a point mutation associated with Crouzon syndrome. J Biol Chem. 1995;270:26037–26040. doi: 10.1074/jbc.270.44.26037. [DOI] [PubMed] [Google Scholar]
- 46.Ibrahimi OA, et al. Biochemical analysis of pathogenic ligand-dependent FGFR2 mutations suggests distinct pathophysiological mechanisms for craniofacial and limb abnormalities. Hum Mol Genet. 2004;13:2313–2324. doi: 10.1093/hmg/ddh235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raybaud C, Di Rocco C. Brain malformation in syndromic craniosynostoses, a primary disorder of white matter: a review. Childs Nerv Syst. 2007;23:1379–1388. doi: 10.1007/s00381-007-0474-7. [DOI] [PubMed] [Google Scholar]
- 48.Tanimoto Y, et al. A soluble form of fibroblast growth factor receptor 2 (FGFR2) with S252W mutation acts as an efficient inhibitor for the enhanced osteoblastic differentiation caused by FGFR2 activation in Apert syndrome. J Biol Chem. 2004;279:45926–45934. doi: 10.1074/jbc.M404824200. [DOI] [PubMed] [Google Scholar]
- 49.Antoniou AC, et al. Common breast cancer-predisposition alleles are associated with breast cancer risk in BRCA1 and BRCA2 mutation carriers. Am J Hum Genet. 2008;82:937–948. doi: 10.1016/j.ajhg.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Webster MK, Donoghue DJ. FGFR activation in skeletal disorders: too much of a good thing. Trends Genet. 1997;13:178–182. doi: 10.1016/s0168-9525(97)01131-1. [DOI] [PubMed] [Google Scholar]
- 51.Naski MC, Wang Q, Xu J, Ornitz DM. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nature Genet. 1996;13:233–237. doi: 10.1038/ng0696-233. [DOI] [PubMed] [Google Scholar]
- 52.Passos-Bueno MR, et al. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat. 1999;14:115–125. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 53.Tavormina PL, et al. A novel skeletal dysplasia with developmental delay and acanthosis nigricans is caused by a Lys650Met mutation in the fibroblast growth factor receptor 3 gene. Am J Hum Genet. 1999;64:722–731. doi: 10.1086/302275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rohmann E, et al. Mutations in different components of FGF signaling in LADD syndrome. Nature Genet. 2006;38:414–417. doi: 10.1038/ng1757. [DOI] [PubMed] [Google Scholar]
- 55.Chesi M, et al. Frequent translocation t(4;14) (p16.3; q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nature Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cappellen D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nature Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- 57.Hafner C, Vogt T, Hartmann A. FGFR3 mutations in benign skin tumors. Cell Cycle. 2006;5:2723–2728. doi: 10.4161/cc.5.23.3509. [DOI] [PubMed] [Google Scholar]
- 58.Logie A, et al. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum Mol Genet. 2005;14:1153–1160. doi: 10.1093/hmg/ddi127. [DOI] [PubMed] [Google Scholar]
- 59.Wang J, Stockton DW, Ittmann M. The fibroblast growth factor receptor-4 Arg388 allele is associated with prostate cancer initiation and progression. Clin Cancer Res. 2004;10:6169–6178. doi: 10.1158/1078-0432.CCR-04-0408. [DOI] [PubMed] [Google Scholar]
- 60.Streit S, et al. Involvement of the FGFR4 Arg388 allele in head and neck squamous cell carcinoma. Int J Cancer. 2004;111:213–217. doi: 10.1002/ijc.20204. [DOI] [PubMed] [Google Scholar]
- 61.Meijer D, et al. Fibroblast growth factor receptor 4 predicts failure on tamoxifen therapy in patients with recurrent breast cancer. Endocr Relat Cancer. 2008;15:101–111. doi: 10.1677/ERC-07-0080. [DOI] [PubMed] [Google Scholar]
- 62.Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884–896. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 63.Grand EK, Chase AJ, Heath C, Rahemtulla A, Cross NC. Targeting FGFR3 in multiple myeloma: inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia. 2004;18:962–966. doi: 10.1038/sj.leu.2403347. [DOI] [PubMed] [Google Scholar]
- 64.Meyer AN, McAndrew CW, Donoghue DJ. Nordihydroguaiaretic acid inhibits an activated fibroblast growth factor receptor 3 mutant and blocks downstream signaling in multiple myeloma cells. Cancer Res. 2008;68:7362–7370. doi: 10.1158/0008-5472.CAN-08-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Byron SA, et al. Inhibition of activated fibroblast growth factor receptor 2 in endometrial cancer cells induces cell death despite PTEN abrogation. Cancer Res. 2008;68:6902–6907. doi: 10.1158/0008-5472.CAN-08-0770. [DOI] [PubMed] [Google Scholar]
- 66.Martinez-Torrecuadrada JL, et al. Antitumor activity of fibroblast growth factor receptor 3-specific immunotoxins in a xenograft mouse model of bladder carcinoma is mediated by apoptosis. Mol Cancer Ther. 2008;7:862–873. doi: 10.1158/1535-7163.MCT-07-0394. [DOI] [PubMed] [Google Scholar]
- 67.Trudel S, et al. The inhibitory anti-FGFR3 antibody, PRO-001, is cytotoxic to t(4;14) multiple myeloma cells. Blood. 2006;107:4039–4046. doi: 10.1182/blood-2005-10-4179. [DOI] [PubMed] [Google Scholar]
- 68.Roumiantsev S, et al. Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198–FGFR1 and BCR–FGFR1 fusion genes from 8p11 translocations. Cancer Cell. 2004;5:287–298. doi: 10.1016/s1535-6108(04)00053-4. [DOI] [PubMed] [Google Scholar]
- 69.Miller DL, Ortega S, Bashayan O, Basch R, Basilico C. Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol Cell Biol. 2000;20:2260–2268. doi: 10.1128/mcb.20.6.2260-2268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cuevas P, et al. Hypotensive activity of fibroblast growth factor. Science. 1991;254:1208–1210. doi: 10.1126/science.1957172. One of the original papers on FGF1 physiology that helped lay the groundwork for the extensive study of FGF1 and FGF2 in clinical trials. [DOI] [PubMed] [Google Scholar]
- 71.Cuevas P, et al. Correction of hypertension by normalization of endothelial levels of fibroblast growth factor and nitric oxide synthase in spontaneously hypertensive rats. Proc Natl Acad Sci USA. 1996;93:11996–12001. doi: 10.1073/pnas.93.21.11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou M, et al. Fibroblast growth factor 2 control of vascular tone. Nature Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ware JA, Simons M. Angiogenesis in ischemic heart disease. Nature Med. 1997;3:158–164. doi: 10.1038/nm0297-158. [DOI] [PubMed] [Google Scholar]
- 75.Yanagisawa-Miwa A, et al. Salvage of infarcted myocardium by angiogenic action of basic fibroblast growth factor. Science. 1992;257:1401–1403. doi: 10.1126/science.1382313. [DOI] [PubMed] [Google Scholar]
- 76.Scholz D, Cai WJ, Schaper W. Arteriogenesis, a new concept of vascular adaptation in occlusive disease. Angiogenesis. 2001;4:247–257. doi: 10.1023/a:1016094004084. [DOI] [PubMed] [Google Scholar]
- 77.Fulgham DL, Widhalm SR, Martin S, Coffin JD. FGF-2 dependent angiogenesis is a latent phenotype in basic fibroblast growth factor transgenic mice. Endothelium. 1999;6:185–195. doi: 10.3109/10623329909053409. [DOI] [PubMed] [Google Scholar]
- 78.Khurana R, Simons M. Insights from angiogenesis trials using fibroblast growth factor for advanced arteriosclerotic disease. Trends Cardiovasc Med. 2003;13:116–122. doi: 10.1016/s1050-1738(02)00259-1. [DOI] [PubMed] [Google Scholar]
- 79.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 80.Bosse Y, Rola-Pleszczynski M. FGF2 in asthmatic airway-smooth-muscle-cell hyperplasia. Trends Mol Med. 2008;14:3–11. doi: 10.1016/j.molmed.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 81.Hutley L, et al. Fibroblast growth factor 1: a key regulator of human adipogenesis. Diabetes. 2004;53:3097–3106. doi: 10.2337/diabetes.53.12.3097. [DOI] [PubMed] [Google Scholar]
- 82.Iwakura A, et al. Myocardial ischemia enhances the expression of acidic fibroblast growth factor in human pericardial fluid. Heart Vessels. 2000;15:112–116. doi: 10.1007/pl00007264. [DOI] [PubMed] [Google Scholar]
- 83.Uriel S, Brey EM, Greisler HP. Sustained low levels of fibroblast growth factor-1 promote persistent microvascular network formation. Am J Surg. 2006;192:604–609. doi: 10.1016/j.amjsurg.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 84.Cuevas P, et al. Fibroblast growth factor-1 prevents myocardial apoptosis triggered by ischemia reperfusion injury. Eur J Med Res. 1997;2:465–468. [PubMed] [Google Scholar]
- 85.Schumacher B, Pecher P, von Specht BU, Stegmann T. Induction of neoangiogenesis in ischemic myocardium by human growth factors: first clinical results of a new treatment of coronary heart disease. Circulation. 1998;97:645–650. doi: 10.1161/01.cir.97.7.645. [DOI] [PubMed] [Google Scholar]
- 86.Comerota AJ, et al. Naked plasmid DNA encoding fibroblast growth factor type 1 for the treatment of end-stage unreconstructible lower extremity ischemia: preliminary results of a phase I trial. J Vasc Surg. 2002;35:930–936. doi: 10.1067/mva.2002.123677. [DOI] [PubMed] [Google Scholar]
- 87.Nikol S, et al. Therapeutic angiogenesis with intramuscular NV1FGF improves amputation-free survival in patients with critical limb ischemia. Mol Ther. 2008;16:972–978. doi: 10.1038/mt.2008.33. [DOI] [PubMed] [Google Scholar]
- 88.Ruck A, Sylven C. Therapeutic angiogenesis gains a leg to stand on. Mol Ther. 2008;16:808–810. doi: 10.1038/mt.2008.65. [DOI] [PubMed] [Google Scholar]
- 89.Cheng H, Cao Y, Olson L. Spinal cord repair in adult paraplegic rats: partial restoration of hind limb function. Science. 1996;273:510–513. doi: 10.1126/science.273.5274.510. [DOI] [PubMed] [Google Scholar]
- 90.Lin PH, Cheng H, Huang WC, Chuang TY. Spinal cord implantation with acidic fibroblast growth factor as a treatment for root avulsion in obstetric brachial plexus palsy. J Chin Med Assoc. 2005;68:392–396. doi: 10.1016/S1726-4901(09)70182-0. [DOI] [PubMed] [Google Scholar]
- 91.Lin PH, Chuang TY, Liao KK, Cheng H, Shih YS. Functional recovery of chronic complete idiopathic transverse myelitis after administration of neurotrophic factors. Spinal Cord. 2006;44:254–257. doi: 10.1038/sj.sc.3101809. [DOI] [PubMed] [Google Scholar]
- 92.Cheng H, Liao KK, Liao SF, Chuang TY, Shih YH. Spinal cord repair with acidic fibroblast growth factor as a treatment for a patient with chronic paraplegia. Spine. 2004;29:E284–E288. doi: 10.1097/01.brs.0000131217.61390.2c. [DOI] [PubMed] [Google Scholar]
- 93.Unger EF, et al. Effects of a single intracoronary injection of basic fibroblast growth factor in stable angina pectoris. Am J Cardiol. 2000;85:1414–1419. doi: 10.1016/s0002-9149(00)00787-6. [DOI] [PubMed] [Google Scholar]
- 94.Laham RJ, et al. Intracoronary and intravenous administration of basic fibroblast growth factor: myocardial and tissue distribution. Drug Metab Dispos. 1999;27:821–826. [PubMed] [Google Scholar]
- 95.Simons M, et al. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, controlled clinical trial. Circulation. 2002;105:788–793. doi: 10.1161/hc0802.104407. [DOI] [PubMed] [Google Scholar]
- 96.Sellke FW, Laham RJ, Edelman ER, Pearlman JD, Simons M. Therapeutic angiogenesis with basic fibroblast growth factor: technique and early results. Ann Thorac Surg. 1998;65:1540–1544. doi: 10.1016/s0003-4975(98)00340-3. [DOI] [PubMed] [Google Scholar]
- 97.Ruel M, et al. Long-term effects of surgical angiogenic therapy with fibroblast growth factor 2 protein. J Thorac Cardiovasc Surg. 2002;124:28–34. doi: 10.1067/mtc.2002.121974. [DOI] [PubMed] [Google Scholar]
- 98.Lazarous DF, et al. Basic fibroblast growth factor in patients with intermittent claudication: results of a phase I trial. J Am Coll Cardiol. 2000;36:1239–1244. doi: 10.1016/s0735-1097(00)00882-2. [DOI] [PubMed] [Google Scholar]
- 99.Lederman RJ, et al. Therapeutic angiogenesis with recombinant fibroblast growth factor-2 for intermittent claudication (the TRAFFIC study): a randomised trial. Lancet. 2002;359:2053–2058. doi: 10.1016/s0140-6736(02)08937-7. [DOI] [PubMed] [Google Scholar]
- 100.D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA. 1994;91:4082–4085. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Figg WD, et al. A randomized phase II trial of thalidomide, an angiogenesis inhibitor, in patients with androgen-independent prostate cancer. Clin Cancer Res. 2001;7:1888–1893. [PubMed] [Google Scholar]
- 102.Eisen T, et al. Continuous low dose Thalidomide: a phase II study in advanced melanoma, renal cell, ovarian and breast cancer. Br J Cancer. 2000;82:812–817. doi: 10.1054/bjoc.1999.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Myers C, et al. Suramin: a novel growth factor antagonist with activity in hormone-refractory metastatic prostate cancer. J Clin Oncol. 1992;10:881–889. doi: 10.1200/JCO.1992.10.6.881. [DOI] [PubMed] [Google Scholar]
- 104.Eisenberger MA, et al. Suramin, an active drug for prostate cancer: interim observations in a phase I trial. J Natl Cancer Inst. 1993;85:611–621. doi: 10.1093/jnci/85.8.611. [DOI] [PubMed] [Google Scholar]
- 105.Motzer RJ, et al. Phase II trial of suramin in patients with advanced renal cell carcinoma: treatment results, pharmacokinetics, and tumor growth factor expression. Cancer Res. 1992;52:5775–5779. [PubMed] [Google Scholar]
- 106.Walther MM, Figg WD, Linehan WM. Intravesical suramin: a novel agent for the treatment of superficial transitional-cell carcinoma of the bladder. World J Urol. 1996;14:S8–S11. doi: 10.1007/BF00182057. [DOI] [PubMed] [Google Scholar]
- 107.Danesi R, et al. Suramin inhibits bFGF-induced endothelial cell proliferation and angiogenesis in the chick chorioallantoic membrane. Br J Cancer. 1993;68:932–938. doi: 10.1038/bjc.1993.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang Y, Song S, Yang F, Au JL, Wientjes MG. Nontoxic doses of suramin enhance activity of doxorubicin in prostate tumors. J Pharmacol Exp Ther. 2001;299:426–433. [PubMed] [Google Scholar]
- 109.Song S, Wientjes MG, Gan Y, Au JL. Fibroblast growth factors: an epigenetic mechanism of broad spectrum resistance to anticancer drugs. Proc Natl Acad Sci USA. 2000;97:8658–8663. doi: 10.1073/pnas.140210697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hawkins MJ. Clinical trials of antiangiogenic agents. Curr Opin Oncol. 1995;7:90–93. [PubMed] [Google Scholar]
- 111.Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nature Rev Cancer. 2002;2:521–528. doi: 10.1038/nrc842. [DOI] [PubMed] [Google Scholar]
- 112.Kudchadkar R, Gonzalez R, Lewis KD. PI-88: a novel inhibitor of angiogenesis. Expert Opin Investig Drugs. 2008;17:1769–1776. doi: 10.1517/13543784.17.11.1769. [DOI] [PubMed] [Google Scholar]
- 113.Singh RK, et al. Interferons alpha and beta down-regulate the expression of basic fibroblast growth factor in human carcinomas. Proc Natl Acad Sci USA. 1995;92:4562–4566. doi: 10.1073/pnas.92.10.4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dinney CP, et al. Inhibition of basic fibroblast growth factor expression, angiogenesis, and growth of human bladder carcinoma in mice by systemic interferon-α administration. Cancer Res. 1998;58:808–814. [PubMed] [Google Scholar]
- 115.Wang Y, Becker D. Antisense targeting of basic fibroblast growth factor and fibroblast growth factor receptor-1 in human melanomas blocks intratumoral angiogenesis and tumor growth. Nature Med. 1997;3:887–893. doi: 10.1038/nm0897-887. [DOI] [PubMed] [Google Scholar]
- 116.Presta M, et al. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–178. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 117.Kirkwood JM, et al. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14:7–17. doi: 10.1200/JCO.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- 118.Hamm C, Verma S, Petrella T, Bak K, Charette M. Biochemotherapy for the treatment of metastatic malignant melanoma: a systematic review. Cancer Treat Rev. 2008;34:145–156. doi: 10.1016/j.ctrv.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 119.Rayburn ER, Zhang R. Antisense, RNAi, and gene silencing strategies for therapy: mission possible or impossible? Drug Discov Today. 2008;13:513–521. doi: 10.1016/j.drudis.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Evans SJ, et al. Dysregulation of the fibroblast growth factor system in major depression. Proc Natl Acad Sci USA. 2004;101:15506–15511. doi: 10.1073/pnas.0406788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Turner CA, Calvo N, Frost DO, Akil H, Watson SJ. The fibroblast growth factor system is downregulated following social defeat. Neurosci Lett. 2008;430:147–150. doi: 10.1016/j.neulet.2007.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Turner CA, Gula EL, Taylor LP, Watson SJ, Akil H. Antidepressant-like effects of intracerebroventricular FGF2 in rats. Brain Res. 2008;1224:63–68. doi: 10.1016/j.brainres.2008.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ellman MB, An HS, Muddasani P, Im HJ. Biological impact of the fibroblast growth factor family on articular cartilage and intervertebral disc homeostasis. Gene. 2008;420:82–89. doi: 10.1016/j.gene.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Aviles RJ, Annex BH, Lederman RJ. Testing clinical therapeutic angiogenesis using basic fibroblast growth factor (FGF-2) Br J Pharmacol. 2003;140:637–646. doi: 10.1038/sj.bjp.0705493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kitamura M, et al. Periodontal tissue regeneration using fibroblast growth factor-2: randomized controlled phase II clinical trial. PLoS ONE. 2008;3:e2611. doi: 10.1371/journal.pone.0002611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sugi Y, et al. Fibroblast growth factor (FGF)-4 can induce proliferation of cardiac cushion mesenchymal cells during early valve leaflet formation. Dev Biol. 2003;258:252–263. doi: 10.1016/s0012-1606(03)00099-x. [DOI] [PubMed] [Google Scholar]
- 127.Sun X, Mariani FV, Martin GR. Functions of FGF signalling from the apical ectodermal ridge in limb development. Nature. 2002;418:501–508. doi: 10.1038/nature00902. [DOI] [PubMed] [Google Scholar]
- 128.Feldman B, Poueymirou W, Papaioannou VE, DeChiara TM, Goldfarb M. Requirement of FGF-4 for postimplantation mouse development. Science. 1995;267:246–249. doi: 10.1126/science.7809630. [DOI] [PubMed] [Google Scholar]
- 129.Hebert JM, Rosenquist T, Gotz J, Martin GR. FGF5 as a regulator of the hair growth cycle: evidence from targeted and spontaneous mutations. Cell. 1994;78:1017–1025. doi: 10.1016/0092-8674(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 130.Drogemuller C, Rufenacht S, Wichert B, Leeb T. Mutations within the FGF5 gene are associated with hair length in cats. Anim Genet. 2007;38:218–221. doi: 10.1111/j.1365-2052.2007.01590.x. [DOI] [PubMed] [Google Scholar]
- 131.Housley DJ, Venta PJ. The long and the short of it: evidence that FGF5 is a major determinant of canine ‘hair’-itability. Anim Genet. 2006;37:309–315. doi: 10.1111/j.1365-2052.2006.01448.x. [DOI] [PubMed] [Google Scholar]
- 132.Armand AS, Laziz I, Chanoine C. FGF6 in myogenesis. Biochim Biophys Acta. 2006;1763:773–778. doi: 10.1016/j.bbamcr.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 133.Floss T, Arnold HH, Braun T. A role for FGF-6 in skeletal muscle regeneration. Genes Dev. 1997;11:2040–2051. doi: 10.1101/gad.11.16.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Flynn A, O’Brien T. Alferminogene tadenovec, an angiogenic FGF4 gene therapy for coronary artery disease. IDrugs. 2008;11:283–293. [PubMed] [Google Scholar]
- 135.Grines CL, et al. Angiogenic Gene Therapy (AGENT) trial in patients with stable angina pectoris. Circulation. 2002;105:1291–1297. doi: 10.1161/hc1102.105595. [DOI] [PubMed] [Google Scholar]
- 136.Henry TD, et al. Effects of Ad5FGF-4 in patients with angina: an analysis of pooled data from the AGENT-3 and AGENT-4 trials. J Am Coll Cardiol. 2007;50:1038–1046. doi: 10.1016/j.jacc.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 137.Guo L, Degenstein L, Fuchs E. Keratinocyte growth factor is required for hair development but not for wound healing. Genes Dev. 1996;10:165–175. doi: 10.1101/gad.10.2.165. [DOI] [PubMed] [Google Scholar]
- 138.Qiao J, et al. FGF-7 modulates ureteric bud growth and nephron number in the developing kidney. Development. 1999;126:547–554. doi: 10.1242/dev.126.3.547. [DOI] [PubMed] [Google Scholar]
- 139.Werner S, et al. Large induction of keratinocyte growth factor expression in the dermis during wound healing. Proc Natl Acad Sci USA. 1992;89:6896–6900. doi: 10.1073/pnas.89.15.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Baskin LS, et al. Growth factors in bladder wound healing. J Urol. 1997;157:2388–2395. [PubMed] [Google Scholar]
- 141.Ichimura T, Finch PW, Zhang G, Kan M, Stevens JL. Induction of FGF-7 after kidney damage: a possible paracrine mechanism for tubule repair. Am J Physiol. 1996;271:F967–F976. doi: 10.1152/ajprenal.1996.271.5.F967. [DOI] [PubMed] [Google Scholar]
- 142.Kato S, Sekine K. FGF–FGFR signaling in vertebrate organogenesis. Cell Mol Biol (Noisy-le-grand) 1999;45:631–638. [PubMed] [Google Scholar]
- 143.Umemori H, Linhoff MW, Ornitz DM, Sanes JR. FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell. 2004;118:257–270. doi: 10.1016/j.cell.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 144.Finch PW, Pricolo V, Wu A, Finkelstein SD. Increased expression of keratinocyte growth factor messenger RNA associated with inflammatory bowel disease. Gastroenterology. 1996;110:441–451. doi: 10.1053/gast.1996.v110.pm8566591. [DOI] [PubMed] [Google Scholar]
- 145.Finch PW, Murphy F, Cardinale I, Krueger JG. Altered expression of keratinocyte growth factor and its receptor in psoriasis. Am J Pathol. 1997;151:1619–1628. [PMC free article] [PubMed] [Google Scholar]
- 146.Kovacs D, et al. Immunohistochemical analysis of keratinocyte growth factor and fibroblast growth factor 10 expression in psoriasis. Exp Dermatol. 2005;14:130–137. doi: 10.1111/j.0906-6705.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- 147.Thomson AA, Cunha GR. Prostatic growth and development are regulated by FGF10. Development. 1999;126:3693–3701. doi: 10.1242/dev.126.16.3693. [DOI] [PubMed] [Google Scholar]
- 148.Yan G, Fukabori Y, Nikolaropoulos S, Wang F, McKeehan WL. Heparin-binding keratinocyte growth factor is a candidate stromal-to-epithelial-cell andromedin. Mol Endocrinol. 1992;6:2123–2128. doi: 10.1210/mend.6.12.1491693. [DOI] [PubMed] [Google Scholar]
- 149.Spielberger R, et al. Palifermin for oral mucositis after intensive therapy for hematologic cancers. N Engl J Med. 2004;351:2590–2598. doi: 10.1056/NEJMoa040125. The results of this clinical trial helped bring FGF7 into use for the treatment of oral mucositis. [DOI] [PubMed] [Google Scholar]
- 150.Potten CS, et al. Cell kinetic studies in the murine ventral tongue epithelium: the effects of repeated exposure to keratinocyte growth factor. Cell Prolif. 2002;35 (Suppl 1):22–31. doi: 10.1046/j.1365-2184.35.s1.3.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Potten CS, et al. Cell kinetic studies in the murine ventral tongue epithelium: mucositis induced by radiation and its protection by pretreatment with keratinocyte growth factor (KGF) Cell Prolif. 2002;35 (Suppl 1):32–47. doi: 10.1046/j.1365-2184.35.s1.4.x. [DOI] [PubMed] [Google Scholar]
- 152.Braun S, et al. Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound. Mol Cell Biol. 2002;22:5492–5505. doi: 10.1128/MCB.22.15.5492-5505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ellison CA, et al. Effect of recombinant human keratinocyte growth factor (rHuKGF) on the immunopathogenesis of intestinal graft-vs-host disease induced without a preconditioning regimen. J Clin Immunol. 2004;24:197–211. doi: 10.1023/B:JOCI.0000019785.35850.a5. [DOI] [PubMed] [Google Scholar]
- 154.Panoskaltsis-Mortari A, et al. Keratinocyte growth factor facilitates alloengraftment and ameliorates graft-versus-host disease in mice by a mechanism independent of repair of conditioning-induced tissue injury. Blood. 2000;96:4350–4356. [PubMed] [Google Scholar]
- 155.Beaven AW, Shea TC. The effect of palifermin on chemotherapy and radiation therapy-induced mucositis: a review of the current literature. Support Cancer Ther. 2007;4:188–197. doi: 10.3816/SCT.2007.n.014. [DOI] [PubMed] [Google Scholar]
- 156.van der Velden WJ, Herbers AH, Blijlevens NM. Palifermin in allogeneic HSCT: many questions remain. Bone Marrow Transplant. 2008;43:85–86. doi: 10.1038/bmt.2008.269. [DOI] [PubMed] [Google Scholar]
- 157.Werner S. Keratinocyte growth factor: a unique player in epithelial repair processes. Cytokine Growth Factor Rev. 1998;9:153–165. doi: 10.1016/s1359-6101(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 158.Freytes CO, et al. Phase I/II randomized trial evaluating the safety and clinical effects of repifermin administered to reduce mucositis in patients undergoing autologous hematopoietic stem cell transplantation. Clin Cancer Res. 2004;10:8318–8324. doi: 10.1158/1078-0432.CCR-04-1118. [DOI] [PubMed] [Google Scholar]
- 159.Sandborn WJ, et al. Repifermin (keratinocyte growth factor-2) for the treatment of active ulcerative colitis: a randomized, double-blind, placebo-controlled, dose-escalation trial. Aliment Pharmacol Ther. 2003;17:1355–1364. doi: 10.1046/j.1365-2036.2003.01589.x. [DOI] [PubMed] [Google Scholar]
- 160.Liu A, Joyner AL. Early anterior/posterior patterning of the midbrain and cerebellum. Annu Rev Neurosci. 2001;24:869–896. doi: 10.1146/annurev.neuro.24.1.869. [DOI] [PubMed] [Google Scholar]
- 161.O’Leary DD, Chou SJ, Sahara S. Area patterning of the mammalian cortex. Neuron. 2007;56:252–269. doi: 10.1016/j.neuron.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 162.Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nature Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- 163.Xu J, Liu Z, Ornitz DM. Temporal and spatial gradients of Fgf8 and Fgf17 regulate proliferation and differentiation of midline cerebellar structures. Development. 2000;127:1833–1843. doi: 10.1242/dev.127.9.1833. [DOI] [PubMed] [Google Scholar]
- 164.Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002;16:859–869. doi: 10.1101/gad.965602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ohbayashi N, et al. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002;16:870–879. doi: 10.1101/gad.965702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ellsworth JL, et al. Fibroblast growth factor-18 is a trophic factor for mature chondrocytes and their progenitors. Osteoarthritis Cartilage. 2002;10:308–320. doi: 10.1053/joca.2002.0514. [DOI] [PubMed] [Google Scholar]
- 167.Moore EE, et al. Fibroblast growth factor-18 stimulates chondrogenesis and cartilage repair in a rat model of injury-induced osteoarthritis. Osteoarthritis Cartilage. 2005;13:623–631. doi: 10.1016/j.joca.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 168.Maruyama-Takahashi K, et al. A neutralizing anti-fibroblast growth factor (FGF) 8 monoclonal antibody shows anti-tumor activity against FGF8b-expressing LNCaP xenografts in androgen-dependent and -independent conditions. Prostate. 2008;68:640–650. doi: 10.1002/pros.20728. [DOI] [PubMed] [Google Scholar]
- 169.Shimada N, et al. A neutralizing anti-fibroblast growth factor 8 monoclonal antibody shows potent antitumor activity against androgen-dependent mouse mammary tumors in vivo. Clin Cancer Res. 2005;11:3897–3904. doi: 10.1158/1078-0432.CCR-04-2358. [DOI] [PubMed] [Google Scholar]
- 170.Colvin JS, Green RP, Schmahl J, Capel B, Ornitz DM. Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell. 2001;104:875–889. doi: 10.1016/s0092-8674(01)00284-7. [DOI] [PubMed] [Google Scholar]
- 171.Colvin JS, White AC, Pratt SJ, Ornitz DM. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development. 2001;128:2095–2106. doi: 10.1242/dev.128.11.2095. [DOI] [PubMed] [Google Scholar]
- 172.Lu SY, et al. FGF-16 is required for embryonic heart development. Biochem Biophys Res Commun. 2008;373:270–274. doi: 10.1016/j.bbrc.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.van der Walt JM, et al. Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. Am J Hum Genet. 2004;74:1121–1127. doi: 10.1086/421052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang G, et al. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of α-synuclein. Am J Hum Genet. 2008;82:283–289. doi: 10.1016/j.ajhg.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ohmachi S, Mikami T, Konishi M, Miyake A, Itoh N. Preferential neurotrophic activity of fibroblast growth factor-20 for dopaminergic neurons through fibroblast growth factor receptor-1c. J Neurosci Res. 2003;72:436–443. doi: 10.1002/jnr.10592. [DOI] [PubMed] [Google Scholar]
- 176.Takagi Y, et al. Dopaminergic neurons generated from monkey embryonic stem cells function in a Parkinson primate model. J Clin Invest. 2005;115:102–109. doi: 10.1172/JCI21137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Schuster MW, et al. Safety and tolerability of velafermin (CG53135–05) in patients receiving high-dose chemotherapy and autologous peripheral blood stem cell transplant. Support Care Cancer. 2008;16:477–483. doi: 10.1007/s00520-007-0325-9. [DOI] [PubMed] [Google Scholar]
- 178.Kuro-o M, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. The first paper to describe the discovery of αklotho and its role in ageing in mice. [DOI] [PubMed] [Google Scholar]
- 179.Kurosu H, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Imura A, et al. Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004;565:143–147. doi: 10.1016/j.febslet.2004.03.090. [DOI] [PubMed] [Google Scholar]
- 181.Nabeshima Y. The discovery of α-Klotho and FGF23 unveiled new insight into calcium and phosphate homeostasis. Cell Mol Life Sci. 2008;65:3218–3230. doi: 10.1007/s00018-008-8177-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Imura A, et al. α-Klotho as a regulator of calcium homeostasis. Science. 2007;316:1615–1618. doi: 10.1126/science.1135901. [DOI] [PubMed] [Google Scholar]
- 183.Chang Q, et al. The β-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310:490–493. doi: 10.1126/science.1114245. [DOI] [PubMed] [Google Scholar]
- 184.Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol. 2003;17:2393–2403. doi: 10.1210/me.2003-0048. [DOI] [PubMed] [Google Scholar]
- 185.Shimada T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kurosu H, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–6123. doi: 10.1074/jbc.C500457200. The first evidence that FGF23 requires α-klotho to activate FGFRs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Urakawa I, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. References 186 and 187 showed, for the first time, that FGF23 requires α-klotho to activate FGFR1c. [DOI] [PubMed] [Google Scholar]
- 188.Ito S, et al. Impaired negative feedback suppression of bile acid synthesis in mice lacking βKlotho. J Clin Invest. 2005;115:2202–2208. doi: 10.1172/JCI23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Inagaki T, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 190.Yu C, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275:15482–15489. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 191.Kurosu H, et al. Tissue-specific expression of βKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem. 2007;282:26687–26695. doi: 10.1074/jbc.M704165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lin BC, Wang M, Blackmore C, Desnoyers LR. Liver-specific activities of FGF19 require Klotho beta. J Biol Chem. 2007;282:27277–27284. doi: 10.1074/jbc.M704244200. [DOI] [PubMed] [Google Scholar]
- 193.Wu X, et al. Co-receptor requirements for fibroblast growth factor-19 signaling. J Biol Chem. 2007;282:29069–29072. doi: 10.1074/jbc.C700130200. [DOI] [PubMed] [Google Scholar]
- 194.Kharitonenkov A, et al. FGF-21/FGF-21 receptor interaction and activation is determined by βKlotho. J Cell Physiol. 2008;215:1–7. doi: 10.1002/jcp.21357. [DOI] [PubMed] [Google Scholar]
- 195.Ogawa Y, et al. βKlotho is required for metabolic activity of fibroblast growth factor 21. Proc Natl Acad Sci USA. 2007;104:7432–7437. doi: 10.1073/pnas.0701600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Suzuki M, et al. βKlotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Mol Endocrinol. 2008;22:1006–1014. doi: 10.1210/me.2007-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nishimura T, Utsunomiya Y, Hoshikawa M, Ohuchi H, Itoh N. Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain. Biochim Biophys Acta. 1999;1444:148–151. doi: 10.1016/s0167-4781(98)00255-3. [DOI] [PubMed] [Google Scholar]
- 198.Xie MH, et al. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine. 1999;11:729–735. doi: 10.1006/cyto.1999.0485. [DOI] [PubMed] [Google Scholar]
- 199.Holt JA, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lundasen T, Galman C, Angelin B, Rudling M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J Intern Med. 2006;260:530–536. doi: 10.1111/j.1365-2796.2006.01731.x. This interesting study revealed that FGF19 is induced following feeding in humans. [DOI] [PubMed] [Google Scholar]
- 201.Choi M, et al. Identification of a hormonal basis for gallbladder filling. Nature Med. 2006;12:1253–1255. doi: 10.1038/nm1501. [DOI] [PubMed] [Google Scholar]
- 202.Harmer NJ, et al. Towards a resolution of the stoichiometry of the fibroblast growth factor (FGF)–FGF receptor–heparin complex. J Mol Biol. 2004;339:821–834. doi: 10.1016/j.jmb.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 203.Dostalova I, et al. Plasma concentrations of fibroblast growth factors 19 and 21 in patients with anorexia nervosa. J Clin Endocrinol Metab. 2008;93:3627–3632. doi: 10.1210/jc.2008-0746. [DOI] [PubMed] [Google Scholar]
- 204.Nicholes K, et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol. 2002;160:2295–2307. doi: 10.1016/S0002-9440(10)61177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Desnoyers LR, et al. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene. 2008;27:85–97. doi: 10.1038/sj.onc.1210623. [DOI] [PubMed] [Google Scholar]
- 206.Pai R, et al. Inhibition of fibroblast growth factor 19 reduces tumor growth by modulating β-catenin signaling. Cancer Res. 2008;68:5086–5095. doi: 10.1158/0008-5472.CAN-07-2325. [DOI] [PubMed] [Google Scholar]
- 207.Strack AM, Myers RW. Modulation of metabolic syndrome by fibroblast growth factor 19 (FGF19)? Endocrinology. 2004;145:2591–2593. doi: 10.1210/en.2004-0367. [DOI] [PubMed] [Google Scholar]
- 208.Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta. 2000;1492:203–206. doi: 10.1016/s0167-4781(00)00067-1. [DOI] [PubMed] [Google Scholar]
- 209.Zhang X, et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes. 2008;57:1246–1253. doi: 10.2337/db07-1476. [DOI] [PubMed] [Google Scholar]
- 210.Wente W, et al. Fibroblast growth factor-21 improves pancreatic β-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Diabetes. 2006;55:2470–2478. doi: 10.2337/db05-1435. [DOI] [PubMed] [Google Scholar]
- 211.Izumiya Y, et al. FGF21 is an Akt-regulated myokine. FEBS Lett. 2008;582:3805–3810. doi: 10.1016/j.febslet.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Coskun T, et al. FGF21 corrects obesity in mice. Endocrinology. 2008;149:6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 213.Xu J, et al. FGF21 reverses hepatic steatosis, increases energy expenditure and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Inagaki T, et al. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. 2008;8:77–83. doi: 10.1016/j.cmet.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Badman MK, et al. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 216.Kharitonenkov A, et al. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology. 2007;148:774–781. doi: 10.1210/en.2006-1168. [DOI] [PubMed] [Google Scholar]
- 217.Inagaki T, et al. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. References 215 and 217 describe the role of FGF21 in the fasting response. [DOI] [PubMed] [Google Scholar]
- 218.Palou M, et al. Sequential changes in the expression of genes involved in lipid metabolism in adipose tissue and liver in response to fasting. Pflugers Arch. 2008;456:825–836. doi: 10.1007/s00424-008-0461-1. [DOI] [PubMed] [Google Scholar]
- 219.Reitman ML. FGF21: a missing link in the biology of fasting. Cell Metab. 2007;5:405–407. doi: 10.1016/j.cmet.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 220.Wang H, Qiang L, Farmer SR. Identification of a domain within peroxisome proliferator-activated receptor γ regulating expression of a group of genes containing fibroblast growth factor 21 that are selectively repressed by SIRT1 in adipocytes. Mol Cell Biol. 2008;28:188–200. doi: 10.1128/MCB.00992-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Moyers JS, et al. Molecular determinants of FGF-21 activity-synergy and cross-talk with PPARγ signaling. J Cell Physiol. 2007;210:1–6. doi: 10.1002/jcp.20847. [DOI] [PubMed] [Google Scholar]
- 222.Arner P, et al. FGF21 attenuates lipolysis in human adipocytes — a possible link to improved insulin sensitivity. FEBS Lett. 2008;582:1725–1730. doi: 10.1016/j.febslet.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 223.Muise ES, et al. Adipose fibroblast growth factor 21 is up-regulated by peroxisome proliferator-activated receptorγ and altered metabolic states. Mol Pharmacol. 2008;74:403–412. doi: 10.1124/mol.108.044826. [DOI] [PubMed] [Google Scholar]
- 224.Galman C, et al. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARα activation in man. Cell Metab. 2008;8:169–174. doi: 10.1016/j.cmet.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 225.Chen WW, et al. Circulating FGF-21 levels in normal subjects and in newly diagnose patients with type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 2008;116:65–68. doi: 10.1055/s-2007-985148. [DOI] [PubMed] [Google Scholar]
- 226.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 227.Riminucci M, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–692. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Liu S, et al. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278:37419–37426. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- 229.Bai XY, Miao D, Goltzman D, Karaplis AC. The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J Biol Chem. 2003;278:9843–9849. doi: 10.1074/jbc.M210490200. [DOI] [PubMed] [Google Scholar]
- 230.Larsson T, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the α1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145:3087–3094. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- 231.Fukumoto S. Physiological regulation and disorders of phosphate metabolism — pivotal role of fibroblast growth factor 23. Intern Med. 2008;47:337–343. doi: 10.2169/internalmedicine.47.0730. [DOI] [PubMed] [Google Scholar]
- 232.Saito H, et al. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1α, 25-dihydroxyvitamin D3 production. J Biol Chem. 2003;278:2206–2211. doi: 10.1074/jbc.M207872200. [DOI] [PubMed] [Google Scholar]
- 233.Segawa H, et al. Effect of hydrolysis-resistant FGF23–R179Q on dietary phosphate regulation of the renal type-II Na/Pi transporter. Pflugers Arch. 2003;446:585–592. doi: 10.1007/s00424-003-1084-1. [DOI] [PubMed] [Google Scholar]
- 234.Ben-Dov IZ, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sitara D, et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–432. doi: 10.1016/j.matbio.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hesse M, Frohlich LF, Zeitz U, Lanske B, Erben RG. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol. 2007;26:75–84. doi: 10.1016/j.matbio.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 237.Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. FASEB J. 2006;20:720–722. doi: 10.1096/fj.05-5432fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sitara D, et al. Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am J Pathol. 2006;169:2161–2170. doi: 10.2353/ajpath.2006.060329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens. 2006;15:437–441. doi: 10.1097/01.mnh.0000232885.81142.83. [DOI] [PubMed] [Google Scholar]
- 240.Inoue Y, et al. Role of the vitamin D receptor in FGF23 action on phosphate metabolism. Biochem J. 2005;390:325–331. doi: 10.1042/BJ20041799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Medici D, et al. FGF-23-Klotho signaling stimulates proliferation and prevents vitamin D-induced apoptosis. J Cell Biol. 2008;182:459–465. doi: 10.1083/jcb.200803024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nature Genet. 1995;11:130–136. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- 243.Jonsson KB, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–1663. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 244.Yamazaki Y, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87:4957–4960. doi: 10.1210/jc.2002-021105. [DOI] [PubMed] [Google Scholar]
- 245.Weber TJ, Liu S, Indridason OS, Quarles LD. Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res. 2003;18:1227–1234. doi: 10.1359/jbmr.2003.18.7.1227. [DOI] [PubMed] [Google Scholar]
- 246.Shulman DI, et al. Tumor-induced rickets: usefulness of MR gradient echo recall imaging for tumor localization. J Pediatr. 2004;144:381–385. doi: 10.1016/j.jpeds.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 247.Lyles KW, et al. Genetic transmission of tumoral calcinosis: autosomal dominant with variable clinical expressivity. J Clin Endocrinol Metab. 1985;60:1093–1096. doi: 10.1210/jcem-60-6-1093. [DOI] [PubMed] [Google Scholar]
- 248.Araya K, et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:5523–5527. doi: 10.1210/jc.2005-0301. [DOI] [PubMed] [Google Scholar]
- 249.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–390. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 250.Larsson T, et al. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:2424–2427. doi: 10.1210/jc.2004-2238. [DOI] [PubMed] [Google Scholar]
- 251.Ichikawa S, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Musculoskelet Neuronal Interact. 2007;7:318–319. [PubMed] [Google Scholar]
- 252.Shimada T, et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143:3179–3182. doi: 10.1210/endo.143.8.8795. [DOI] [PubMed] [Google Scholar]
- 253.Kato K, et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem. 2006;281:18370–18377. doi: 10.1074/jbc.M602469200. [DOI] [PubMed] [Google Scholar]
- 254.Topaz O, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nature Genet. 2004;36:579–581. doi: 10.1038/ng1358. [DOI] [PubMed] [Google Scholar]
- 255.Imanishi Y, et al. FGF-23 in patients with end-stage renal disease on hemodialysis. Kidney Int. 2004;65:1943–1946. doi: 10.1111/j.1523-1755.2004.00604.x. [DOI] [PubMed] [Google Scholar]
- 256.Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- 257.Razzaque MS. Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrol Dial Transplant. 2009;24:4–7. doi: 10.1093/ndt/gfn620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Gutierrez OM, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592. doi: 10.1056/NEJMoa0706130. This study shows a correlation between serum FGF23 levels and chronic kidney disease mortality. This area requires further research, as the role of FGF23 in chronic kidney disease is poorly understood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Nakanishi S, et al. Serum fibroblast growth factor-23 levels predict the future refractory hyperparathyroidism in dialysis patients. Kidney Int. 2005;67:1171–1178. doi: 10.1111/j.1523-1755.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- 260.Aono Y, et al. The neutralization of FGF-23 ameliorates hypophosphatemia and rickets in Hyp mice. J Bone Miner Res. 2003;18:S16. [Google Scholar]
- 261.Yamazaki Y, et al. Anti-FGF23 neutralizing antibodies demonstrate the physiological role and structural features of FGF23. J Bone Miner Res. 2008;23:1509–1518. doi: 10.1359/jbmr.080417. [DOI] [PubMed] [Google Scholar]
- 262.Turner CA, Akil H, Watson SJ, Evans SJ. The fibroblast growth factor system and mood disorders. Biol Psychiatry. 2006;59:1128–1135. doi: 10.1016/j.biopsych.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 263.Goldfarb M. Fibroblast growth factor homologous factors: evolution, structure, and function. Cytokine Growth Factor Rev. 2005;16:215–220. doi: 10.1016/j.cytogfr.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Dor Y, et al. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J. 2002;21:1939–1947. doi: 10.1093/emboj/21.8.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Bush MA, et al. Pharmacokinetics and pharmacodynamics of recombinant FGF-2 in a phase I trial in coronary artery disease. J Clin Pharmacol. 2001;41:378–385. doi: 10.1177/00912700122010230. [DOI] [PubMed] [Google Scholar]
- 266.Nugent MA, Edelman ER. Kinetics of basic fibroblast growth factor binding to its receptor and heparan sulfate proteoglycan: a mechanism for cooperactivity. Biochemistry. 1992;31:8876–8883. doi: 10.1021/bi00152a026. [DOI] [PubMed] [Google Scholar]
- 267.Ortega S, et al. Conversion of cysteine to serine residues alters the activity, stability, and heparin dependence of acidic fibroblast growth factor. J Biol Chem. 1991;266:5842–5846. [PubMed] [Google Scholar]
- 268.Dubey VK, Lee J, Somasundaram T, Blaber S, Blaber M. Spackling the crack: stabilizing human fibroblast growth factor-1 by targeting the N and C terminus β-strand interactions. J Mol Biol. 2007;371:256–268. doi: 10.1016/j.jmb.2007.05.065. [DOI] [PubMed] [Google Scholar]
- 269.Rajanayagam MA, et al. Intracoronary basic fibroblast growth factor enhances myocardial collateral perfusion in dogs. J Am Coll Cardiol. 2000;35:519–526. doi: 10.1016/s0735-1097(99)00550-1. [DOI] [PubMed] [Google Scholar]
- 270.Lazarous DF, et al. Pharmacodynamics of basic fibroblast growth factor: route of administration determines myocardial and systemic distribution. Cardiovasc Res. 1997;36:78–85. doi: 10.1016/s0008-6363(97)00142-9. [DOI] [PubMed] [Google Scholar]
- 271.Post MJ, Laham R, Sellke FW, Simons M. Therapeutic angiogenesis in cardiology using protein formulations. Cardiovasc Res. 2001;49:522–531. doi: 10.1016/s0008-6363(00)00216-9. [DOI] [PubMed] [Google Scholar]
- 272.Yla-Herttuala S, Martin JF. Cardiovascular gene therapy. Lancet. 2000;355:213–222. doi: 10.1016/S0140-6736(99)04180-X. [DOI] [PubMed] [Google Scholar]
- 273.Kornowski R, Fuchs S, Leon MB, Epstein SE. Delivery strategies to achieve therapeutic myocardial angiogenesis. Circulation. 2000;101:454–458. doi: 10.1161/01.cir.101.4.454. [DOI] [PubMed] [Google Scholar]
- 274.Lee RJ, et al. VEGF gene delivery to myocardium: deleterious effects of unregulated expression. Circulation. 2000;102:898–901. doi: 10.1161/01.cir.102.8.898. [DOI] [PubMed] [Google Scholar]
- 275.Celletti FL, et al. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nature Med. 2001;7:425–429. doi: 10.1038/86490. [DOI] [PubMed] [Google Scholar]
- 276.Simons M, et al. Clinical trials in coronary angiogenesis: issues, problems, consensus: an expert panel summary. Circulation. 2000;102:E73–E86. doi: 10.1161/01.cir.102.11.e73. [DOI] [PubMed] [Google Scholar]
- 277.Presta M, et al. Heparin derivatives as angiogenesis inhibitors. Curr Pharm Des. 2003;9:553–566. doi: 10.2174/1381612033391379. [DOI] [PubMed] [Google Scholar]
- 278.Goetz R, et al. Crystal structure of a fibroblast growth factor homologous factor defines conserved surface for binding and modulation of voltage-gated sodium channels. J Biol Chem. doi: 10.1074/jbc.M109.001842. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]