

Abstract
A combination of genetic, environmental, and metabolic factors contribute to development and recurrence of acute and chronic pancreatitis; information on all of these is required to manage patients effectively. For example, variants that affect regulation of the protease, serine (PRSS)1-PRSS2 and claudin (CLDN)2 loci, rather than their coding sequences, interact with other genetic and environmental factors to affect disease development. New strategies are needed to use these data and determine their contribution to pathogenesis, because these variants differs from previously studied, rare variants in exons (coding regions) of genes such as PRSS1, SPINK1, cystic fibrosis transmembrane conductance regulator (CFTR), chymotrypsin (CTR)C, and calcium-sensing receptor (CASR). Learning how various genetic factors affect pancreatic cells and systems could lead to etiology-based therapies, rather than treatment of symptoms.
Keywords: cystic fibrosis, alcoholism, hereditary pancreatitis, complex traits, chronic disease, personalized medicine
Introduction
Genetic analysis will soon be central to management of patients with complex pancreatic disorders 1. The effects of genetic variations, however, cannot be understood in isolation; they must be considered in the context of environmental, metabolic, epigenetic, and other genetic factors that influence the dynamics of disease activity. A combination of all these factors should be considered in development of disease interventions.
In 1996, a breakthrough in understanding recurrent acute pancreatitis (RAP) and chronic pancreatitis (CP) came with the discovery that gain-of-function mutations in the gene that encodes cationic trypsinogen (PRSS1) cause hereditary pancreatitis 2, 3, a syndrome characterized by RAP and later CP. Using candidate gene approaches, mutations in the coding regions of 4 additional genes linked to the control of trypsin in the pancreas have been associated with pancreatitis. RAP and CP have also been associated with loss-of-function mutations in genes that encode the serine peptidase inhibitor Kazal type 1 (SPINK1) and the cystic fibrosis transmembrane conductance regulator (ATP-binding cassette sub-family C, member 7; CFTR). Mutations in the chymotrypsin C (caldecrin) gene (CTRC) and the calcium-sensing receptor gene (CASR) (see recent reviews 4-8) were associated with smaller increases in risk. These genetic factors have greater levels of association with early-onset pancreatitis than alcohol- and smoking-induced pancreatitis.
More recent breakthroughs are helping us to understand the complex risk factors for RAP and CP. These were led by the discovery that variants in non-coding regions of the PRSS1-PRSS2 and CLDN2 loci affect risk for sporadic and alcoholic pancreatitis 9. In this review, we integrate the knowledge gained from these recent advances into our understanding and management of pancreatitis.
Genetics of Pancreatitis
The pancreas is a simple retroperitoneal digestive gland that secretes zymogens to digest intraluminal nutrients, bicarbonate to neutralize gastric acid, and hormones to regulate nutrient assimilation and utilization. Located in the back of the abdomen, the pancreas is well protected from mechanical injury and from direct interactions with the environment, toxins, and infectious agents. As a result, pancreatic diseases are relatively rare, with the exception of type 1 diabetes mellitus (T1DM), which results from autoimmune-associated destruction of the pancreatic islet β cells 10. AP and CP are the most common disorders of the exocrine pancreas, and in contrast to T1DM, the mechanism of injury or destruction is generally not autoimmune-mediated. The major risk for the development of these diseases lies with the risk of premature activation of trypsin, followed by zymogen activation, tissue auto-digestion, and the generation of a robust immune response and its sequelae 11.
AP and CP are clinical syndromes, 12 defined by signs and symptoms that together form a pathologic condition. The problem with these clinical-pathologic definitions, however, is that they are descriptive rather than mechanistic, which limits the diagnosis of mild and early-stage disease and the design of targeted treatments. Additional challenges for physicians include the variability and unpredictability of disease onset, severity, complications, and clinical course. As a result, therapies are primarily directed at symptoms, rather than pathogenic mechanisms. A new approach is needed to better understand and manage patients with pancreatic diseases (reviewed in 1).
Models
In a simple model, CP results from 2 general hits. 13, 14. The first hit, AP, activates the immune system and starts the process14, 15. Susceptibility to pancreatic injury is linked to premature activation of trypsin—either in acinar cells or pancreatic ducts. In addition, there are trypsin-independent mechanisms of injury that can activate the inflammatory process, such as direct trauma, toxic agents, or immune-mediated mechanisms, such as in autoimmune pancreatitis (AIP). Factors that determine the severity of AP will not be addressed here.
Inflammation is a normal response to injury that usually leads to tissue repair and regeneration. The second hit that contributes to CP appears to be modification of the normal inflammatory response, leading to sustained activation of pancreatic stellate cells and fibrosis (or other irreversible structural or functional changes; see reviews 13, 14). So the first hit comes from factors that cause injury, whereas the second hit involves factors that promote inflammation and inflammation-associated complications, such as fibrosis and sclerosis, failed acinar cell regeneration, distorted tissue architecture, progressive loss of the normal parenchyma, metaplasia, dysplasia, and pain syndromes. This second hit could include various responses of the immune and autonomic and sensory nervous systems, acinar and duct cell stress responses, cell regeneration and trans-differentiation, tissue remodeling, dysplasia, altered anatomy, and other factors 15 (Figure 1). Different genetic variants can affect each of these systems.
Figure 1. General Models of Inflammation.

Chronic pancreatitis is illustrated as a syndrome that develops over time (left to right). A. Individuals may live for years with multiple susceptibility factors. Patients with very high risk may be candidates for new prevention strategies such as avoiding alcohol. B. When a stochastic event leads to pancreatic injures it initiates the Sentinel Acute Pancreatitis Event (SAPE) with intra-pancreatic immune system activation.
Management includes identification of actionable risk and prevention of RAP. C. Factors such as alcohol, smoking and genetic mutations or presently unidentified factors affect the specialized cells of the pancreas and infiltrating cells to cause various complications. Research efforts are focusing on identifying risk, mechanisms and biomarkers of the progression pathways.
The development of models is important for studying and identifying treatment strategies for disorders that arise based on dozens of variables. Modeling allows for the organization of known risk factors and disease-modifying factors into compartments, which in turn provides information about how multiple risk factors interact. Additionally, modeling organizes sequential events, in which responses depend on initiating conditions, facilitating the anticipation of potential effects when initial conditions are met. Finally, risk categories can constructed based on therapeutic approaches, linking risk factors with specific therapies.
Modeling provides a framework for understanding complex diseases1 and therefore a basis for personalized medicine. Treatment approaches must be specifically designed for disorders that have multiple etiologies with similar pathologies, those that cause organ dysfunction without tissue pathology, and those with unpredictable behaviors, such as RAP and CP.
Susceptibility to AP and RAP
Disorders of the pancreas can be congenital; acquired through trauma, infection, or gallstones; linked to alcohol, smoking, or genetic factors; or result from unknown factors (idiopathic disease). The fact that most people do not have pancreatic disease indicates that the system is robust and has multiple adaptive and protective mechanisms in place.
One approach to understanding complex genetics is to focus on the specialized cells that mediate organ function and response to injury. This approach is similar to reverse engineering, in which individual components are studied, and the information is used to determine the mechanisms of a larger system. Acinar and duct cells are the primary effector cells of the pancreas and the likely initiators of AP.
Acinar Cells
The acinus, comprising acinar cells and upstream duct cells, are the primary functional unit of the exocrine pancreas (Figure 2). Acinar cells rapidly produce large amounts of pancreatic digestive pro-enzymes (zymogens) that are delivered to the duodenum, where they are activated to digest ingested nutrients. Trypsin is a protease and digestive enzyme that breaks peptide chains at arginine (Arg, R) or lysine (Lys, L) residues; it activates all of the other pancreatic zymogens in the duodenum under normal conditions. Like the mast cell enzyme tryptase, trypsin can also initiate a prolonged immune response if it is activated in the wrong location as in pancreatitis16. Therefore, it is important for pancreatic homeostasis (and human health) that trypsin remain in its zymogen form (trypsinogen) until its activity is needed.
Figure 2. Genetic and Environmental Factors that Affect Acinar Cells or Ducts.
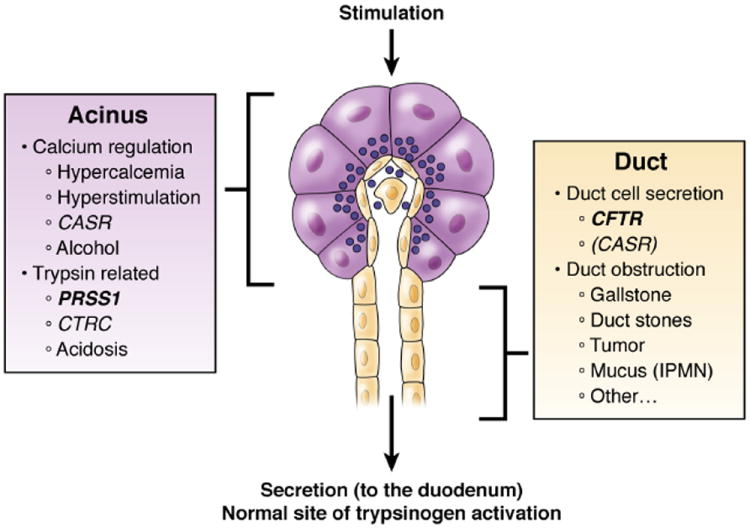
Premature trypsin activation may occur within the acinar cell or within the duct to initiate pancreatitis. The majority of known risk factors can be classified as primarily affecting the acinar cells or pancreatic ducts. Understanding the site of likely trypsin activation and mechanism may guide preventative strategies in the future. (from Solomon and Whitcomb76).
Calcium signaling is an important regulator of acinar cell function 17. Release of calcium from internal stores is required for excitation to be coupled with secretion and expulsion of zymogen granule contents from inside the acinar cells to the duct lumen. Calcium levels are tightly regulated; ATP is required to pump calcium into internal stores and out of the cell 18. Excessive alcohol intake can damage mitochondria, which normally produce large amounts of ATP 19, 20, and thereby disrupt the energy source for calcium regulation in acinar cells.
Multiple lines of evidence indicate that premature activation of trypsin in acinar cells damages the cells to cause an inflammatory response, recognized clinically as acute pancreatitis (see reviews 18, 19, 21-23). The most abundant forms of trypsinogen in the pancreas are cationic trypsinogen (PRSS1) and anionic trypsinogen (PRSS2). PRSS1 is more easily activated, from trypsinogen to trypsin; 24 this occurs via cleavage of trypsinogen activation peptide (TAP), an 8 amino acid N-terminus extension that forms a calcium binding site 25-27. When the calcium concentration increases, the activation site is stabilized, allowing cleavage of the TAP. Therefore, activation of trypsin is also regulated by the calcium concentration 25. Super-physiological concentrations of calcium in acinar cells have been associated with trypsin activation and initiation of pancreatitis 19, 20.
Cleavage of TAP alone does not activate trypsin—its N-terminus must fold back into the globular molecule and interact with the internal sites 28. This process is pH dependent 29, with highest levels of activation occurring at pH values between 7 and 8 29, 30. Low acinar cell pH (<7.15), observed, also promotes activation of trypsin, and can lead to pancreatitis as observed in diabetic patients with ketoacidosis 31-33, possibly in conjunction with activation of cathepsin B 34.
Acinar and duct cells have many mechanisms that protect against trypsin activation, including trypsin self destruction (autolysis), in which other trypsins and CTRC (another proteolytic zymogen activated by trypsin) digest trypsinogen and trypsin at specific sites (Arg-122–Val-123 and Leu-81–Glu-82) to inactivate them 35, 36. However, there is a second calcium-binding site on the trypsin molecule, near the 2 cleavage sites; when calcium concentrations increase, the cleavage sites are blocked, and trypsin is protected 26, 36, 37. Furthermore, during the inflammatory response, the trypsin inhibitor SPINK1 is activated 38-40. Therefore, pancreatitis can arise from defects in acinar cell regulation of trypsin activity or calcium concentration, changes in pH, or alterations in trypsin structure.
Genetic Risk for Acinar Cell Susceptibility
Several genetic factors that affect risk for pancreatitis affect acinar cell function. Mutations in the trypsinogen gene (such as PRSS1 N29I, N29T, V39A, R122C, and R122H; see http://www.pancreasgenetics.org) prevent cleavage of CTRC by trypsin. CTRC is likely to function in acinar cells, and loss-of-function mutations in CTRC are associated with pancreatitis 41, 42. Finally, mutations in the trypsin inhibitor SPINK1 (such as N34S) are associated with increased risk of pancreatitis 43-45. SPINK1 is synthesized by acinar cells 46 and follows trypsinogen from synthesis to secretion, so it is likely to be involved in protecting both acinar cell and the duct from prematurely activated trypsin.
A new finding is that a variant on a haplotype in the non-coding region of the PRSS1-PRSS2 locus significantly reduces expression of PRSS1 and reduces the risk of pancreatitis 9. A common haplotype in linkage with rs10273639T (present in about 40% of population alleles) is associated with partial protection against pancreatitis. This finding again indicates the importance of the trypsinogen genes in RAP and CP.
Alcohol
Multiple studies have linked alcohol consumption with an increased risk of developing AP and CP (see reviews 14, 47, 48). Animal studies have indicated that the increased risk is related to a lower threshold for hyperstimulation-associated pancreatic injury 49, 50. Other studies have indicated that chronic alcohol consumption alters the neuro-hormonal mechanisms of pancreatic activation, with hyperstimulation occurring during alcohol withdrawal (disinhibiting excitatory nerves that adapted to alcohol-associated inhibition) and nutrient feeding (resulting in hyperstimulation) 51, consistent with clinical observations 52. However, the conditions needed for alcohol to initiate AP are so specific that alcoholic AP is uncommon even among heavy drinkers. Genetic risk factors that link alcohol to susceptibility to AP have not been clearly defined.
Epidemiology studies have provided additional insight. Less than 3% of heavy users of alcohol develop CP 53, and the risk of alcoholic pancreatitis, when adjusted for smoking in regression analysis, is low 54. Furthermore, a threshold of >5 drinks a day (or 35 drinks a week) must be reached before there is an associated risk of pancreatitis 14, 54. These data indicate that alcohol intake is a weak susceptibility factor (first hit), but could be a strong modifier factor (second hit), especially with smoking54 and the CLDN risk variant9.
Duct Biology and Risk
The primary function of duct cells is to secrete a bicarbonate-rich fluid that flushes the zymogens out of the pancreas into the duodenum. The most important molecule within the duct is CFTR, an anion channel that transports chloride and bicarbonate. The electrochemical mechanism of pancreatic chloride and bicarbonate secretion has been well defined in animal and mathematical models 55, 56.
Risk for pancreatitis is linked with the zymogens within the duct rather than within the duct cell. The duct cells express multiple sensors on the luminal surface that detect trypsin activity (such as protease activated receptors PAR 1 and PAR2 57, 58) and are therefore protective, whereas other molecules (such as the purinergic receptors P2Y2, P2X4, and P2X7 59) sense calcium concentration and ATP release as signals of injury. Activation of these sensors results in opening of CFTR channels, secretion of bicarbonate-rich fluid, and flushing of the duct contents into the duodenum 59. There are also sensors inside duct cells that change the permeability of CFTR to promote conductance of bicarbonate when intracellular concentrations of chloride are low 55, 60. In addition, there are multiple types of duct cells, with different characteristics—for example, the duct cells nearest the acinar cells have the highest concentration of CFTR molecules 55. The duct also contains mucus-secreting cells that protect the pancreatic duct from zymogens. So, there are different sensors and mechanisms that protect the pancreas from different types of risk; defects in 1 protective mechanism might be only relevant to 1 type of stress or injury, but not others.
Duct Cell Dysfunction
Mutations in CFTR cause cystic fibrosis, an autosomal recessive disease characterized by the development of chronic pancreatitis (beginning in utero), demonstrating the importance of CFTR in pancreatic physiology. Nearly 2000 variants in CFTR have been identified (http://www.genet.sickkids.on.ca/app), but little is known about their functional effects (CFTR2 project - http://www.cftr2.org). The more-common variants are classified clinically as severe or mild, based on their effect on pancreatic function. An accepted molecular classification strategy organizes the variants by their effects on CFTR function, with Class I–III variants causing severe dysfunction, and Class IV–V causing reduced or altered function 61, 62. Class IV–V variants have mild–variable effects on the pancreas and other organs, often leaving sufficient function for basic physiological needs but not enough to handle stress. The effects of CFTR Class IV–V variants have been reported on borderline pancreatic exocrine sufficiency in patients with CF, and their risk of pancreatitis 63.
In 1998, 2 studies reported that patients with idiopathic pancreatitis, and some with alcohol-associated CP, had more variants in CFTR than could be explained by chance 64, 65. These findings have been replicated in studies from the United States (US) 66, Europe 67-70, India 71, and China and Taiwan 72, 73. What was not clear was whether these patients had mild or atypical CF, or whether they had CFTR-related disorders with complex, pancreas-specific mechanisms 63, 74-76. What is clear is that proper CFTR activity is required for normal duct cell function, and that variants in CFTR are associated with susceptibility to pancreatitis.
There have been no genetic factors associated with duct obstruction, sphincter of Oddi dysfunction, or pancreatic divisum. The effect of genetic factors on formation of pancreatic duct stones, gallstones, and pancreatic neoplasia have not been fully evaluated in patients with pancreatitis.
Genetic Risk of Autoimmune Disease
AIP is identified based on imaging analysis of the pancreatic parenchyma and duct, serologic analysis of immunoglobulin (Ig)G4 levels, features of other organs, histologic analysis of the pancreas, and sometimes response to steroid therapy 77. Two types of AIP have been described: 77, 78 Type 1 AIP is more prevalent in Japan and Asia that in the United States and Europe, and occurs at an older age that Type 2, which is associated with higher levels of IgG4 and more proximal biliary, retroperitoneal, renal, or salivary disease. Type 2 AIP is rarely observed in Asians, is more often associated with inflammatory bowel disease, and has unique pathology features 77, 79.
Differences in the racial and geographic distribution of AIP indicate that environmental or genetic risk factors are important. To date, no compelling evidence has been reported to establish or refute either type of risk. Challenges in doing so include the complex interaction of AIP with the involvement of other organs and immune-mediated syndromes and the relatively small number of patients with this disease. Human and animal models have focused on the immune system, including the HLA region 80, 81 and other loci linked with autoimmunity in rodent models 82, but little is known about the pathogenesis of AIP.
Progression from RAP to CP
There is no single etiology for CP. It could be considered as a disorder that progresses over years from an initial injury to complete gland destruction under the influence of 1 or more etiologic pathways, so treatments might be selected to interrupt the process and allow healing and regeneration. The Sentinel Acute Pancreatitis Event (SAPE) model 13, 14, 83, recently tested in a population-based study from Allegheny County (Pittsburgh), PA 84 (Figure 3), proposes that patients with AP progress to CP at different rates, based on etiology, with alcohol etiology having the highest risk (Figure 3A), and that the mechanism for progression is linked to RAP (Figure 3B).
Figure 3. Risks of RAP and CP.

Data collected in population-based analysis of 7456 residents of Allegheny County, PA following their first (sentinel) episode of AP. A. Risk of developing RAP based on etiology, B. Risk of CP based on the presence or absence of documented RAP. From Yadav, O’Connell and Papachristou. 84.
The SAPE model is built on the observation that individuals live for many years with multiple risk factors for CP—then suddenly the process begins and CP develops 13, 14. The premise is that the development of CP requires activation of the immune response, as happens during an episode of AP. The outcome of an episode of AP can be either complete recovery, necrosis–fibrosis following severe AP, or the initiation of progressive inflammation and fibrosis, leading to CP over time. During the interval between AP and CP (Figure 1), there is opportunity to intervene and stop progression. The approach selected to prevent RAP and/or progression should be based on etiology, such as a cholecystectomy for patients with gallstone pancreatitis but not for patients with alcohol or genetic etiologies. Risk factors for RAP and progression of fibrosis must therefore be addressed.
Trypsin Activation and Disease Progression
Studies in patients with hereditary pancreatitis have shown how a major susceptibility factors, such as PRSS1 R122H, become risk factors for AP and CP through RAP 2, 85, 86. On the average, carriers of PRSS1 R122H or N29I have their first episode of AP at a median age of 10 years, with evidence of CP developing within the next decade. This provides some of the most compelling data that RAP could eventually lead to CP.
SPINK1 variants could contribute to pancreatic disease via different mechanisms than PRSS1 or CFTR variants. Heterozygous mutations or variants are likely to modify CP development, rather increase susceptibility 43, 87. This is based on the observation that the SPINK1 N34S high-risk haplotype is common (1%–3% of most populations), whereas CP is uncommon (42/100,000 persons 88). However, the SPINK1 N34S variant increases the risk of alcoholic CP 5-fold, idiopathic CP 15-fold, and tropical CP 19-fold45. It is unlikely that SPINK1 mutations are a susceptibility factor for AP, but rather for RAP and CP 89, 90. SPINK1 could be effective in controlling the effects of recurrent intra-pancreatic trypsin activation from a variety of etiologies, but mutations in SPINK1 might allow recurrent trypsin-associated injury to lead to development of fibrosis (Figure 4).
Figure 4. Factors that Contribute to Pancreatic Fibrosis.

A combination of factors contribute to pancreatitis. The first hit increases susceptibility to injury, whereas the second hit affects the immune response (and includes leukocytes, such as M2 macrophages) to promote stellate cell-associated fibrosis. Alcohol could injure the pancreas via its effects on acinar cells, but it is probably more important in the second group, as a modifier of the immune response. For example it could promote a Th17-cell response, or act directly on stellate cells (dashed lines). Altered trypsin functions in acinar cells or ducts could also initiate disease. In either case, similar second hit factors then contribute to development of fibrosis (via variants in SPINK1 and/or CTRC). Severe acute pancreatitis involves widespread pancreatic necrosis (about 90% of tissue), which leads directly to scaring and fibrosis in the recovery phase. Obese patients could have areas of adipose tissue that contain local necrosis of acinar tissue, which has been linked to lipotoxicity. Progressive fibrosis could occur in patients with RAP. Other pathways include autoimmune pancreatitis in which the pathway to fibrosis is less well understood.
In support of this hypothesis, increasing evidence indicates that heterozygous SPINK1 mutations are only associated with RAP or CP when patients have a mutation in a susceptibility gene associated with recurrent trypsin activation, such as PRSS1, CFTR, CASR, or CTRC 66, 69, 71. Thus, heterozygous SPINK1 mutations do not cause pancreatitis, but instead exacerbate the clinical outcome of patients with recurrent pancreatic injury from trypsin activation. Of note, SPINK1 mutations are only slightly more common in patients with alcoholic pancreatitis patients than in the general population 91-94. The progression from pancreatic injury to CP therefore appears to differ from that of alcohol-associated CP 45, 95 (Figure 4).
Complex genetics—CTRC and CASR
Mutations in CTRC and CASR are associated with CP 7, 8. Unlike CFTR and SPINK1 mutations, homozygous or compound heterozygous mutations in CTRC and CASR have not been found in family clusters of CP or patients with sporadic pancreatitis, indicating that, independently, they are not sufficient to cause RAP or CP. Instead, variants in CTRC and CASR genes identified in patients with CP almost always occur with heterozygous variants in PRSS1, CFTR, or SPINK1 7, 69, 96, indicating that combinations of genetic factors are required to increase the risk for RAP and CP. Table 1 summarizes various combinations of genetic risk factors and clinical phenotypes.
Table 1.
Genotype–Phenotype Correlations in Multi-Organ Syndromes
| Genotype (variants) | Phenotype (syndrome) | Comment |
|---|---|---|
| PRSS1 | HP | Genetic counseling recommended |
| PRSS1 / any | HP, worse clinical course | Genetic counseling recommended |
| CFTRsev/CFTRsev | CF | Manage with a CF center |
| CFTRsev/CFTRm-v | atypical CF | Manage with a CF center |
| SPINK1/ SPINK1 | familial pancreatitis | Usually progresses to severe CP |
| CFTRbicarb/CFTRany | Pancreas/Sinus/CBAVD | Newly defined syndrome |
| CFTRany/SPINK1 | RAP / CP | Pancreas only |
| CTRC / SPINK1 | RAP / CP | Pancreas only – not well studied |
| CASR+ / alcohol | RAP/ CP | Pancreas only – not well studied |
| CASR- / SPINK1 | RAP/ CP, familial CP | CP in FHH and sporadic CP |
| CASR- / CFTR | RAP/ CP | Pancreas only – not well studied |
CFTR: sev=severe mutations (typically functional class I-III), m-v = mild-variable mutations, (typically CFTR functional class IV), bicarb = bicarbonate conductance disrupting variant (e.g. R75Q), any= either severe, mild-variable or bicarbonate disrupting variants. CASR+, gain of function mutations; CASR-, loss of function mutations; FHH, familial hypocalciuric hypercalcemia; HP, hereditary pancreatitis
Although the mechanisms by which CTRC protects against pancreatitis are established 35, 97, the importance of CTRC variants in terms of risk for RAP and CP is less clear. Rosendhal et al. 42 demonstrated that the rare p.R254W and p.K247_R254del variants were significantly overrepresented in pancreatitis cases from Germany but not among individuals with CP in India. Masson et al. 41 did not associate p.R254W or p.K247_R254del with CP in patients in France, but did associate them with multiple rare, newly identified mutations in CTRC. Chang et al. 98 evaluated a Chinese cohort of CP cases and associated additional mutations with CP, but did not replicate the previous findings. Paliwal et al. reported multiple variants in CTRC in among patients with CP in India, with p.V235I being the most prevalent96.
In the North American Pancreatitis Study (NAPS)-2, CP in the US cohort identified a few of the variants in exons 2, 3, or 7 that were seen in Germany, but these were too rare in the NAPS-2 population for the finding to be statically significant (D. Whitcomb, unpublished observations, 2009, 2012). However, functional studies have shown that rare mutations do affect CTRC function35, 97 and therefore are likely of clinically important based on mechanistic evidence rather than statistical evidence alone.,. Taken together, it has been difficult to prove a role for specific CTRC variants in CP using population-based genetic studies, even though mechanistic studies have shown how defects in CTRC could contribute to the disease. If CTRC variants can be associated with CP risk, they can be used to design personalized treatment approaches.
Mutations in the CASR gene have also been associated with CP 7. The association was initially reported in patients with familial hypocalciuric hypercalcemia, a condition caused by loss-of-function mutations in CASR. Felderbauer et al. 99 identified 2 patients with CP that each carried the CASR and SPINK1 mutations. A screen of 19 families with idiopathic CP revealed multiple additional cases with loss-of-function variants in CASR together with SPINK1 mutations 100. In a study of 338 subjects, Muddana et al. 101 associated pancreatitis with the gain-of-function R990G variant of CASR 102. Risk of CP was not associated with SPINK1 variants, but was associated with moderate to heavy alcohol consumption.
Over 70 CASR variants have been classified and are included in the calcium-sensing receptor locus-specific database (CASRdb); they have been associated with familial (benign) hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia 103. There is evidence that that loss-of-function variants of CASR, in association with SPINK1 and CFTR variants, affect duct cell function (Figure 2), whereas gain-of-function variants in CASR that are associated with alcoholic pancreatitis affect acinar cell functions. Further research is needed to test this hypothesis.
Alcoholic Pancreatitis and Progression
Patients with alcoholic AP are at high risk of RAP 104 and progression from RAP to CP, if they continue to drink alcohol and/or smoke 84, 105-107. In the NAPS2, the prevalence of CFTR variants was equal among subjects with RAP and CP suggesting that CFTR variants increase susceptibility to pancreatic injury, yet only 25% of RAP and 46% of CP subjects drank amounts of alcohol associated with disease risk 54. This could mean that alcohol (especially with smoking) is a disease modifier rather than a typical susceptibility factor and is associated with rapid transition from RAP to CP. The predicted result would be lowering the prevalence of alcohol-associated pancreatitis among individuals with RAP while increasing it among those with CP. Several recent discoveries provide some insight as to how alcohol might promote this transition.
Alcohol Causes RAP
Recurrent pancreatitis itself promotes fibrosis (Figure 3). In humans, this initially became evident in patients with hereditary pancreatitis, 2, 3 and was supported by data from animal studies 108, 109. Alcohol could affect regulation of calcium by acinar cells by injuring the mitochondria and limiting the ability of the acinar cells to pump calcium out of the cytoplasm 19, 20. In support of this concept, gain-of-function mutations in CASR the risk increase risk for alcoholic pancreatitis 7, 101. Preventing RAP through cessation of alcohol and smoking 105, 106 is important in limiting progression to CP.
Alcohol Alters the Immune Response
Alcohol increases the rate of fibrosis development in animal models beyond what is expected from RAP alone 110, 111. The mechanism appears involve alterations in the immune response to recurrent injury110. In humans, preliminary studies link alcohol-associated CP, but not other types of CP, with the T-helper (Th)17 cell response. Mutations in the interleukin (IL)-23 receptor that reduce its function protect against Crohn’s disease, 112, 113 were also reported to protect against alcoholic pancreatitis 114.
The CLDN2 Locus and Risk for Alcoholic Pancreatitis
Recently, a genetic locus was found to be associated with a marked increase in the risk of alcohol-related RAP and CP 9. In population controls, 25.8% of men carried a single copy of the X-linked CLDN2 risk allele T (rs12688220) and 6.9% of women were homozygous for the variant. The variant was found to have a significant effect of risk of pancreatitis with a non-alcohol etiology, with a single-copy frequency in men of 38.5% (expected 26%) and homozygous frequency in women of 10.0% (expected 7%). Only 4% of men are expected to have both at-risk levels of drinking (about 16% prevalence115) and the CLDN2 risk allele T (25.8% prevalence in male controls). However, 47.6% of men with alcohol-associated CP were found to have the CLDN2 risk allele T, indicating that these factors, in combination, increase risk for CP. Furthermore, the X-chromosome linkage could partially explain the higher prevalence of alcohol-associated pancreatitis among men.
CLDN2 is normally expressed at low levels by the duct cells of the exocrine pancreas and by the islets. It differs from other claudins in the pancreas in that it forms low-resistance, cation-selective ion and water channels between endothelial cells116, 117, which facilitate sodium and water movement into the duct lumen. Variants in the coding region of CLDN2 have not been associated with pancreatitis. However, immunohistolochemical analysis of pancreatic tissues from patients with CP, collected during pancreatic surgery, found increased levels and altered localization of CLDN2 in acinar cells of patients with the high-risk CLDN2 genotype. Expression of CLDN2 increases in in porcine acinar cells during development of AP 118, linking acinar cell stress to CLDN2 levels. The CLDN2 promoter includes a binding site for the nuclear factor-κB 119, and CLDN2 expression increases in other cells under conditions of injury or stress120-122. In inflamed tissues, acinar cells might interact with or be injured by M2-type macrophages that express CLDN2,123 which promote the inflammatory process. Further studies are needed to fully define the functional variant in the CLDN2-locus and the mechanisms by which it could contribute to CP pathogenesis.
Future Directions
Insights gained from studies of genetic variations and/or environmental factors have transformed our understanding of CP. We now know that CP begins with an injury that activates the immune response, and progression occurs with immune system activation, in the context of additional risk and disease modifiers, which leads to fibrosis and other complications. Pathogenesis of CP is multi-factorial, and involves altered activities of acinar cells, duct cells, pancreatic ducts, and the immune system (autoimmunity). Treatment will therefore likely require a combination of different therapeutic approaches. In the future, patients with AP or early signs of pancreatic disease will undergo a systematic evaluation that includes genetic analysis, to provide information about disease etiology, and then receive therapies selected to target specific factors, based on the genetic information. A model pancreas clinic has already been developed 1 and will continue to integrate new findings from genetic analyses into therapeutic approaches for pancreatic disorders. Someday, the effectiveness of treatment could be monitored with biomarkers; therapeutics could be continually adjusted to maintain pancreatic function and prevent progression of disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Whitcomb DC. What is personalized medicine and should does it replace? Nat Rev Gastroenterol Hepatol. 2012;9:418–24. doi: 10.1038/nrgastro.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorry MC, Gabbaizedeh D, Furey W, et al. Mutations in the cationic trypsinogen gene are associated with recurrent acute and chronic pancreatitis. Gastroenterology. 1997;113:1063–8. doi: 10.1053/gast.1997.v113.pm9322498. [DOI] [PubMed] [Google Scholar]
- 3.Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nature Genetics. 1996;14:141–5. doi: 10.1038/ng1096-141. [DOI] [PubMed] [Google Scholar]
- 4.Witt H, Apte MV, Keim V, et al. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology. 2007;132:1557–73. doi: 10.1053/j.gastro.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Chen JM, Ferec C. Chronic pancreatitis: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2009;10:63–87. doi: 10.1146/annurev-genom-082908-150009. [DOI] [PubMed] [Google Scholar]
- 6.Whitcomb DC. Genetic aspects of pancreatitis. Annu Rev Med. 2010;61:413–24. doi: 10.1146/annurev.med.041608.121416. [DOI] [PubMed] [Google Scholar]
- 7.Larusch J, Whitcomb DC. Genetics of pancreatitis. Current opinion in gastroenterology. 2011;27:467–74. doi: 10.1097/MOG.0b013e328349e2f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen JM, Ferec C. Genetics and pathogenesis of chronic pancreatitis: The 2012 update. Clinics and research in hepatology and gastroenterology. 2012;36(4):334–40. doi: 10.1016/j.clinre.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Whitcomb DC, LaRusch J, Krasinskas AM, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet. 2012 doi: 10.1038/ng.2466. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coppieters KT, Wiberg A, Tracy SM, et al. Immunology in the clinic review series: focus on type 1 diabetes and viruses: the role of viruses in type 1 diabetes: a difficult dilemma. Clinical and experimental immunology. 2012;168:5–11. doi: 10.1111/j.1365-2249.2011.04554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitcomb DC. Mechanisms of disease: Advances in understanding the mechanisms leading to chronic pancreatitis. Nat Clin Pract Gastroenterol Hepatol. 2004;1:46–52. doi: 10.1038/ncpgasthep0025. [DOI] [PubMed] [Google Scholar]
- 12.Etemad B, Whitcomb DC. Chronic pancreatitis: Diagnosis, classification, and new genetic developments. Gastroenterology. 2001;120:682–707. doi: 10.1053/gast.2001.22586. [DOI] [PubMed] [Google Scholar]
- 13.Whitcomb DC. Hereditary Pancreatitis: New insights into acute and chronic pancreatitis. Gut. 1999;45:317–322. doi: 10.1136/gut.45.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yadav D, Whitcomb DC. The role of alcohol and smoking in pancreatitis. Nat Rev Gastroenterol Hepatol. 2010;7:131–45. doi: 10.1038/nrgastro.2010.6. [DOI] [PubMed] [Google Scholar]
- 15.Whitcomb DC, Barmada MM. A systems biology approach to genetic studies of pancreatitis and other complex diseases. Cell Mol Life Sci. 2007;64:1763–77. doi: 10.1007/s00018-007-7055-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacob C, Yang PC, Darmoul D, et al. Mast cell tryptase controls paracellular permeability of the intestine. Role of protease-activated receptor 2 and beta-arrestins. J Biol Chem. 2005;280:31936–48. doi: 10.1074/jbc.M506338200. [DOI] [PubMed] [Google Scholar]
- 17.Mogami H, Nakano K, Tepikin AV, et al. Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell. 1997;88:49–55. doi: 10.1016/s0092-8674(00)81857-7. [DOI] [PubMed] [Google Scholar]
- 18.Raraty M, Ward J, Erdemli G, et al. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci U S A. 2000;97:13126–31. doi: 10.1073/pnas.97.24.13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sutton R, Criddle D, Raraty MG, et al. Signal transduction, calcium and acute pancreatitis. Pancreatology. 2003;3:497–505. doi: 10.1159/000075581. [DOI] [PubMed] [Google Scholar]
- 20.Criddle DN, Raraty MG, Neoptolemos JP, et al. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc Natl Acad Sci U S A. 2004;101:10738–43. doi: 10.1073/pnas.0403431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lerch MM, Gorelick FS. Early trypsinogen activation in acute pancreatitis. Med Clin North Am. 2000;84:549–63. doi: 10.1016/s0025-7125(05)70239-x. [DOI] [PubMed] [Google Scholar]
- 22.Mithofer K, Fernandez-Del Castillo C, Frick TW, et al. Acute hypercalcemia causes acute pancreatitis and ectopic trypsinogen activation in the rat. Gastroenterology. 1995;109:239–46. doi: 10.1016/0016-5085(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 23.Frick TW, Fernandez-del Castillo C, Bimmler D, et al. Elevated calcium and activation of trypsinogen in rat pancreatic acini. Gut. 1997;41:339–43. doi: 10.1136/gut.41.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kukor Z, Tóth M, Sahin-Tóth M. Human anionic trypsinogen. Eur J Biochem. 2003;270:2047–58. doi: 10.1046/j.1432-1033.2003.03581.x. [DOI] [PubMed] [Google Scholar]
- 25.Liu JH, Wang ZX. Kinetic analysis of ligand-induced autocatalytic reactions. The Biochemical journal. 2004;379:697–702. doi: 10.1042/BJ20031365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colomb E, Figarella C. Comparative studies on the mechanism of activation of the two human trypsinogens. Biochimica et biophysica acta. 1979;571:343–51. doi: 10.1016/0005-2744(79)90104-9. [DOI] [PubMed] [Google Scholar]
- 27.Guy O, Lombardo D, Bartelt DC, et al. Two human trypsinogens. Purification, molecular properties, and N-terminal sequences. Biochemistry. 1978;17:1669–75. doi: 10.1021/bi00602a014. [DOI] [PubMed] [Google Scholar]
- 28.Bennett WS, Huber R. Structural and functional aspects of domain motions in proteins. CRC Crit Rev Biochem. 1984;15:291–384. doi: 10.3109/10409238409117796. [DOI] [PubMed] [Google Scholar]
- 29.Nemoda Z, Sahin-Toth M. The tetra-aspartate motif in the activation peptide of human cationic trypsinogen is essential for autoactivation control but not for enteropeptidase recognition. The Journal of biological chemistry. 2005;280:29645–52. doi: 10.1074/jbc.M505661200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rinderknecht H, Renner IG, Abramson SB, et al. Mesotrypsin: a new inhibitor-resistant protease from a zymogen in human pancreatic tissue and fluid. Gastroenterology. 1984;86:681–92. [PubMed] [Google Scholar]
- 31.Bhoomagoud M, Jung T, Atladottir J, et al. Reducing extracellular pH sensitizes the acinar cell to secretagogue-induced pancreatitis responses in rats. Gastroenterology. 2009;137:1083–92. doi: 10.1053/j.gastro.2009.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yadav D, Nair S, Norkus EP, et al. Nonspecific hyperamylasemia and hyperlipasemia in diabetic ketoacidosis: incidence and correlation with biochemical abnormalities. The American journal of gastroenterology. 2000;95:3123–8. doi: 10.1111/j.1572-0241.2000.03279.x. [DOI] [PubMed] [Google Scholar]
- 33.Nair S, Yadav D, Pitchumoni CS. Association of diabetic ketoacidosis and acute pancreatitis: observations in 100 consecutive episodes of DKA. The American journal of gastroenterology. 2000;95:2795–800. doi: 10.1111/j.1572-0241.2000.03188.x. [DOI] [PubMed] [Google Scholar]
- 34.Kukor Z, Mayerle J, Kruger B, et al. Presence of cathepsin B in the human pancreatic secretory pathway and its role in trypsinogen activation during hereditary pancreatitis. The Journal of biological chemistry. 2002;277:21389–96. doi: 10.1074/jbc.M200878200. [DOI] [PubMed] [Google Scholar]
- 35.Beer S, Zhou J, Szabo A, et al. Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut. 2012 doi: 10.1136/gutjnl-2012-303090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szabo A, Sahin-Toth M. Determinants of chymotrypsin C cleavage specificity in the calcium binding loop of human cationic trypsinogen. The FEBS journal. 2012 doi: 10.1111/febs.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitcomb DC, Lowe ME. Human pancreatic digestive enzymes. Dig Dis Sci. 2007;52:1–17. doi: 10.1007/s10620-006-9589-z. [DOI] [PubMed] [Google Scholar]
- 38.Khalid A, Finkelstein S, Thompson B, et al. A 93 year old man with the PRSS1 R122H mutation, low SPINK1 expression, and no pancreatitis: insights into phenotypic non-penetrance. Gut. 2006;55:728–31. doi: 10.1136/gut.2005.067959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lasson A, Borgstrom A, Ohlsson K. Elevated pancreatic secretory trypsin inhibitor levels during severe inflammatory disease, renal insufficiency, and after various surgical procedures. Scand J Gastroenterol. 1986;21:1275–80. doi: 10.3109/00365528608996455. [DOI] [PubMed] [Google Scholar]
- 40.Ogawa M. Pancreatic secretory trypsin inhibitor as an acute phase reactant. Clin Biochem. 1988;21:19–25. doi: 10.1016/s0009-9120(88)80107-3. [DOI] [PubMed] [Google Scholar]
- 41.Masson E, Chen JM, Scotet V, et al. Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum Genet. 2008;123:83–91. doi: 10.1007/s00439-007-0459-3. [DOI] [PubMed] [Google Scholar]
- 42.Rosendahl J, Witt H, Szmola R, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40:78–82. doi: 10.1038/ng.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfutzer RH, Barmada MM, Brunskill AP, et al. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology. 2000;119:615–23. doi: 10.1053/gast.2000.18017. [DOI] [PubMed] [Google Scholar]
- 44.Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nature Genetics. 2000;25:213–6. doi: 10.1038/76088. [DOI] [PubMed] [Google Scholar]
- 45.Aoun E, Chang CC, Greer JB, et al. Pathways to injury in chronic pancreatitis: decoding the role of the high-risk SPINK1 N34S haplotype using meta-analysis. PLoS ONE. 2008;3:e2003. doi: 10.1371/journal.pone.0002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marks WH, Ohlsson K, Polling A. Immunocytochemical distribution of trypsinogen and pancreatic secretory trypsin inhibitor in normal and neoplastic tissues in man. Scandinavian journal of gastroenterology. 1984;19:673–6. [PubMed] [Google Scholar]
- 47.Sand J, Lankisch PG, Nordback I. Alcohol consumption in patients with acute or chronic pancreatitis. Pancreatology : official journal of the International Association of Pancreatology. 2007;7:147–56. doi: 10.1159/000104251. [DOI] [PubMed] [Google Scholar]
- 48.Apte M, Pirola R, Wilson J. New insights into alcoholic pancreatitis and pancreatic cancer. Journal of gastroenterology and hepatology. 2009;24(Suppl 3):S51–6. doi: 10.1111/j.1440-1746.2009.06071.x. [DOI] [PubMed] [Google Scholar]
- 49.Lu Z, Karne S, Kolodecik T, et al. Alcohols enhance caerulein-induced zymogen activation in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2002;282:G501–7. doi: 10.1152/ajpgi.00388.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pandol SJ, Periskic S, Gukovsky I, et al. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology. 1999;117:706–16. doi: 10.1016/s0016-5085(99)70465-8. [DOI] [PubMed] [Google Scholar]
- 51.Deng X, Wood PG, Eagon PK, et al. Chronic alcohol-induced alterations in the pancreatic secretory control mechanisms. Dig Dis Sci. 2004;49:805–19. doi: 10.1023/b:ddas.0000030093.25897.61. [DOI] [PubMed] [Google Scholar]
- 52.Nordback I, Pelli H, Lappalainen-Lehto R, et al. Is it long-term continuous drinking or the post-drinking withdrawal period that triggers the first acute alcoholic pancreatitis? Scand J Gastroenterol. 2005;40:1235–9. doi: 10.1080/00365520510023413. [DOI] [PubMed] [Google Scholar]
- 53.Yadav D, Eigenbrodt ML, Briggs MJ, et al. Pancreatitis: prevalence and risk factors among male veterans in a detoxification program. Pancreas. 2007;34:390–8. doi: 10.1097/mpa.0b013e318040b332. [DOI] [PubMed] [Google Scholar]
- 54.Yadav D, Hawes RH, Brand RE, et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med. 2009;169:1035–45. doi: 10.1001/archinternmed.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee MG, Ohana E, Park HW, et al. Molecular Mechanism of Pancreatic and Salivary Gland Fluid and HCOFormula Secretion. Physiological reviews. 2012;92:39–74. doi: 10.1152/physrev.00011.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whitcomb DC, Ermentrout GB. A mathematical model of the pancreatic duct cell generating high bicarbonate concentrations in pancreatic juice. Pancreas. 2004;29:E30–E40. doi: 10.1097/00006676-200408000-00016. [DOI] [PubMed] [Google Scholar]
- 57.Hansen KK, Sherman PM, Cellars L, et al. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc Natl Acad Sci U S A. 2005;102:8363–8. doi: 10.1073/pnas.0409535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharma A, Tao X, Gopal A, et al. Protection against acute pancreatitis by activation of protease-activated receptor-2. Am J Physiol Gastrointest Liver Physiol. 2005;288:G388–95. doi: 10.1152/ajpgi.00341.2004. [DOI] [PubMed] [Google Scholar]
- 59.Steward MC, Ishiguro H. Molecular and cellular regulation of pancreatic duct cell function. Current opinion in gastroenterology. 2009;25:447–53. doi: 10.1097/MOG.0b013e32832e06ce. [DOI] [PubMed] [Google Scholar]
- 60.Park HW, Nam JH, Kim JY, et al. Dynamic regulation of CFTR bicarbonate permeability by [Cl-]i and its role in pancreatic bicarbonate secretion. Gastroenterology. 2010;139:620–31. doi: 10.1053/j.gastro.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 61.Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Ann Hum Genet. 2003;67:471–85. doi: 10.1046/j.1469-1809.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 62.Kerem E. Atypical CF and CF related diseases. Paediatr Respir Rev. 2006;7(Suppl 1):S144–6. doi: 10.1016/j.prrv.2006.04.219. [DOI] [PubMed] [Google Scholar]
- 63.Ooi CY, Dorfman R, Cipolli M, et al. Type of CFTR mutation determines risk of pancreatitis in patients with cystic fibrosis. Gastroenterology. 2011;140:153–61. doi: 10.1053/j.gastro.2010.09.046. [DOI] [PubMed] [Google Scholar]
- 64.Sharer N, Schwarz M, Malone G, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. New England Journal of Medicine. 1998;339:645–652. doi: 10.1056/NEJM199809033391001. [DOI] [PubMed] [Google Scholar]
- 65.Cohn JA, Friedman KJ, Noone PG, et al. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998;339:653–658. doi: 10.1056/NEJM199809033391002. [DOI] [PubMed] [Google Scholar]
- 66.Schneider A, Larusch J, Sun X, et al. Combined Bicarbonate Conductance-Impairing Variants in CFTR and SPINK1 Variants Are Associated With Chronic Pancreatitis in Patients Without Cystic Fibrosis. Gastroenterology. 2011;140:162–71. doi: 10.1053/j.gastro.2010.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Audrezet MP, Chen JM, Le Marechal C, et al. Determination of the relative contribution of three genes-the cystic fibrosis transmembrane conductance regulator gene, the cationic trypsinogen gene, and the pancreatic secretory trypsin inhibitor gene-to the etiology of idiopathic chronic pancreatitis. Eur J Hum Genet. 2002;10:100–6. doi: 10.1038/sj.ejhg.5200786. [DOI] [PubMed] [Google Scholar]
- 68.Sobczynska-Tomaszewska A, Bak D, Oralewska B, et al. Analysis of CFTR, SPINK1, PRSS1 and AAT mutations in children with acute or chronic pancreatitis. J Pediatr Gastroenterol Nutr. 2006;43:299–306. doi: 10.1097/01.mpg.0000232570.48773.df. [DOI] [PubMed] [Google Scholar]
- 69.Rosendahl J, Landt O, Bernadova J, et al. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut. 2012 doi: 10.1136/gutjnl-2011-300645. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 70.de Cid R, Ramos MD, Aparisi L, et al. Independent contribution of common CFTR variants to chronic pancreatitis. Pancreas. 2010;39:209–15. doi: 10.1097/MPA.0b013e3181bab679. [DOI] [PubMed] [Google Scholar]
- 71.Midha S, Khajuria R, Shastri S, et al. Idiopathic chronic pancreatitis in India: phenotypic characterisation and strong genetic susceptibility due to SPINK1 and CFTR gene mutations. Gut. 2010;59:800–7. doi: 10.1136/gut.2009.191239. [DOI] [PubMed] [Google Scholar]
- 72.Chang YT, Chang MC, Su TC, et al. Association of cystic fibrosis transmembrane conductance regulator (CFTR) mutation/variant/haplotype and tumor necrosis factor (TNF) promoter polymorphism in hyperlipidemic pancreatitis. Clin Chem. 2008;54:131–8. doi: 10.1373/clinchem.2007.093492. [DOI] [PubMed] [Google Scholar]
- 73.Chang MC, Chang YT, Wei SC, et al. Spectrum of mutations and variants/haplotypes of CFTR and genotype-phenotype correlation in idiopathic chronic pancreatitis and controls in Chinese by complete analysis. Clin Genet. 2007;71:530–9. doi: 10.1111/j.1399-0004.2007.00813.x. [DOI] [PubMed] [Google Scholar]
- 74.Noone PG, Zhou Z, Silverman LM, et al. Cystic fibrosis gene mutations and pancreatitis risk: relation to epithelial ion transport and trypsin inhibitor gene mutations. Gastroenterology. 2001;121:1310–9. doi: 10.1053/gast.2001.29673. [DOI] [PubMed] [Google Scholar]
- 75.Bombieri C, Claustres M, De Boeck K, et al. Recommendations for the classification of diseases as CFTR-related disorders. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2011;10(Suppl 2):S86–102. doi: 10.1016/S1569-1993(11)60014-3. [DOI] [PubMed] [Google Scholar]
- 76.Solomon S, Whitcomb DC. Genetics of Pancreatitis: An Update for Clinicians and Genetic Counselors. Current gastroenterology reports. 2012;14:112–117. doi: 10.1007/s11894-012-0240-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shimosegawa T, Chari ST, Frulloni L, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011;40:352–8. doi: 10.1097/MPA.0b013e3182142fd2. [DOI] [PubMed] [Google Scholar]
- 78.Sah RP, Chari ST, Pannala R, et al. Differences in Clinical Profile and Relapse Rate of Type 1 Versus Type 2 Autoimmune Pancreatitis. Gastroenterology. 2010 doi: 10.1053/j.gastro.2010.03.054. [DOI] [PubMed] [Google Scholar]
- 79.Kloppel G, Detlefsen S, Chari ST, et al. Autoimmune pancreatitis: the clinicopathological characteristics of the subtype with granulocytic epithelial lesions. J Gastroenterol. 2010 doi: 10.1007/s00535-010-0265-x. [DOI] [PubMed] [Google Scholar]
- 80.Freitag TL, Cham C, Sung HH, et al. Human risk allele HLA-DRB1*0405 predisposes class II transgenic Ab0 NOD mice to autoimmune pancreatitis. Gastroenterology. 2010;139:281–91. doi: 10.1053/j.gastro.2010.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ota M, Katsuyama Y, Hamano H, et al. Two critical genes (HLA-DRB1 and ABCF1)in the HLA region are associated with the susceptibility to autoimmune pancreatitis. Immunogenetics. 2007;59:45–52. doi: 10.1007/s00251-006-0178-2. [DOI] [PubMed] [Google Scholar]
- 82.Asghari F, Fitzner B, Holzhuter SA, et al. Identification of quantitative trait loci for murine autoimmune pancreatitis. Journal of medical genetics. 2011;48:557–62. doi: 10.1136/jmg.2011.089730. [DOI] [PubMed] [Google Scholar]
- 83.Stevens T, Conwell DL, Zuccaro G. Pathogenesis of chronic pancreatitis: an evidence-based review of past theories and recent developments. Am J Gastroenterol. 2004;99:2256–70. doi: 10.1111/j.1572-0241.2004.40694.x. [DOI] [PubMed] [Google Scholar]
- 84.Yadav D, O’Connell M, Papachristou GI. Natural history following the first attack of acute pancreatitis. The American journal of gastroenterology. 2012;107:1096–103. doi: 10.1038/ajg.2012.126. [DOI] [PubMed] [Google Scholar]
- 85.Howes N, Lerch MM, Greenhalf W, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2:252–61. doi: 10.1016/s1542-3565(04)00013-8. [DOI] [PubMed] [Google Scholar]
- 86.Rebours V, Boutron-Ruault MC, Schnee M, et al. The natural history of hereditary pancreatitis: a national series. Gut. 2009;58:97–103. doi: 10.1136/gut.2008.149179. [DOI] [PubMed] [Google Scholar]
- 87.Threadgold J, Greenhalf W, Ellis I, et al. The N34S mutation of SPINK1 (PSTI) is associated with a familial pattern of idiopathic chronic pancreatitis but does not cause the disease. Gut. 2002;50:675–81. doi: 10.1136/gut.50.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yadav D, Timmons L, Benson JT, et al. Incidence, prevalence, and survival of chronic pancreatitis: a population-based study. The American journal of gastroenterology. 2011;106:2192–9. doi: 10.1038/ajg.2011.328. [DOI] [PubMed] [Google Scholar]
- 89.Aoun E, Muddana V, Papachristou GI, et al. SPINK1 N34S is strongly associated with recurrent acute pancreatitis but is not a risk factor for the first or sentinel acute pancreatitis event. Am J Gastroenterol. 2010;105:446–51. doi: 10.1038/ajg.2009.630. [DOI] [PubMed] [Google Scholar]
- 90.Masamune A, Ariga H, Kume K, et al. Genetic background is different between sentinel and recurrent acute pancreatitis. Journal of gastroenterology and hepatology. 2011;26:974–8. doi: 10.1111/j.1440-1746.2011.06691.x. [DOI] [PubMed] [Google Scholar]
- 91.Schneider A, Pfützer RH, Barmada MM, et al. Limited contribution of the SPINK1 N34S mutation to the risk and severity of alcoholic chronic pancreatitis – a preliminary report from the United States. Dig Dis Sci. 2003;48:1110–1115. doi: 10.1023/a:1023768829772. [DOI] [PubMed] [Google Scholar]
- 92.Lee KH, Ryu JK, Yoon WJ, et al. Mutation analysis of SPINK1 and CFTR gene in Korean patients with alcoholic chronic pancreatitis. Dig Dis Sci. 2005;50:1852–6. doi: 10.1007/s10620-005-2950-9. [DOI] [PubMed] [Google Scholar]
- 93.Shimosegawa T, Kume K, Masamune A. SPINK1 gene mutations and pancreatitis in Japan. J Gastroenterol Hepatol. 2006;21(Suppl 3):S47–51. doi: 10.1111/j.1440-1746.2006.04594.x. [DOI] [PubMed] [Google Scholar]
- 94.Masamune A, Kume K, Shimosegawa T. Differential roles of the SPINK1 gene mutations in alcoholic and nonalcoholic chronic pancreatitis. J Gastroenterol. 2007;42(Suppl 17):135–40. doi: 10.1007/s00535-006-1921-z. [DOI] [PubMed] [Google Scholar]
- 95.Bertin C, Pelletier AL, Vullierme MP, et al. Pancreas divisum is not a cause of pancreatitis by itself but acts as a partner of genetic mutations. The American journal of gastroenterology. 2012;107:311–7. doi: 10.1038/ajg.2011.424. [DOI] [PubMed] [Google Scholar]
- 96.Paliwal S, Bhaskar S, Mani KR, et al. Comprehensive screening of chymotrypsin C (CTRC) gene in tropical calcific pancreatitis identifies novel variants. Gut. 2012 doi: 10.1136/gutjnl-2012-302448. [DOI] [PubMed] [Google Scholar]
- 97.Szmola R, Sahin-Toth M. Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: identity with Rinderknecht’s enzyme Y. Proc Natl Acad Sci U S A. 2007;104:11227–32. doi: 10.1073/pnas.0703714104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chang MC, Chang YT, Wei SC, et al. Association of novel chymotrypsin C gene variations and haplotypes in patients with chronic pancreatitis in Chinese in Taiwan. Pancreatology. 2009;9:287–92. doi: 10.1159/000199437. [DOI] [PubMed] [Google Scholar]
- 99.Felderbauer P, Hoffmann P, Einwachter H, et al. A novel mutation of the calcium sensing receptor gene is associated with chronic pancreatitis in a family with heterozygous SPINK1 mutations. BMC Gastroenterol. 2003;3:34. doi: 10.1186/1471-230X-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Felderbauer P, Klein W, Bulut K, et al. Mutations in the calcium-sensing receptor: a new genetic risk factor for chronic pancreatitis? Scand J Gastroenterol. 2006;41:343–8. doi: 10.1080/00365520510024214. [DOI] [PubMed] [Google Scholar]
- 101.Muddana V, Lamb J, Greer JB, et al. Association between calcium sensing receptor gene polymorphisms and chronic pancreatitis in a US population: Role of serine protease inhibitor Kazal 1type and alcohol. World J Gastroenterol. 2008;14:4486–91. doi: 10.3748/wjg.14.4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vezzoli G, Terranegra A, Arcidiacono T, et al. R990G polymorphism of calcium-sensing receptor does produce a gain-of-function and predispose to primary hypercalciuria. Kidney Int. 2007;71:1155–62. doi: 10.1038/sj.ki.5002156. [DOI] [PubMed] [Google Scholar]
- 103.Pidasheva S, D’Souza-Li L, Canaff L, et al. CASRdb: calcium-sensing receptor locus-specific database for mutations causing familial (benign) hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat. 2004;24:107–11. doi: 10.1002/humu.20067. [DOI] [PubMed] [Google Scholar]
- 104.Pelli H, Lappalainen-Lehto R, Piironen A, et al. Risk factors for recurrent acute alcohol-associated pancreatitis: a prospective analysis. Scand J Gastroenterol. 2008;43:614–21. doi: 10.1080/00365520701843027. [DOI] [PubMed] [Google Scholar]
- 105.Nordback I, Pelli H, Lappalainen-Lehto R, et al. The recurrence of acute alcohol-associated pancreatitis can be reduced: a randomized controlled trial. Gastroenterology. 2009;136:848–55. doi: 10.1053/j.gastro.2008.11.044. [DOI] [PubMed] [Google Scholar]
- 106.Talamini G, Bassi C, Falconi M, et al. Smoking cessation at the clinical onset of chronic pancreatitis and risk of pancreatic calcifications. Pancreas. 2007;35:320–6. doi: 10.1097/mpa.0b013e31812e965e. [DOI] [PubMed] [Google Scholar]
- 107.Lankisch PG, Breuer N, Bruns A, et al. Natural history of acute pancreatitis: a long-term population-based study. Am J Gastroenterol. 2009;104:2797–805. doi: 10.1038/ajg.2009.405. quiz 2806. [DOI] [PubMed] [Google Scholar]
- 108.Van Laethem J, Robberecht P, Resibois A, et al. Transforming growth factor beta promotes development of fibrosis after repeated courses of acute pancreatitis in mice. Gastroenterology. 1996;110:576–82. doi: 10.1053/gast.1996.v110.pm8566606. [DOI] [PubMed] [Google Scholar]
- 109.Neuschwander-Tetri BA, Burton FR, Presti ME, et al. Repetitive self-limited acute pancreatitis induces pancreatic fibrogenesis in the mouse. Dig Dis Sci. 2000;45:665–74. doi: 10.1023/a:1005423122127. [DOI] [PubMed] [Google Scholar]
- 110.Deng X, Wang L, Elm MS, et al. Chronic alcohol consumption accelerates fibrosis in response to cerulein-induced pancreatitis in rats. Am J Pathol. 2005;166:93–106. doi: 10.1016/S0002-9440(10)62235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Perides G, Tao X, West N, et al. A mouse model of ethanol dependent pancreatic fibrosis. Gut. 2005;54:1461–7. doi: 10.1136/gut.2004.062919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–90. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barrie A, Lamb J, Sandhu B, et al. Susceptibility to alcoholic pancreatitis, but not sporadic pancreatitis, is significantly reduced by the IL23R R381Q gene variant. Am J Gastroenterol. 2012 in press. [Google Scholar]
- 115.Alcohol use and alcohol use disorders in the United States: Main findings from teh 2001-2002 National Epidemiological Survey on Alcohol and Related Conditions (NESARC) U S Alcohol Epidemiologic Data Reference Manual. 2006;8 NIH Publication No. 05-5737. [Google Scholar]
- 116.Van Itallie CM, Holmes J, Bridges A, et al. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. Journal of cell science. 2008;121:298–305. doi: 10.1242/jcs.021485. [DOI] [PubMed] [Google Scholar]
- 117.Amasheh S, Meiri N, Gitter AH, et al. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. Journal of cell science. 2002;115:4969–76. doi: 10.1242/jcs.00165. [DOI] [PubMed] [Google Scholar]
- 118.Merilainen S, Makela J, Anttila V, et al. Acute edematous and necrotic pancreatitis in a porcine model. Scandinavian journal of gastroenterology. 2008;43:1259–68. doi: 10.1080/00365520802158580. [DOI] [PubMed] [Google Scholar]
- 119.Sakaguchi T, Gu X, Golden HM, et al. Cloning of the human claudin-2 5’-flanking region revealed a TATA-less promoter with conserved binding sites in mouse and human for caudal-related homeodomain proteins and hepatocyte nuclear factor-1alpha. The Journal of biological chemistry. 2002;277:21361–70. doi: 10.1074/jbc.M110261200. [DOI] [PubMed] [Google Scholar]
- 120.Mankertz J, Amasheh M, Krug SM, et al. TNFalpha up-regulates claudin-2 expression in epithelial HT-29/B6 cells via phosphatidylinositol-3-kinase signaling. Cell and tissue research. 2009;336:67–77. doi: 10.1007/s00441-009-0751-8. [DOI] [PubMed] [Google Scholar]
- 121.Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. The Journal of biological chemistry. 2011;286:31263–71. doi: 10.1074/jbc.M111.238147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mankertz J, Hillenbrand B, Tavalali S, et al. Functional crosstalk between Wnt signaling and Cdx-related transcriptional activation in the regulation of the claudin-2 promoter activity. Biochemical and biophysical research communications. 2004;314:1001–7. doi: 10.1016/j.bbrc.2003.12.185. [DOI] [PubMed] [Google Scholar]
- 123.Van den Bossche J, Laoui D, Morias Y, et al. Claudin-1, claudin-2 and claudin-11 genes differentially associate with distinct types of anti-inflammatory macrophages in vitro and with parasite- and tumor-elicited macrophages in vivo. Scandinavian journal of immunology. 2012 doi: 10.1111/j.1365-3083.2012.02689.x. [DOI] [PubMed] [Google Scholar]