

Abstract
Objective
To report on the clinical response to canakinumab in a patient with sporadic nucleotide-binding oligomerization domain–containing protein 2 (NOD-2)–associated pediatric granulomatous arthritis (Blau syndrome) and severe resistant panuveitis, and to describe gene expression profile changes throughout such treatment.
Methods
A 4-year-old boy was diagnosed as having Blau syndrome on the basis of typical clinical features, histologic evidence of noncaseating granulomas, and a NOD2 mutation. Ocular involvement was initially controlled by topical and oral corticosteroids, but over the years visual impairment and complications, such as macular edema and retinal detachment, progressed. Ocular disease remained persistently active despite treatment with multiple different immunosuppressants; therefore, canakinumab treatment was started. Before and during the first 6 months of treatment, the gene expression profile was determined each month.
Results
Canakinumab treatment was well tolerated and led to rapid quiescence of uveitis, which had been continuously active before this treatment. Gene expression profiling analysis of the patient's blood prior to initiation of interleukin-1 (IL-1) blockade revealed differential expression of 1,993 transcripts when compared to healthy controls, and among the up-regulated transcripts, pathway analysis showed that the predominant network consisted of innate immunity–related transcripts. The transcriptional signature of the patient overlapped with the transcriptional signature of patients with systemic-onset juvenile idiopathic arthritis, and canakinumab treatment led to the normalization of most of these transcriptional changes.
Conclusion
The pathogenesis of Blau syndrome may be mediated by IL-1, and canakinumab may be useful when this disorder is unresponsive to more conventional treatments.
Sporadic nucleotide-binding oligomerization domain–containing protein 2 (NOD-2)–associated pediatric granulomatous arthritis (Blau syndrome) (MIM ID #186580) is the genetic form of what was previously known as early-onset sarcoidosis, now better defined as pediatric granulomatous arthritis (1,2). It is characterized clinically by the triad of arthritis, skin rash, and uveitis, and histologically by the presence of noncaseating epithelioid granulomas in the affected sites (3). Genetic studies have shown that the disease is due to mutations in the nucleotide-binding domain of the NOD-2 (CARD-15) gene (4).
The course of the disease is variable, but in many cases the prognosis is poor, with severe disabilities and sequelae in a high percentage of patients (5). Eye involvement is frequently progressive and can lead to panuveitis and severe complications up to legal blindness (5,6). The pathogenesis of Blau syndrome is not well known, but the disease is believed to belong to the spectrum of autoinflammatory disorders, for which interleukin-1 (IL-1) inhibition is now an effective treatment option.
Canakinumab is an anti–IL-1β monoclonal antibody that is effective in the treatment of cryopyrin-associated periodic syndromes (7) and currently approved for that indication. Herein we report on the case of a patient with Blau syndrome whom we have followed up since early infancy (8). The patient developed sight-threatening, drug-resistant panuveitis, which responded quite well to canakinumab. During the first 6 months of treatment we also determined his gene expression profile, which showed down-regulation of several IL-1–related transcripts over time.
PATIENT AND METHODS
Case description
The patient, a 4-year-old boy, was diagnosed as having Blau syndrome based on the manifestation of typical clinical features (ankle and wrist arthritis/tenosynovitis, diffuse eczematous rash, and uveitis), histologic evidence of noncaseating granulomas, and a heterozygous NOD2 mutation (p.R334W). Ocular involvement was initially controlled by topical and oral corticosteroids, but over the years visual impairment progressed. Other manifestations of Blau syndrome (arthritis and rash) subsided over time. Bilateral panuveitis progressed after age 5 years, and was initially treated with methotrexate. However, ocular inflammation persisted despite the addition of local steroid injections and repeated intravenous (IV) bolus methylprednisolone treatment; therefore, when the patient was age 10 years, infliximab (initially at 5 mg/kg increased to 10 mg/kg IV every 4 to 6 weeks) was initiated. Although there was an initial improvement, 1 year after this treatment was started uveitis worsened, and at age 12 years infliximab was discontinued. Adalimumab (24 mg/m2 every 2 weeks) was then initiated and the dosage of methotrexate (15 mg/m2/weekly) was increased. However, ocular disease remained active. Mycophenolate mofetil (750 mg/m2) and then abatacept (10 mg/kg/month IV) were sequentially administered, without significant improvement.
At age 16 years the patient still had granulomatous retinal lesions and anterior chamber inflammation, and macular edema developed, which led to retinal detachment. In addition to the other steroid therapy, corticosteroid pulse therapy was necessary to control disease flares, with an average of 3 boluses/month for 6 consecutive months. Because of the supposed autoinflammatory nature of Blau syndrome, we initiated a trial of IL-1 antibody administration (2 mg/kg/month of canakinumab). During the 6 months that followed, no ocular flare occurred and no steroid pulse therapy was necessary. Concomitant treatment with oral methotrexate and low-dose prednisone (0.2 mg/kg/day) remained unchanged. Figure 1 shows fluorangiograms before treatment and after the first 6 injections. The drug was well tolerated with no side effects, and findings on laboratory tests (performed monthly) were normal.
Figure 1.

Serial retinal angiograms showing the evolution of peripapillary granulomas in both eyes. Fluorescein angiography reveals severe leakage from the granulomas before treatment, while after treatment the inflammatory leakage is reduced, although residual fluorescein staining in the granulomas is still evident. A and B, Early-phase retinal angiography (at 43 seconds) (A) and late-phase retinal angiography (at 220 seconds) (B) of the right eye, prior to canakinumab treatment. E and F, Early-phase retinal angiography (at 26 seconds) (E) and late-phase retinal angiography (at 231 seconds) (F) of the left eye, prior to canakinumab treatment. C and D, Early-phase retinal angiography (at 38 seconds) (C) and late-phase retinal angiography (at 251 seconds) (D) of the right eye, after canakinumab treatment. G and H, Early-phase retinal angiography (at 48 seconds) (G) and late-phase retinal angiography (at 267 seconds) (H) of the left eye, after canakinumab treatment.
Methods
Blood was collected into Tempus Vacutainer tubes (Applied Biosystems) during routine venipuncture before the first canakinumab injection and monthly thereafter for 5 months (immediately before the scheduled canakinumab dose). Four demographically matched healthy subjects served as controls. Parents provided informed consent to the study authors and for the off-label use of the medication, and approval of the Meyer Children's Hospital Ethics Committee was also obtained. Total RNA was isolated from the whole blood lysate using a MagMax total RNA extraction kit (Applied Biosystems), and globin messenger RNA was removed with a GLOBINclear Whole Blood Globin Reduction kit (Applied Biosystems). RNA was further amplified and labeled with an Illumina TotalPrep RNA Amplification kit (Applied Biosystems). Complementary RNA was hybridized to a Human HT12 V4 BeadChip array (IIlumina) and scanned on an Illumina BeadStation 500. Fluorescent hybridization signals were assessed with GenomeStudio software (Illumina).
After background subtraction and average normalization, microarray data were analyzed using GeneSpring 11.2 software (Agilent). Expression values of probes were log2-transformed and normalized to the median value in healthy controls. Fold change analysis was performed to compare the patient samples with those of healthy controls. IPA (Ingenuity Systems) was used to perform pathway analysis of the genes of interest. For modular interpretation, a set of 260 transcriptional modules was used as a preexisting framework for the analysis. The approach used for the construction of such a framework has been previously reported (9). Briefly, genes with coordinate expression in whole blood within or across 9 disease data sets were selected in multiple rounds of clique and paraclique clustering to form a 260–transcriptional module framework. Within each module, the percentage of significant probes was assessed by t-test with an assumption of equal variance in an error model.
RESULTS
Gene expression profiling analysis of the patient's blood prior to initiation of IL-1 blockade revealed the differential expression of 1,993 transcripts, in which 496 transcripts were up-regulated and 1,497 transcripts were down-regulated (Figure 2A). Using our previously described modular approach to data interpretation (9), inflammation and interferon-inducible modules were shown to be up-regulated, while adaptive immunity–related modules were highly suppressed. Interestingly, IL-1 blockade led to the normalization of most of these transcriptional changes except for those related to neutrophils, for which up-regulation was maintained throughout the followup period (Figure 2B). Among the up-regulated transcripts, pathway analysis confirmed that the predominant network consisted of innate immunity–related transcripts (Figure 2C). Transcripts encoding inflammasome-related molecules (i.e., AIM2, NLRC4, NAIP, NLRP1, CASP4, and CASP5) were up-regulated together with IL-1, Toll-like receptor 2, and neutrophil-related transcripts.
Figure 2.

Whole blood transcriptional profile of the patient with Blau syndrome compared to controls. A, Expression values of transcripts from 1 time point pretreatment and 1, 2, 3, 4, and 5 months after initiation of canakinumab therapy (V1–5) were log2 transformed and normalized to the median value in 4 healthy controls. Fold change analysis of the sample from the untreated patient with Blau syndrome and 4 healthy controls resulted in 1,993 differentially expressed transcripts. Hierarchical clustering of these probes was generated using the Pearson correlation method. B, Modular analysis of samples from the patient with Blau syndrome was performed. The results revealed up-regulation of neutrophil and inflammation modules and down-regulation of adaptive immune responses. A t-test was performed for each (baseline and followup) time point and for the healthy controls, with an assumption of equal variance in an error model. Each column corresponds to 1 time point. Within each module, numbers of significant genes were identified and their percentage represented as up-regulated or down-regulated. The transcript composition for each module has been described elsewhere (9). DC = dendritic cell; NK = natural killer. In A and B, red signifies up-regulation and blue signifies down-regulation. C, In the sample from the untreated patient with Blau syndrome, 496 up-regulated transcripts were analyzed by IPA. An inflammatory response network centered around Toll-like receptor 2, inflammasome components, interleukin-1/interleukin-18 signaling, and neutrophil-encoded transcripts was predominant.
To identify changes that could be specific to NOD pathway dysregulation, we compared the transcriptional profile of the patient's blood at baseline (prior to therapy) with the profiles of blood samples from 21 patients with systemic-onset juvenile idiopathic arthritis (JIA), a disease known to respond to IL-1 and/or IL-6 blockade (10,11). The transcriptional signature of the patient mostly overlapped with that of the patients with systemic-onset JIA (Figure 3A). Among transcripts that were up-regulated in the patient with Blau syndrome but not in patients with systemic-onset JIA, we identified a network centered around acylcoenzyme A oxidase 1, leptin receptor, and retinoic acid receptor (RXRG), that normalized upon initiation of IL-1 blockade (Figure 3B).
Figure 3.
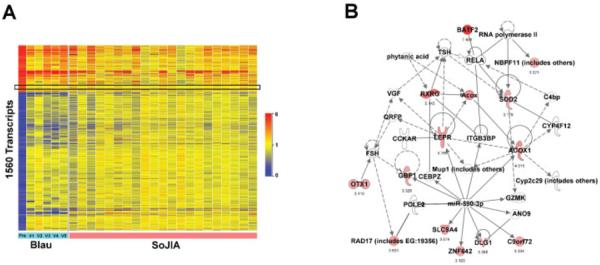
Whole blood transcriptional profile of the patient with untreated Blau syndrome overlaps with that of systemic-onset juvenile idiopathic arthritis (SoJIA) patients. A, Expression values of transcripts from the patient with Blau syndrome at baseline (as in Figure 1A) were compared with those from patients with systemic-onset JIA (normalized to values in 18 matched healthy controls). In this analysis, 1,560 transcripts were included as they were common to the 2 Illumina bead array chips used in original analyses of Blau syndrome and systemic-onset JIA. As shown in the boxed area, only 46 transcripts were up-regulated in Blau syndrome but not in systemic-onset JIA. B, The 46 genes were analyzed by IPA. A leptin receptor–centered network was revealed. See Figure 2 for other definitions.
DISCUSSION
Blau syndrome is usually a progressive disorder, which, if untreated, is often associated with severe sequelae. In particular, ocular involvement can be devastating and resistant to multiple treatments. Our patient had severe panuveitis, which did not respond to treatment with corticosteroids or multiple different immunosuppressants, including infliximab, adalimumab, mycophenolate mofetil, and abatacept, and which led to retinal detachment at age 16 years. His disease required repeated corticosteroid pulse therapy in order to control frequent flares of posterior uveitis. Soon after the initiation of canakinumab treatment, inflammatory eye involvement subsided and steroid pulse treatment, which had been almost continuous for the previous 6 months, was no longer needed.
The rationale for treating with canakinumab was based on the presumed autoinflammatory nature of Blau syndrome, as well as on a previous report that noted clinical improvement in a patient with Blau syndrome who was treated with anakinra (12). In that study, Arostegui et al also analyzed cytokine patterns, and showed increased levels of IL-6 and tumor necrosis factor (TNF) in plasma, and a slight increase in the IL-1β level in the more severely affected patient, when compared with controls. The patient was then treated with anakinra, and analyses of cytokine levels in plasma showed a marked decrease in IL-6, TNF, and IL-1β levels during treatment, which occurred simultaneously with the patient's clinical improvement. This therefore suggests that IL-1β dysregulation might occur upstream of IL-6 and TNF dysregulation in some patients with Blau syndrome.
Moreover, there is some rationale for the use of IL-1 in NOD-2–related disorders, since mutations in a related gene (NLRP3) are responsible for excess IL-1β in cryopyrinopathies, and functional studies of the inflammasome have shown that caspase 1–mediated release of IL-1β also involves NOD-2 (13). However, subsequent studies failed to demonstrate increased IL-1β production by peripheral blood mononuclear cells in patients with Blau syndrome, and anakinra was ineffective in 2 such patients (14,15). In both of these studies, IL-1β in supernatants was measured by enzyme-linked immunosorbent assay (ELISA), a less sensitive method than whole blood gene expression profiling. The difference between these 2 methods of cytokine detection has been evident in systemic-onset JIA as well, where studies have failed to detect elevated IL-1β levels by ELISA, while the role of this cytokine has been proven by gene expression studies and clinical trials to be important in the disease pathogenesis (10,16).
Our patient showed high expression levels of innate immunity–related genes at baseline, and substantial overlap with the profile of patients with systemic-onset JIA, consistent with our hypothesis of an autoinflammatory, IL-1–responding disorder. Moreover, almost all of these up-regulated transcripts normalized after the first canakinumab injection. This normalization remained stable during the months that followed, concomitant with the clinical improvement. The only module that remained up-regulated through treatment was the neutrophil-related module; we are not able to fully explain this phenomenon but speculate that NOD2 mutation induces additional cytokine dysregulation, other than dysregulation of IL-1β, that might be responsible for this.
Gene expression in Blau syndrome has been rarely studied, and to our knowledge there is only one such earlier report (17). This is the first study to demonstrate the differential gene expression profile in Blau syndrome at baseline and upon effective treatment with canakinumab. To date, the patient has had 1 full year of treatment, without further relapses of uveitis. Given the lack of sustained response to all other treatments received, and the temporal association of remission soon after initiation of canakinumab, together with evidence of the rapid normalization of innate immunity–related transcripts, we are confident that the disease activity in this patient was due to excessive IL-1β production and that the use of IL-1 blockade is warranted in this disease.
Acknowledgments
Dr. Pascual's work was supported by NIH grant AR-050770-02.
Footnotes
AUTHOR CONTRIBUTIONS All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Cimaz had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Simonini, De Libero, Pascual, Cimaz.
Acquisition of data. Simonini, Xu, Caputo, Pagnini, Cimaz.
Analysis and interpretation of data. Xu, Pagnini, Pascual, Cimaz.
REFERENCES
- 1.Rose CD, Martin TM, Wouters CH. Blau syndrome revisited. Curr Opin Rheumatol. 2011;23:411–8. doi: 10.1097/BOR.0b013e328349c430. [DOI] [PubMed] [Google Scholar]
- 2.Rose CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis Rheum. 2006;54:3337–44. doi: 10.1002/art.22122. [DOI] [PubMed] [Google Scholar]
- 3.Janssen CE, Rose CD, De Hertogh G, Martin TM, Bader Meunier B, Cimaz R, et al. Morphologic and immunohistochemical characterization of granulomas in the nucleotide oligomerization domain 2-related disorders Blau syndrome and Crohn disease. J Allergy Clin Immunol. 2012;129:1076–84. doi: 10.1016/j.jaci.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- 5.Fink CW, Cimaz R. Early onset sarcoidosis: not a benign disease. J Rheumatol. 1997;24:174–7. [PubMed] [Google Scholar]
- 6.Raiji VR, Miller MM, Jung LK. Uveitis in Blau syndrome from a de novo mutation of the NOD2/CARD15 gene. J AAPOS. 2011;15:205–7. doi: 10.1016/j.jaapos.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360:2416–25. doi: 10.1056/NEJMoa0810787. [DOI] [PubMed] [Google Scholar]
- 8.Falcini F, Battini ML, Ceruso M, Cimaz R. A 4-year-old with a rash. Lancet. 1999;354:40. doi: 10.1016/S0140-6736(99)03274-2. [DOI] [PubMed] [Google Scholar]
- 9.Chaussabel D, Quinn C, Shen J, Patel P, Glaser C, Baldwin N, et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity. 2008;29:150–64. doi: 10.1016/j.immuni.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial) Ann Rheum Dis. 2011;70:747–54. doi: 10.1136/ard.2010.134254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, Takei S, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371:998–1006. doi: 10.1016/S0140-6736(08)60454-7. [DOI] [PubMed] [Google Scholar]
- 12.Arostegui JI, Arnal C, Merino R, Modesto C, Antonia Carballo M, Moreno P, et al. NOD2 gene–associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56:3805–13. doi: 10.1002/art.22966. [DOI] [PubMed] [Google Scholar]
- 13.Pan Q, Mathison J, Fearns C, Kravchenko VV, Da Silva Correia J, Hoffman HM, et al. MDP-induced interleukin-1β processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol. 2007;82:177–83. doi: 10.1189/jlb.1006627. [DOI] [PubMed] [Google Scholar]
- 14.Martin TM, Zhang Z, Kurz P, Rose CD, Chen H, Lu H, et al. The NOD2 defect in Blau syndrome does not result in excess interleukin-1 activity. Arthritis Rheum. 2009;60:611–8. doi: 10.1002/art.24222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masumoto J, Yamazaki T, Ohta K, Nakayama J, Agematsu K. Interleukin-1β suppression in Blau syndrome: comment on the article by Martin et al [letter] Arthritis Rheum. 2009;60:2544–5. doi: 10.1002/art.24691. [DOI] [PubMed] [Google Scholar]
- 16.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–86. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foley KP, Wang L, Cooper D, Magid-Slav M, Lipshutz D, Connor J, et al. Identification of Blau syndrome disease signatures. Pediatr Rheumatol. 2011;9(Suppl 1):O25. [Google Scholar]