

Abstract
In response to ligand binding to the Toll-like receptor 4 (TLR4) and myeloid differentiation-2 (MD-2) receptor complex, two major signaling pathways are activated that involve different adaptor proteins. One pathway depends on myeloid differentiation marker 88 (MyD88), which elicits proinflammatory responses, whereas the other depends on Toll–IL-1 receptor (TIR) domain–containing adaptor inducing interferon-β (TRIF), which elicits type I interferon production. Here, we showed that the TLR4 agonist and vaccine adjuvant CRX-547, a member of the aminoalkyl glucosaminide 4-phosphate (AGP) class of synthetic lipid A mimetics, displayed TRIF-selective signaling in human cells, which was dependent on a minor structural modification to the carboxyl bioisostere corresponding to the 1-phosphate group on most lipid A types. CRX-547 stimulated little or no activation of MyD88-dependent signaling molecules or cytokines, whereas its ability to activate the TRIF-dependent pathway was similar to that of a structurally related inflammatory AGP and of lipopolysaccharide from Salmonella minnesota. This TRIF-selective signaling response resulted in the production of substantially less of the proinflammatory mediators that are associated with MyD88 signaling, thereby potentially reducing toxicity and improving the therapeutic index of this synthetic TLR4 agonist and vaccine adjuvant.
INTRODUCTION
Toll-like receptors (TLRs) are pattern recognition receptors that recognize conserved microbial motifs, including peptidoglycan (which is recognized by TLR2), CpG DNA (TLR9), viral RNA (TLR3, TLR7, and TLR8), bacterial flagellin (TLR5), and lipopolysaccharide (LPS, TLR4). TLRs are characterized by an extracellular ligand-binding leucine-rich repeat domain and a cytoplasmic Toll–interleukin-1 (IL-1) receptor (TIR) homology domain that recruits intracellular signaling adaptor proteins. Several ligands for TLR4 are known, including LPS, lipoteichoic acid, fibronectin, the fusion protein of respiratory syncytial virus (RSV), and taxol. LPS and its active component, lipid A, are well-known TLR4-dependent immunostimulatory molecules, but they also stimulate the production of inflammatory mediators of toxicity, which limits their therapeutic use in humans (1–6). Detoxification of the LPS from Salmonella minnesota by chemical modification produces a TLR4-active lipid A derivative known as mono-phosphoryl lipid A (MPL) adjuvant, which displays greatly reduced toxicity while maintaining most of the beneficial immunostimulatory activity of LPS. Note that MPL adjuvant refers to the clinical-grade (GMP) MPL adjuvant that is produced by GlaxoSmithKline Biologicals. MPL from Salmonella minnesota Re595 (MLA) or synthetic Escherichia coli MPL (sMLA), used later in this study, is available from InvivoGen. The safety and efficacy of MPL adjuvant are exemplified by the recent approval of Cervarix, a human papillomavirus vaccine that contains MPL adsorbed to alum, for the prevention of cervical cancer (7).
Lipid A is the hydrophobic moiety of LPS that binds to the membrane-bound TLR4–myeloid differentiation-2 (MD-2) receptor complex (8). The structures of lipid A molecules from various Gram-negative bacteria show considerable variability, and some species can even modulate the structure of the lipid A on their surface. Structural differences in lipid A can substantially affect the immune response of the host, which results in varying degrees of inflammation (9, 10). Previously, we showed that changes in the number and length of acyl chains as well as the character of the functional groups at the bioisosteric position of the 1-phosphate of lipid A substantially altered the activities of lipid A mimetics from those of highly potent agonists to those of antagonists (11). Thus, structural differences can affect both the potency and the character of the immunological response to lipid A (7, 11–13).
Agonist binding to TLR4–MD-2 receptor complexes and their subsequent dimerization on the cell surface lead to the activation of two major downstream signaling pathways, one of which depends on the adaptor protein myeloid differentiation marker 88 (MyD88), whereas the other depends on the adaptor protein Toll–interleukin-1 (IL-1) receptor (TIR) domain–containing adaptor inducing interferon-β (TRIF) (11, 14–16). MyD88-dependent signaling results from sequential or simultaneous binding of the two TIR domain–containing adaptor proteins, Mal [also known as TIR domain–containing adaptor protein (TIRAP)] and MyD88, to the cytoplasmic TIR domain of TLR4. Activation of the signaling cascade downstream of MyD88 results in the early activation and nuclear translocation of the transcription factor nuclear factor κB (NF-κB), which induces the expression of genes encoding proinflammatory cytokines and chemokines, such as tumor necrosis factor–α (TNF-α), interleukin-1β (IL-1β), and macrophage inflammatory protein 1α (MIP-1α). TRIF-dependent signaling requires the sequential or simultaneous binding of the TIR domain–containing adaptor proteins TRIF-related adaptor molecule [TRAM, also known as (TICAM-2)] and TRIF to the TIR domain of TLR4 (17, 18). Signaling through the TRIF-dependent pathway results in decreased and delayed activation of NF-κB compared to that by MyD88-dependent signaling and involves an alternative pathway using receptor-interacting protein 1 (RIP1) (19–21). TRIF-dependent signaling also causes the activation and nuclear translocation of the transcription factors interferon regulatory factor 3 (IRF3) and IRF7 (21, 22), which induce expression of the gene encoding interferon-β (IFN-β) and its subsequent release from the cell. Autocrine or paracrine binding of IFN-β to the IFN-α/β receptor (IFNAR), in turn, activates the Janus-activated kinase (JAK)–signal transducer and activator of transcription (STAT) pathway, leading to increased production of IFN-α and IFN-β, as well as of IFN-inducible chemokines, such as interferon-inducible protein-10 (IP-10, also known as CXCL10), RANTES (regulated on activation, normal T cell–expressed and –secreted, also known as CCL5), and macrophage chemotactic protein-1 (MCP-1, also known as CCL2) (13, 18, 22, 23). A study showed that treatment of murine cells with MLA results in reduced MyD88-dependent signaling activity, but similar TRIF-dependent signaling activity as occurs in response to LPS (24, 25). This TRIF-biased response could be responsible for the increased therapeutic index, reduced toxicity, and sustained adjuvant activity observed with MPL adjuvant (26).
Previously, we described the design and synthesis of a library of monosaccharide lipid A mimetics, the aminoalkyl glucosaminide 4-phosphates (AGPs), which had TLR4 agonist and antagonist activities (11, 27, 28). Here, we compared the stimulation of the MyD88- and TRIF-dependent signaling pathways by two diastereomeric AGPs, CRX-527 and CRX-547, which differ only with respect to the configuration of the seryl stereocenter. CRX-527 and CRX-547 (Fig. 1A) each contains three (R)-3decanoyloxytetradecanoyl residues that are N- or O-linked to an O-glucosaminyl serine backbone, wherein the seryl unit is a conformationally flexible substitute for the reducing sugar of lipid A, and the seryl carboxyl group is bioisosteric with the anomeric phosphate of lipid A (Fig. 1A). CRX-547, which contains a D-seryl aglycon unit, is referred herein as the “D-isomer” of CRX-527, whereas CRX-527 contains L-serine. We demonstrated that this small stereochemical change in the structure of a lipid A mimetic substantially reduced the extent of MyD88-dependent downstream signaling, which resulted in the decreased release of proinflammatory cytokines by human monocytic cells, but only minimally affected TRIF-dependent signaling through the TLR4–MD-2 receptor complex. These results suggest that the nature of the interaction of charged groups on the hydrophilic portion of lipid A with the TLR4–MD-2 receptor complex can substantially influence the character of downstream signaling and the subsequent immune response, which has implications for the design of rational vaccine adjuvants.
Fig. 1.

CRX-547 stimulates production of similar amounts of TRIF-dependent cytokines and chemokines by human primary monocytes and monocyte-derived DCs to those stimulated by CRX-527, but reduced amounts of MyD88-dependent cytokines. (A) Structures of the lipid A mimetics CRX-527 and CRX-547. The molecules differ only in the configuration of the aglycon stereocenter (indicated by an arrow): CRX-527 has the L configuration, whereas CRX-547 has the D configuration. (B and C) The abilities of CRX-527, CRX-547, and S. minnesota Re595 LPS to stimulate the production of MyD88-dependent (TNF-α) and TRIF-dependent (RANTES and IP-10) cytokines and chemokines were compared in (B) human PBMC–derived monocytes and (C) monocyte-derived DCs. When data from three independent experiments with monocytes and monocyte-derived DCs were analyzed, the difference in induction of TNF-α between CRX-527 and CRX-547 was significantly higher than the difference in the induction of RANTES and IP-10 [P < 0.01; analysis of variance (ANOVA) with Tukey post hoc testing]. Graphs are from one donor and are representative of three independent donors with three replicate samples from each.
RESULTS
The AGP D-isomer CRX-547 stimulates the production of lower amounts of MyD88-dependent cytokines from human monocytes and dendritic cells than does the L-isomer CRX-527
To compare the activities of the AGP diastereomers, we measured the amounts of cytokines and chemokines produced by IFN-γ–stimulated human primary peripheral blood mononuclear cell (PBMC)–derived monocytes and dendritic cells (DCs) that were treated with a range of concentrations of CRX-547, CRX-527, and LPS. Prestimulation of these cells with IFN-γ ensured that they exhibited more robust cytokine and chemokine responses to the various treatments. CRX-527 stimulated the production of similar amounts of TNF-α from monocytes and DCs (Fig. 1, B and C). In contrast, CRX-547 stimulated the production of lower amounts of TNF-α; however, it also stimulated the production of similar or slightly reduced amounts of the chemokines IP-10 (from DCs) and RANTES (from monocytes) to those stimulated by CRX-527 (Fig. 1, B and C). The production of TNF-α by human monocytic cells is primarily dependent on signaling through MyD88, whereas the production of IP-10 and RANTES primarily depends on TRIF-dependent signaling (22, 29). When we compared data from experiments with monocytes and DCs from three human donors, we found that the difference between CRX-527 and CRX-547 in their ability to stimulate TNF-α was significantly higher (P < 0.01) than the differences in their abilities to stimulate production of RANTES and IP-10. Data from these experiments suggested that CRX-547 selectively stimulated the production of TRIF-dependent, rather than MyD88-dependent, cytokines and chemokines by these cell types.
A TRIF-biased signaling pattern for MLA was shown in experiments with murine cells (24, 25). Embry et al. (30) suggested that a defect in IL-1β maturation after stimulation of TLR4 by sMLA, linked to the TRIF-biased signaling pattern of MLA, is in part responsible for the reduced toxicity of MLA compared to that of LPS. A preliminary evaluation of mature IL-1β production by human PBMCs demonstrated that CRX-547 induced substantially less IL-1β than did CRX-527 (fig. S2), a finding consistent with the previously published work with sMLA (30). When we compared the signaling of CRX-547 and sMLA in human PBMC–derived monocytes, we found that the agonists stimulated similar patterns and extents of TRIF-dependent RANTES production (fig. S1). However, although CRX-547 and sMLA stimulated the production of similar amounts of TNF-α and MIP-1α, which are MyD88-dependent, at low concentrations (0.00016 to 0.1 μM), sMLA stimulated the production of greater amounts of MyD88-dependent cytokines than did CRX-547 when both agonists were used at concentrations greater than 0.1 μM. We found an intermediate phenotype when we measured the production of the cytokine IL-12p70, which depends on both MyD88 and TRIF. Treatment with CRX-547 resulted in the production of slightly less IL-12p70 through the concentration range, whereas sMLA resulted in the production of lower amounts of IL-12p70 at concentrations below 1 μM. These data suggested that sMLA was TRIF-selective at lower, but not higher, concentrations, whereas CRX-547 was TRIF-selective at all of the concentrations used.
Stimulation of RANTES production by CRX-547 depends on endocytosis and TRIF
Signaling through MyD88 and TRIF leads to the activation of NF-κB and subsequent production of cytokines. Therefore, comparisons of the cytokines and chemokines produced in response to TLR4 agonists may be difficult to interpret because of the potential contributions of both MyD88-dependent and TRIF-dependent signaling pathways to the final outcome. To more closely define the contributions of the TRIF and MyD88 pathways to the production of TNF-α and RANTES, we transiently transfected THP-1 cells, a human monocytic cell line, with plasmids expressing dominant-negative forms of human MyD88 (hMyD88) and hTRIF and assessed the ability of the cells to produce cytokines after stimulation with CRX-527 or CRX-547. The dominant-negative MyD88 construct (MyD88-DN) lacks the death domain that is responsible for the binding of MyD88 to downstream signaling kinases, such as IL-1 receptor (IL-1R)–associated kinase (IRAK1) and IRAK4, but it retains the TIR domain that enables it to bind to and block the cytoplasmic TIR domain of TLR4. The dominant-negative mutant of TRIF (TRIF-DN) harbors only the TIR domain and it blocks TRIF-dependent signaling through TLR4 in a similar manner.
TNF-α production by cells containing MyD88-DN in response to CRX-527 was significantly reduced compared to that in control cells (P < 0.01), whereas the ability of CRX-547 to stimulate TNF-α production (albeit in small quantities) was unaffected (Fig. 2A). TNF-α production by cells containing TRIF-DN in response to either CRX-527 or CRX-547 was unaffected. Similarly, RANTES production by cells containing MyD88-DN was only slightly, but not significantly, reduced after treatment with CRX-527 and was unchanged in response to CRX-547 (Fig. 2B). In cells containing TRIF-DN, RANTES production by either CRX-547 or CRX-527 was significantly and similarly reduced compared to that in untransfected or plasmid control transfected cells (P < 0.05) (Fig. 2B). Whereas TNF-α production in response to CRX-527 was substantially reduced in cells containing MyD88-DN compared to that in untransfected and plasmid control transfected cells, the amounts of TNF-α and RANTES produced in response to CRX-547 were unaffected, which suggested that CRX-547 might exclusively signal through the TRIF-dependent pathway.
Fig. 2.
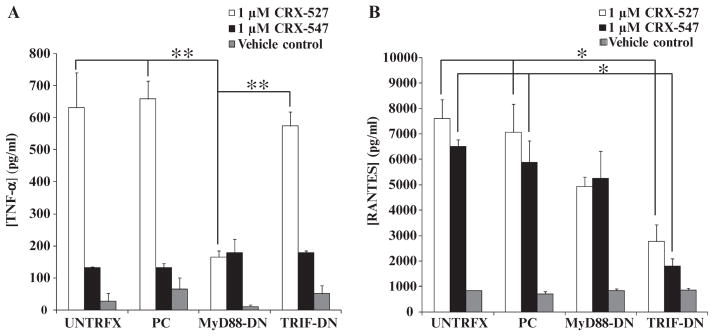
Dominant-negative adaptor protein mutants inhibit MyD88- and TRIF-dependent cytokine and chemokine production by human macrophages. THP-1 cells were differentiated into macrophages in culture and then were mock-transfected (UNTRFX) or were transfected with an empty plasmid control (PC), a plasmid expressing dominant-negative MyD88 (MyD88-DN), or a plasmid encoding dominant-negative TRIF (TRIF-DN). (A and B) The transfected cells were then treated with vehicle, CRX-527 (1 μM), or CRX-547 (1 μM) for 14 hours and then supernatants were analyzed by Luminex for the presence of (A) TNF-α and (B) RANTES. Data are means ± SEM from three independent experiments with three replicates each and were analyzed by ANOVA with Tukey post hoc testing (*P < 0.05; **P < 0.01).
Kagan et al. demonstrated that inhibiting endocytosis in macrophages with the small-molecule endocytosis (dynamin) inhibitor Dynasore specifically inhibits TRIF-dependent TLR4 signaling, suggesting that TRIF signaling originates specifically from early endosomal membranes (31). Thus, inhibition of endocytosis with Dynasore provided us with a method to evaluate the dependence of cytokine and chemokine production by CRX-547 on TRIF signaling. We found that Dynasore significantly inhibited RANTES production by THP-1 cells in response to S. minnesota LPS, CRX-527, or CRX-547 at concentrations above 0.01 μM, but did not significantly inhibit TNF-α production in response to CRX-527 or CRX-547 (Fig. 3, A and B). LPS-stimulated production of TNF-α appeared to be slightly inhibited by Dynasore, but only at the highest concentration of LPS tested, which suggested that the stimulation of TNF-α production by LPS might involve the TRIF pathway to a greater extent than that by CRX-527 or CRX-547; however, this inhibition did not reach statistical significance over three replicate experiments. These results confirmed the published findings that suggest that RANTES production by TLR4 agonists is at least partially dependent on endocytosis (TRIF-dependent signaling), whereas production of the MyD88-dependent cytokine TNF-α is not as dependent on endocytosis. These results also demonstrated that, similar to LPS and CRX-527, TRIF-selective signaling by CRX-547 was also at least partially dependent on endocytosis.
Fig. 3.
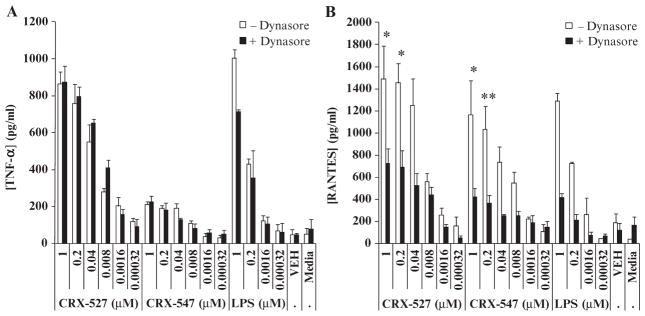
Stimulation of RANTES production by CRX-547 depends on endocytosis. THP-1 cells were differentiated into macrophages in culture, pretreated with the endocytosis (dynamin) inhibitor Dynasore (80 μM) for 60 min in serum-free medium with rhLBP (20 ng/ml), and then treated with the indicated range of concentrations of CRX-527, CRX-547, or vehicle for 14 hours. (A and B) Supernatants were collected and analyzed by Luminex for the presence of (A) TNF-α and (B) RANTES. Data are means ± SEM from three replicates from three independent experiments and were analyzed by ANOVA with Tukey post hoc testing (*P < 0.05; **P < 0.01).
To prevent the inhibition of Dynasore activity by serum components, we performed reactions under serum-free conditions. We added soluble human lipid-binding protein (LBP) to aid in the proper extraction and presentation of agonists to the TLR4–MD-2 receptor complex under serum-free conditions (32). In the absence of serum, the extent of production of RANTES in these cultures was reduced relative to that of cultures in the presence of serum (Figs. 2 and 3), whereas TNF-α production was not affected. The absence of membrane-bound CD14 (mCD14) or soluble CD14 (sCD14) was probably not responsible for this reduction in the amount of RANTES produced, because phorbol 12-myristate 13-acetate (PMA) stimulates an increase in the abundance of mCD14 on THP-1 cells (33). The mechanism involved is currently unknown, but this effect suggests that a serum protein other than sCD14 is required for the full activation of the endocytic TRIF pathway.
Stimulation of the nuclear translocation and transcriptional activity of NF-κB by CRX-547 is diminished compared to that by CRX-527
Signaling through the MyD88- and TRIF-dependent pathways results in activation of the canonical NF-κB (p50/p65) pathway. This pathway culminates in the phosphorylation and degradation of inhibitor of NF-κB (IκBα) and the release of active NF-κB, which then translocates to the nucleus and mediates transcription of genes encoding inflammatory mediators (34, 35). Activation of NF-κB after TLR4 stimulation is temporally controlled; stimulation of the MyD88 pathway results in a rapid increase in NF-κB activity, whereas TRIF-induced NF-κB activity is delayed by 15 to 30 min (21, 22, 36) and does not reach the same extent of activity during long-term exposure to LPS (37, 38). Therefore, the MyD88- and TRIF-dependent pathways can be distinguished by the kinetics and ultimate extent of NF-κB activation after the stimulation of TLR4.
We performed ImageStream analysis of the nuclear translocation of NF-κB in CRX-527– or CRX-547–treated MonoMac6 cells to determine whether CRX-547 differed from CRX-527 in its ability to alter the kinetics and extent of NF-κB activation. ImageStream is a high-throughput imaging flow cytometry system that uses multidimensional pixel analysis of individual cells passed through a flow cell, which enables a statistically robust method for measuring relative amounts of cytoplasmic and nuclear components (39). During the analysis, we analyzed only live focused cells for the nuclear translocation of NF-κB to eliminate the incidence of false positives (Fig. 4A), and we then performed similarity analysis with 3000 to 5000 total live focused cells for each condition at each time point. We defined nuclear translocation as the colocalization of NF-κB with the fluorescent DNA dye DRAQ5 (Fig. 4A). Both AGPs stimulated the nuclear translocation of NF-κB by 15 min after treatment in a dose-dependent manner; however, the magnitudes of the responses differed substantially. CRX-527 stimulated a rapid (within 5 min) increase in the extent of NF-κB nuclear translocation, which peaked by 30 min after treatment and was sustained at a high extent for up to 2 hours. The nuclear translocation of NF-κB stimulated by CRX-547 also peaked by 30 min after treatment, but the extent was lower and more diminished by 2 hours after treatment. The amount of NF-κB that translocated to the nucleus in response to CRX-527 was greater than that stimulated by CRX-547 at all of the time points tested (Fig. 4B).
Fig. 4.

CRX-547 induces less nuclear translocation of NF-κB than CRX-527 in MonoMac6 cells. (A) Gating strategy used for the analysis of NF-κB nuclear translocation (top panels). Live cells were distinguished from dead cells by the presence of a single, well-formed DRAQ5-containing nucleus (Live Cells) after which we gated on live cells (R1) and NF-κB–containing cells (Focused R1) (bottom panels). Nuclear translocation was defined by the colocalization of NF-κB (green) with the nuclear stain DRAQ5 (red), which is indicated by a bright yellow nucleus in the overlaid image of the R1- and NF-κB–containing (Focused R1) cells. (B) Analysis of the nuclear translocation of NF-κB in MonoMac6 cells after incubation with increasing concentrations of CRX-527, CRX-547, vehicle (VEH), or medium for 5, 15, 30, or 120 min. Data are representative of three separate experiments with 3000 to 5000 live focused cells recorded in each experiment.
To measure the sustained induction of total NF-κB activity after stimulation of cells with AGPs, we compared the effects of CRX-527 and CRX-547 on human embryonic kidney (HEK) 293 cells transfected with plasmids encoding human TLR4 (hTLR4), MD-2 (hMD-2), CD14 (hCD14), and an NF-κB reporter plasmid. As was observed in the nuclear translocation assay, CRX-527 induced significantly (P < 0.05) greater activation of the NF-κB promoter than did CRX-547 24 hours after stimulation (Fig. 5). Because the early and sustained stimulation of NF-κB activity depends on activation of the MyD88- and TRIF-dependent signaling pathways (37), these results are consistent with the interpretation that CRX-547 may induce the activation of NF-κB primarily through the TRIF-dependent pathway.
Fig. 5.

CRX-547 stimulates less NF-κB–inducible promoter activity in HEK 293–hTLR4/hMD-2/hCD14 cells than does CRX-527 or S. minnesota Re595 LPS. Stably transfected HEK 293 cells were transiently transfected with a plasmid encoding an NF-κB–inducible reporter gene (encoding SEAP) and were treated with the indicated concentrations of CRX-527, CRX-547, or LPS for 24 hours. Supernatants were assayed for normalized SEAP activity. Data are means ± SEM for three replicates from three independent experiments and were analyzed by ANOVA with Tukey post hoc analysis (*P < 0.05).
CRX-547 activates TRIF-dependent, but not MyD88-dependent, signaling molecules in human monocytes
The recruitment, activation, ubiquitination, and subsequent degradation of the serine and threonine kinase IL-1R–associated kinase–1 (IRAK1) is specific to the MyD88-dependent signaling pathway (40, 41). Alternatively, TRIF-mediated TLR4 signaling specifically results in the activation of the transcription factor IRF3, which is required for the expression of genes encoding the type I IFNs and of interferon-inducible genes such as those encoding RANTES and IP-10 (18, 42). Therefore, if the MyD88 pathway was involved in signaling downstream of TLR4 stimulation, we would expect to observe the polyubiquitination and/or degradation of IRAK1. Alternatively, if TRIF-dependent signaling was involved, we would observe an increase in the amount of phosphorylated IRF3 (pIRF3). We treated human primary monocytes with either CRX-547 or CRX-527 and then prepared cell lysates that we analyzed by Western blotting for the presence of IRAK1 and pIRF3 (Fig. 6). Treatment of the monocytes with CRX-527 resulted in a rapid decrease in the amount of IRAK1 protein detected within 15 min. In contrast, stimulation with CRX-547 resulted in reduced disappearance of IRAK1 relative to that induced by CRX-527 (P < 0.05) up to 60 min after treatment, which suggested that there was little or no activation of the MyD88 signaling pathway by the D-isomer CRX-547. An increase in the abundance of the TRIF-dependent signaling protein pIRF3 was detectable for 30 to 60 min after treatment of the cells with either CRX-527 or CRX-547, which decreased by 120 min after activation. We detected slightly lower amounts of pIRF3 in cells stimulated with CRX-547 compared to those in cells stimulated with CRX-527, consistent with the TRIF-dependent cytokine profiles of both compounds in primary human monocytes (Fig. 1B). Together, these data indicated that both CRX-527 and CRX-547 stimulated TRIF-dependent signaling, whereas CRX-547 stimulated little or no MyD88-dependent signaling.
Fig. 6.

CRX-547, but not CRX-527, selectively activates TRIF-dependent rather than MyD88-dependent signaling proteins. (A and B) Lysates from human primary PBMC-derived, IFN-γ–prestimulated monocytes that were treated with CRX-547, CRX-527, or vehicle for 4 hours were analyzed by Western blotting for the presence of pIRF3 and total IRF3 (TRIF pathway) as well as for IRAK1 (MyD88 pathway). (A) Samples from CRX-547–treated cells showed similar kinetics and extent of activation of IRF3, as assessed by measurement of pIRF3 amounts, but less disappearance of IRAK1 than did samples from CRX-527–stimulated cells. Densitometric analysis (ImageJ) was used to calculate (B) the fold increase in the amount of pIRF3 as a percentage of total IRF3 (pIRF + IRF) and (C) the fold decrease in IRAK1 after treatment with CRX-527 and CRX-547. Data are the average ± SEM from three independent donors and were analyzed by one-way ANOVA with Tukey post hoc analysis (*P < 0.05).
CRX-547 inhibits MyD88-dependent cytokine induction by CRX-527
Differences in the abilities of CRX-527 and CRX-547 to stimulate the production of TNF-α and RANTES suggested that CRX-547 might be a partial agonist or an antagonist of TLR4. To test this, we added increasing concentrations of CRX-547 to human PBMC–derived adherent monocytes treated with a range of concentrations of CRX-527 and compared the characteristics of the inhibition of MyD88-dependent TNF-α production with the inhibition of TRIF-dependent RANTES production (Fig. 7, A to C). The resulting dose-response curves were fit with a four-parameter logistic equation. As expected for a partial agonist, increasing concentrations of CRX-547 shifted the TNF-α response curves to the right (potency shift), whereas the basal level (bottom asymptote) reflected the intrinsic response of CRX-547 alone (with or without CRX-527) (Fig. 7A). Schild regression analysis of the results of the inhibition studies enabled us to make a direct comparison of the calculated affinity (Kb,app) with the median effective concentration (EC50) for CRX-547 as calculated from the dose-response curves for CRX-547 and CRX-527 (Fig. 7C). The values confirmed that the affinity of CRX-547 approached the EC50 values of both CRX-547 and CRX-527 and suggested that CRX-547 acted as a competitive inhibitor of CRX-527 for signaling through the TLR4-MyD88 pathway.
Fig. 7.
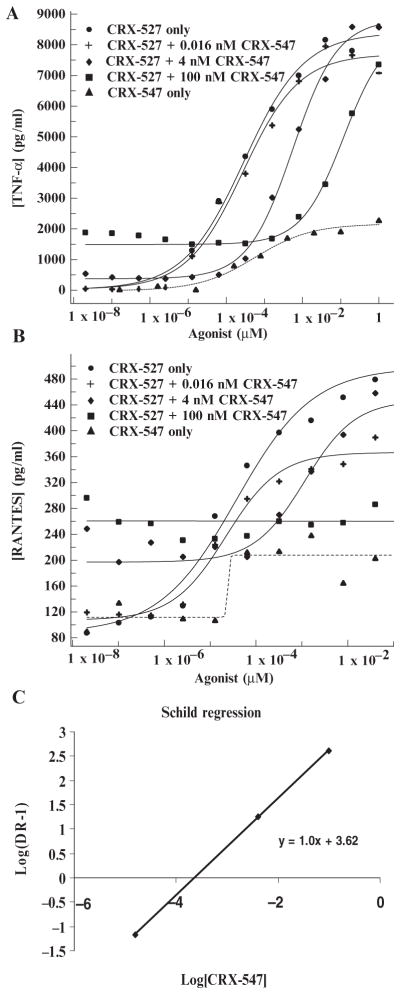
CRX-547 is a competitive inhibitor of CRX-527 for signaling through the TLR4-MyD88 pathway. Human primary monocytes were treated with a range of concentrations of CRX-527 after they were pretreated for 15 min with one of three concentrations (0.016, 4, or 100 nM) of CRX-547. (A and B) The concentrations of (A) TNF-α and (B) RANTES in the culture media were determined by Luminex analysis. (C) A Schild regression analysis plot of the shift in log EC50 (DR-1) for each concentration of CRX-547 versus the log of the concentration of CRX-547 was used to calculate the affinity of CRX-547 on the basis of inhibition of CRX-527–induced TNF-α production (Kb,app).
CRX-547 also inhibited the maximal extent of RANTES production by CRX-527; however, this pattern of inhibition was more complex. The addition of CRX-547 also shifted the dose-response curves (EC50) for CRX-527 to the right, but limited the maximal response such that it was reduced to the extent seen with CRX-547 alone (Fig. 7A). This pattern fits the general model for noncompetitive inhibition, whereby the antagonist (CRX-547) binds to the receptor and either precludes agonist binding (through an allosteric mechanism) or inactivates the receptor for subsequent full agonist responses. Our analysis does not immediately suggest an obvious mechanism for this complex, “selective” competitive or noncompetitive inhibition at the receptor, but it suggests that the interaction between the TRIF-selective agonist CRX-547 and TLR4 differs substantially from that of CRX-527.
DISCUSSION
The toxicity of most LPS species is determined by the interaction of the lipid A portion of these molecules with the TLR4–MD-2 receptor complex (8). Differences in the number, length, and arrangement of acyl chains, as well as in the number and position of charged groups, all substantially affect both the level and the character of the immunological response to lipid A (8, 11, 13, 43). Downstream of the lipid A–receptor interaction, specific TLR4-dependent signaling pathways, the MyD88-and TRIF-dependent pathways, determine the cellular response to receptor activation. MyD88-dependent signaling favors an inflammatory response, which is characterized by the synthesis and secretion of inflammatory mediators such as TNF-α and IL-1β (8, 11, 13, 43, 44). TRIF-dependent signaling favors the production of immune mediators such as the type I IFNs, some IL-12 family members, and chemokines that promote the maturation of DCs and influence the maturation of T cells. Both pathways are involved in linking innate and adaptive immunity (22, 45).
Here, we describe a minor structural change in a synthetic lipid A mimetic that substantially reduced conventional MyD88-dependent signaling while affecting TRIF-dependent signaling to a much lesser extent. Modification of the L-seryl AGP, CRX-527, to the D-isomer, CRX-547, significantly reduced or eliminated MyD88-dependent signaling as demonstrated by Western blotting analysis, analysis of the nuclear translocation of NF-κB, and measurement of cytokine production in primary human cells and cell lines. In contrast, signal transduction through the TRIF-dependent pathway was only slightly reduced. The structural change in CRX-547 affects the position of the aglycon carboxyl group relative to that of CRX-527. The most obvious ramifications of this change would be to affect the interaction of this charged group with the TLR4–MD-2 complex. LPS mimetics interact with the TLR4 receptor complex through MD-2 (46), and therefore, the diastereomeric change in CRX-547 may disrupt interactions of the carboxyl group, a lipid A phosphate bioisostere, with MD-2 or TLR4. This modification could alter receptor dimerization or oligomerization or trigger a conformational change in TLR4 that prevents binding by MyD88 and subsequent downstream signaling. On the basis of the data presented here, this modification to the receptor after the binding of CRX-547 binding does not substantially alter TRIF-dependent signaling.
Published crystal structures of the hMD-2–lipid IVa (46), hTLR4–hMD-2–Eritoran (47), and hTLR4–hMD-2–LPS dimer complexes (14) suggest that the phosphate groups that are attached at the 4′-(lipid IVa, Eritoran) and 1- (LPS) positions of the glucosamines interact with cationic residues near the opening of the hydrophobic binding pocket in MD-2. Structural modeling of TLR4–MD-2 dimerization and the crystal structure of the TLR4–MD-2–LPS dimer complex suggest that the face of MD-2 that interacts with the 1-phosphate of LPS also interacts with TLR4 of the dimer partner (14, 48). The bioisosteric carboxyl group of CRX-527 could maintain interactions similar to those of the 1-phosphate of LPS, whereas the change to the diastereomeric CRX-547 may disrupt these interactions, resulting in altered receptor dimerization, signaling, or both.
A similar selectivity for TRIF-dependent signaling in mice was reported for MLA (24, 25). MLA lacks a phosphate on the reducing sugar at a similar position relative to the bioisosteric carboxyl of CRX-527 and CRX-547. The authors of these studies postulated that differences in MyD88- and TRIF-dependent gene expression downstream of TLR4 not only are related to differential activation of NF-κB and IRF3 but also may involve the differential activation of either phosphoinositide 3-kinase or p38 mitogen-activated protein kinase pathways. A recent study by Embry et al. (30) suggests that a defect in IL-1β maturation after stimulation of TLR4 by sMLA is, in part, responsible for the reduced toxicity of MLA compared to that of LPS. Cells activated with sMLA are deficient in inducing the NLRP3 inflammasome (which is MyD88-dependent), as well as that of subsequent inflammasome assembly, caspase-1 activation, and release of mature IL-1β release. Comparison of the effects of synthetic sMLA with those of CRX-547 on human monocytes indicated that CRX-547 was TRIF-selective throughout the range of concentrations tested, whereas sMLA was TRIF-selective only at lower concentrations. Previous studies describing TRIF-dependent signaling by MLA have been conducted in murine systems (24, 25). These data suggest that important signaling differences may exist for MLA and CRX-547 in human versus murine cells. Overall, our preliminary evaluation of mature IL-1β production by human PBMCs indicated that CRX-547 induced substantially less IL-1β than did CRX-527 (fig. S2), which is consistent with the inefficient activation by CRX-547 of the MyD88-dependent events that are needed for the inflammasome to function.
The selective induction of TRIF-dependent signaling by synthetic lipid A mimetics suggests that further structure-activity investigations of these TLR4 agonists may provide opportunities for a better understanding of the underlying mechanisms that regulate TLR4 signaling pathways. The ability to selectively target either MyD88- or TRIF-dependent signaling downstream of TLR4 with synthetic receptor agonists may also provide valuable insights into the mechanism by which different lipid A structures differentially induce innate and adaptive immune responses. Such studies may be helpful in the development of synthetic vaccine adjuvants or new immunomodulators that selectively alter innate immune responses while simultaneously mitigating the potentially toxic side effects that are associated with the induction of inflammatory cytokines and chemokines.
MATERIALS AND METHODS
Cell lines and plasmids
The HEK 293 cell line expressing hTLR4, hMD-2, and hCD14; the pNiFTy2-SEAP, pSELECT-LacZ, pDeNy-hMyD88, and pDeNy-hTRIF plasmids and the pDeNy-mcs control plasmid; synthetic E. coli MPL (sMLA); and ultrapure Re595 LPS from S. minnesota were purchased from InvivoGen. The human monocytic cell line THP-1 was purchased from the American Type Culture Collection. MonoMac6 cells were obtained from the laboratory of G. Riethmüller (Institute for Immunology, University of Munich, Germany). HEK 293–hTLR4/hMD-2/hCD14 cells were cultured in RPMI 1640 with 10% fetal bovine serum (FBS, Hyclone), blasticidin (10 μg/ml), and HygroGold (50 μg/ml, InvivoGen). THP-1 and MonoMac6 cells were cultured in THP-1 medium containing RPMI 1640 with penicillin (100 U/ml, Sigma) and streptomycin (100 μg/ml, Sigma).
Preparation of AGPs
The AGPs CRX-527 and CRX-547 were synthesized by a highly convergent method as described previously for CRX-527 (12). For the preparation of CRX-547, the corresponding D-seryl glycosyl acceptor was used in the initial glycosylation step (12). Both AGPs were purified by flash chromatography on silica gel (to >95% purity) and analyzed as their triethylammonium salts by standard analytical methods.
Primary human cell culture
Human whole blood was collected from normal healthy donors at GlaxoSmithKline Biologicals (Hamilton, MT) with an Institutional Review Board–approved protocol or was shipped overnight from commercial suppliers (Key Biologics, Innovative Research, and Novi). PBMCs were isolated from a Ficoll-Hypaque 1.077 gradient. PBMC-derived monocytes were selected by the adherence of PBMCs (3 × 106 cells per well) to 48-well plates (Nunc) for 2 hours in 0.5 ml of monocyte medium [RPMI 1640, 10% human AB serum (Lonza/BioWhittaker), penicillin (100 U/ml), streptomycin (100 μg/ml), and 2-mercaptoethanol (50 μM, Sigma)] as previously described (49). Nonadherent cells were washed off with two washes with phosphate-buffered saline (PBS, Sigma) and one wash with RPMI 1640 medium. Adherent cells were prestimulated for 24 hours in medium containing rhIFN-γ (20 ng/ml, R&D Systems). Monocyte-derived DCs were produced by incubating adherent monocytes in 48-well plates for 7 days in monocyte medium containing recombinant human granulocyte macrophage colony-stimulating factor (rhGM-CSF, 50 ng/ml) and rhIL-4 (10 ng/ml, R&D Systems) with media changes on days 3 and 6. On day 7, half of the medium was replaced with medium containing rhGM-CSF (50 ng/ml), rhIL-4 (10 ng/ml), and rhIFN-γ (20 ng/ml) for 24 hours. Adherent monocytes and DCs were treated with the agonists in 0.05 ml of vehicle (2% glycerol) for 24 hours at 37°C in 5% CO2. Supernatants were removed after 24 hours and stored at −80°C for subsequent analysis of cytokines and chemokines by multiplex sandwich enzyme-linked immunosorbent assay (ELISA, R&D Systems Fluorokine MAP) with the Luminex 100 instrument. For pharmacological analysis, human PBMC–derived monocytes and DCs were treated with a wide range of concentrations of CRX-527 or CRX-547, and the resulting dose-response curves were fit with a four-parameter logistic equation by means of XLFit software (IDBS) to determine ED50 (median effective dose or half-maximal effective dose) values and the extents of response. For inhibition experiments, cells were pretreated with constant concentrations of CRX-547, followed by a wide range of concentrations of CRX-527. To perform Schild regression (50), we determined an equi-active dose ratio (DR) for each inhibition curve by dividing the ED50 for the curve by the ED50 of the CRX-527–only response curve. Regression of log(DR-1) upon log[CRX-547] was used to fit a straight line with the intercept representing an estimate of the Kb for CRX-547.
Dominant-negative mutants of MyD88 and TRIF
THP-1 cells were seeded in 12-well plates (5 × 105 cells per well) and incubated in 0.5 ml of THP-1 medium containing PMA (20 ng/ml, InvivoGen) for 48 hours to enable their differentiation into macrophages. The medium was changed to THP-1 medium containing PMA (5 ng/ml), and the cells were transiently transfected with 0.5 μg per well of the pDeNy-hMyD88/pSELECT-LacZ, pDeNy-TRIF/pSELECT-LacZ, or pDeNy-mcs (control)/pSELECT-LacZ plasmids in 3 μl of Fugene 6 transfection reagent per well and incubated for 48 hours. The cell cultures were aspirated and fresh medium containing the TLR4 agonists was added for 14 hours. Supernatants were assayed to determine cytokine amounts by multiplex sandwich ELISA (R&D Systems Fluorokine Map) with the Luminex 100 Instrument. Control wells were assayed for transfection efficiency with the InvivoGen LacZ Detection Kit.
Endocytosis inhibition by Dynasore treatment
THP-1 cells (5 × 105 cells per well in 48-well plates) were differentiated for 48 hours in THP-1 medium containing PMA (20 ng/ml). Differentiated THP-1 cells were pretreated with Dynasore (80 μM, ChemBridge) in dimethyl sulfoxide (DMSO) (or an equivalent volume of DMSO for controls) for 60 min in Macrophage Serum-Free Media (Invitrogen) with rhLBP (20 ng/ml, InvivoGen). Cells were then treated with specified concentrations of TLR4 agonists for 14 hours. Supernatants were collected and stored at −80°C for subsequent analysis by multiplex sandwich ELISA as described earlier.
Analysis of NF-κB nuclear translocation
MonoMac6 cells in exponential growth in THP-1 medium were stimulated with increasing concentrations of CRX-527 or CRX-547 for the indicated times. Cells were fixed in 2% paraformalin overnight, treated with permeabilization buffer (PBS, 2% FBS, and 0.1% Triton X), and incubated with the following primary and secondary antibodies: antibody against NF-κB (p65) (Santa Cruz Biotechnology), antibody against IRF3 (BD Biosciences), fluorescein isothiocyanate (FITC)–conjugated antibody against rabbit antibody (Jackson Labs), and phycoerythrin (PE)–conjugated antibody against mouse antibody (BD Biosciences). Similarity scores of signaling protein localization with the nuclear stain DRAQ5 (Alexis Biochemicals) were used for ImageStream analysis of nuclear translocation with IDEAS software as previously described (Fig. 4A) (39). Briefly, similarity scores were calculated as correlations between the pixel intensities of the nuclear (DRAQ5) and NF-κB images of stained cells that were hydrodynamically focused, excited by laser, and imaged on a charge-coupled device (CCD) camera. The similarity score is a derivation of Pearson’s correlation coefficient and achieves a higher positive value as the pixel intensities for NF-κB and nuclear staining become colocalized. A minimum of 3000 to 5000 cells were collected and analyzed for each condition and time point tested.
Western blotting analysis
For Western blotting analysis of total and phosphorylated proteins, primary human monocytes were isolated from PBMCs (1 × 107) by adherence to six-well culture plates for 2 hours at 37°C and in 5% CO2 and were treated with CRX-527 or CRX-547 (0.1 μM each) for the indicated times. After treatment, the cells were lysed with Cell Lysis Buffer (Cell Signaling Technology) containing Protease Inhibitor Cocktail (Sigma). Proteins were resolved by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane (Millipore). Membranes were incubated with primary monoclonal antibody against β-actin, antibody against pIRF3 (Ser396), primary polyclonal antibody against IRAK1, or antibody against IRF3 (Cell Signaling Technology) followed by incubation with horseradish peroxidase (HRP)–conjugated secondary antibodies against rabbit or mouse antibodies (KPL). Bands were detected with an ECL Advance Western Blot kit (GE Healthcare).
Transfection of HEK 293 cells
HEK 293–hTLR4/hMD-2/hCD14 cells were seeded in 12-well plates (at 4 × 105 cells per well) for 2 days and then were transfected with 100 ng of NF-κB reporter plasmid encoding SEAP (pNiFty2-SEAP) and a constitutive thymidine kinase promoter–driven luciferase reporter plasmid (pRLuc, Promega) in 0.05 ml of Lyovec transfection reagent (InvivoGen). Twenty-four hours later, cells were stimulated for 14 hours with agonists, and the supernatants were analyzed for SEAP activity (SEAP Reporter Assay Kit, InvivoGen). Cell lysates were assayed for luciferase activity (Renilla Luciferase Assay Kit, Promega) to normalize for transfection efficiency.
Supplementary Material
Acknowledgments
We thank J. Ward for technical assistance with laboratory assays and data analysis, L. Walsh for AGP compound formulation and analytical characterization, and J. Baldridge for expert advice and critical review of the manuscript. Cervarix is a trademark of the GlaxoSmithKline group of companies.
Funding: The research was funded through NIH/National Institute of Allergy and Infectious Diseases (NIAID) contracts HHSN266200400008C and HHSN272200900008C for Adjuvant Discovery and Development. Any opinions, findings, and conclusions or recommendations expressed in this article are those of the authors and do not necessarily reflect the views of NIAID.
Footnotes
Author contributions: W.S.B. and L.A.M. performed all the experiments and prepared the manuscript; D.A.J. conceptually designed the AGPs and directed synthetic chemistry efforts; and T.C.M., M.M.H., and J.T.E. assisted in experimental design, data analysis, manuscript revisions, and project management.
Competing interests: Use of the compounds CRX-527 and CRX-547 requires a materials transfer agreement (MTA). GlaxoSmithKline Biologicals has a patent application pending for the synthetic TRIF-selective compound described in this work. W.S.B., L.A.M., D.A.J., M.M.H., and J.T.E. are employees of GlaxoSmithKline Biologicals.
www.sciencesignaling.org/cgi/content/full/5/211/ra13/DC1
Fig. S1. Comparison of the production of MyD88- and TRIF-dependent cytokines and chemokines by human monocytes treated with CRX-527, CRX-547, Re595 LPS, or sMLA.
Fig. S2. CRX-547 stimulates the production of lower amounts of IL-1β by human PBMCs than does CRX-527.
REFERENCES AND NOTES
- 1.Airhart CL, Rohde HN, Hovde CJ, Bohach GA, Deobald CF, Lee SS, Minnich SA. Lipid A mimetics are potent adjuvants for an intranasal pneumonic plague vaccine. Vaccine. 2008;26:5554–5561. doi: 10.1016/j.vaccine.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bortolatto J, Borducchi E, Rodriguez D, Keller AC, Faquim-Mauro E, Bortoluci KR, Mucida D, Gomes E, Christ A, Schnyder-Candrian S, Schnyder B, Ryffel B, Russo M. Toll-like receptor 4 agonists adsorbed to aluminium hydroxide adjuvant attenuate ovalbumin-specific allergic airway disease: Role of MyD88 adaptor molecule and interleukin-12/interferon-γ axis. Clin Exp Allergy. 2008;38:1668–1679. doi: 10.1111/j.1365-2222.2008.03036.x. [DOI] [PubMed] [Google Scholar]
- 3.Fransen F, Boog CJ, van Putten JP, van der Ley P. Agonists of Toll-like receptors 3, 4, 7, and 9 are candidates for use as adjuvants in an outer membrane vaccine against Neisseria meningitidis serogroup B. Infect Immun. 2007;75:5939–5946. doi: 10.1128/IAI.00846-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishizaka ST, Hawkins LD. E6020: A synthetic Toll-like receptor 4 agonist as a vaccine adjuvant. Expert Rev Vaccines. 2007;6:773–784. doi: 10.1586/14760584.6.5.773. [DOI] [PubMed] [Google Scholar]
- 5.Johnson DA. Synthetic TLR4-active glycolipids as vaccine adjuvants and stand-alone immunotherapeutics. Curr Top Med Chem. 2008;8:64–79. doi: 10.2174/156802608783378882. [DOI] [PubMed] [Google Scholar]
- 6.Sheng KC, Kalkanidis M, Pouniotis DS, Wright MD, Pietersz GA, Apostolopoulos V. The adjuvanticity of a mannosylated antigen reveals TLR4 functionality essential for subset specialization and functional maturation of mouse dendritic cells. J Immunol. 2008;181:2455–2464. doi: 10.4049/jimmunol.181.4.2455. [DOI] [PubMed] [Google Scholar]
- 7.Schwarz TF. Clinical update of the AS04-adjuvanted human papillomavirus-16/18 cervical cancer vaccine, Cervarix. Adv Ther. 2009;26:983–998. doi: 10.1007/s12325-009-0079-5. [DOI] [PubMed] [Google Scholar]
- 8.Viriyakosol S, Kirkland T, Soldau K, Tobias P. MD-2 binds to bacterial lipo-polysaccharide. J Endotoxin Res. 2000;6:489–491. [PubMed] [Google Scholar]
- 9.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CR. Transfer of palmitate from phospholipids to lipid A in outer membranes of Gram-negative bacteria. EMBO J. 2000;19:5071–5080. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, Miller SI. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell. 1998;95:189–198. doi: 10.1016/s0092-8674(00)81750-x. [DOI] [PubMed] [Google Scholar]
- 11.Stöver AG, Da Silva Correia J, Evans JT, Cluff CW, Elliott MW, Jeffery EW, Johnson DA, Lacy MJ, Baldridge JR, Probst P, Ulevitch RJ, Persing DH, Hershberg RM. Structure-activity relationship of synthetic Toll-like receptor 4 agonists. J Biol Chem. 2004;279:4440–4449. doi: 10.1074/jbc.M310760200. [DOI] [PubMed] [Google Scholar]
- 12.Bazin HG, Murray TJ, Bowen WS, Mozaffarian A, Fling SP, Bess LS, Livesay MT, Arnold JS, Johnson CL, Ryter KT, Cluff CW, Evans JT, Johnson DA. The ‘Ethereal’ nature of TLR4 agonism and antagonism in the AGP class of lipid A mimetics. Bioorg Med Chem Lett. 2008;18:5350–5354. doi: 10.1016/j.bmcl.2008.09.060. [DOI] [PubMed] [Google Scholar]
- 13.Teghanemt A, Zhang D, Levis EN, Weiss JP, Gioannini TL. Molecular basis of reduced potency of underacylated endotoxins. J Immunol. 2005;175:4669–4676. doi: 10.4049/jimmunol.175.7.4669. [DOI] [PubMed] [Google Scholar]
- 14.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 15.Saitoh S, Akashi S, Yamada T, Tanimura N, Kobayashi M, Konno K, Matsumoto F, Fukase K, Kusumoto S, Nagai Y, Kusumoto Y, Kosugi A, Miyake K. Lipid A antagonist, lipid IVa, is distinct from lipid A in interaction with Toll-like receptor 4 (TLR4)-MD-2 and ligand-induced TLR4 oligomerization. Int Immunol. 2004;16:961–969. doi: 10.1093/intimm/dxh097. [DOI] [PubMed] [Google Scholar]
- 16.Visintin A, Latz E, Monks BG, Espevik T, Golenbock DT. Lysines 128 and 132 enable lipopolysaccharide binding to MD-2, leading to Toll-like receptor-4 aggregation and signal transduction. J Biol Chem. 2003;278:48313–48320. doi: 10.1074/jbc.M306802200. [DOI] [PubMed] [Google Scholar]
- 17.Rowe DC, McGettrick AF, Latz E, Monks BG, Gay NJ, Yamamoto M, Akira S, ONeill LA, Fitzgerald KA, Golenbock DT. The myristoylation of TRIF-related ’ adaptor molecule is essential for Toll-like receptor 4 signal transduction. Proc Natl Acad Sci USA. 2006;103:6299–6304. doi: 10.1073/pnas.0510041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4–mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 19.Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA. Rip1 mediates the Trif-dependent Toll-like receptor 3- and 4-induced NF-κB activation but does not contribute to interferon regulatory factor 3 activation. J Biol Chem. 2005;280:36560–36566. doi: 10.1074/jbc.M506831200. [DOI] [PubMed] [Google Scholar]
- 20.Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J. RIP1 is an essential mediator of Toll-like receptor 3–induced NF-κB activation. Nat Immunol. 2004;5:503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 22.Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]
- 23.Serbina NV, Kuziel W, Flavell R, Akira S, Rollins B, Pamer EG. Sequential MyD88-independent and -dependent activation of innate immune responses to intra-cellular bacterial infection. Immunity. 2003;19:891–901. doi: 10.1016/s1074-7613(03)00330-3. [DOI] [PubMed] [Google Scholar]
- 24.Cekic C, Casella CR, Eaves CA, Matsuzawa A, Ichijo H, Mitchell TC. Selective activation of the p38 MAPK pathway by synthetic monophosphoryl lipid A. J Biol Chem. 2009;284:31982–31991. doi: 10.1074/jbc.M109.046383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 26.Baldridge JR, Yorgensen Y, Ward JR, Ulrich JT. Monophosphoryl lipid A enhances mucosal and systemic immunity to vaccine antigens following intranasal administration. Vaccine. 2000;18:2416–2425. doi: 10.1016/s0264-410x(99)00572-1. [DOI] [PubMed] [Google Scholar]
- 27.Johnson DA, Sowell CG, Johnson CL, Livesay MT, Keegan DS, Rhodes MJ, Ulrich JT, Ward JR, Cantrell JL, Brookshire VG. Synthesis and biological evaluation of a new class of vaccine adjuvants: Aminoalkyl glucosaminide 4-phosphates (AGPs) Bioorg Med Chem Lett. 1999;9:2273–2278. doi: 10.1016/s0960-894x(99)00374-1. [DOI] [PubMed] [Google Scholar]
- 28.Persing DH, Coler RN, Lacy MJ, Johnson DA, Baldridge JR, Hershberg RM, Reed SG. Taking toll: Lipid A mimetics as adjuvants and immunomodulators. Trends Microbiol. 2002;10:S32–S37. doi: 10.1016/s0966-842x(02)02426-5. [DOI] [PubMed] [Google Scholar]
- 29.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 30.Embry CA, Franchi L, Nuñez G, Mitchell TC. Mechanism of impaired NLRP3 inflammasome priming by monophosphoryl lipid A. Sci Signal. 2011;4:ra28. doi: 10.1126/scisignal.2001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ulevitch RJ, Tobias PS. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu Rev Immunol. 1995;13:437–457. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 33.Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm Res. 2007;56:45–50. doi: 10.1007/s00011-007-6115-5. [DOI] [PubMed] [Google Scholar]
- 34.Doyle SL, ONeill LA. Toll-like receptors: From the discovery of NFκB to new insights into transcriptional regulations in innate immunity. Biochem Pharmacol. 2006;72:1102–1113. doi: 10.1016/j.bcp.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 35.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 36.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 37.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-κB activation. Science. 2005;309:1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 38.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 39.George TC, Fanning SL, Fitzgerald-Bocarsly P, Medeiros RB, Highfill S, Shimizu Y, Hall BE, Frost K, Basiji D, Ortyn WE, Morrissey PJ, Lynch DH. Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J Immunol Methods. 2006;311:117–129. doi: 10.1016/j.jim.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 40.Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD. Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and Toll-like receptor-mediated NF-κB activation. Mol Cell Biol. 2008;28:3538–3547. doi: 10.1128/MCB.02098-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neumann D, Kollewe C, Resch K, Martin MU. The death domain of IRAK-1: An oligomerization domain mediating interactions with MyD88, Tollip, IRAK-1, and IRAK-4. Biochem Biophys Res Commun. 2007;354:1089–1094. doi: 10.1016/j.bbrc.2007.01.104. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: A novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 43.Schromm AB, Brandenburg K, Loppnow H, Moran AP, Koch MH, Rietschel ET, Seydel U. Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. Eur J Biochem. 2000;267:2008–2013. doi: 10.1046/j.1432-1327.2000.01204.x. [DOI] [PubMed] [Google Scholar]
- 44.Akira S. Toll-like receptors: Lessons from knockout mice. Biochem Soc Trans. 2000;28:551–556. doi: 10.1042/bst0280551. [DOI] [PubMed] [Google Scholar]
- 45.Kaisho T, Akira S. Regulation of dendritic cell function through Toll-like receptors. Curr Mol Med. 2003;3:373–385. doi: 10.2174/1566524033479726. [DOI] [PubMed] [Google Scholar]
- 46.Ohto U, Fukase K, Miyake K, Satow Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science. 2007;316:1632–1634. doi: 10.1126/science.1139111. [DOI] [PubMed] [Google Scholar]
- 47.Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 48.Walsh C, Gangloff M, Monie T, Smyth T, Wei B, McKinley TJ, Maskell D, Gay N, Bryant C. Elucidation of the MD-2/TLR4 interface required for signaling by lipid IVa. J Immunol. 2008;181:1245–1254. doi: 10.4049/jimmunol.181.2.1245. [DOI] [PubMed] [Google Scholar]
- 49.Probst P, Skeiky YA, Steeves M, Gervassi A, Grabstein KH, Reed SG. A Leishmania protein that modulates interleukin (IL)-12, IL-10 and tumor necrosis factor-α production and expression of B7-1 in human monocyte-derived antigen-presenting cells. Eur J Immunol. 1997;27:2634–2642. doi: 10.1002/eji.1830271024. [DOI] [PubMed] [Google Scholar]
- 50.Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.