

Abstract
Purpose.
Retinitis pigmentosa (RP) is a highly heterogeneous genetic disease; therefore, an accurate molecular diagnosis is essential for appropriate disease treatment and family planning. The prevalence of RP in China had been reported at 1 in 3800, resulting in an estimated total of 340,000 Chinese RP patients. However, genetic studies of Chinese RP patients have been very limited. To date, no comprehensive molecular diagnosis has been done for Chinese RP patients. With the emergence of next-generation sequencing (NGS), comprehensive molecular diagnosis of RP is now within reach. The purpose of this study was to perform the first NGS-based comprehensive molecular diagnosis for Chinese RP patients.
Methods.
Thirty-one well-characterized autosomal recessive RP (arRP) families were recruited. For each family, the DNA sample from one affected member was sequenced using our custom capture panel, which includes 163 retinal disease genes. Variants were called, filtered, and annotated by our in-house automatic pipeline.
Results.
Twelve arRP families were successfully molecular diagnosed, achieving a diagnostic rate of approximately 40%. Interestingly, approximately 63% of the pathogenic mutations we identified are novel, which is higher than that observed in a similar study on European descent (45%). Moreover, the clinical diagnoses of two families were refined based on the pathogenic mutations identified in the patients.
Conclusions.
We conclude that comprehensive molecular diagnosis can be vital for an accurate clinical diagnosis of RP. Applying this tool on patients from different ethnic groups is essential for enhancing our knowledge of the global spectrum of RP disease-causing mutations.
Keywords: retinitis pigmentosa, next-generation sequencing, molecular diagnosis, Chinese population
Next-generation sequencing–based comprehensive molecular diagnosis of 31 families with autosomal recessive retinitis pigmentosa (arRP) was performed. This is the first such study of a Chinese arRP patient cohort, revealing a total of 10 novel putatively pathogenic mutations.
Introduction
Retinitis pigmentosa (RP; Mendelian Inheritance in Man [MIM] #268000) is an inherited retinal degenerative disease that is highly heterogeneous both clinically and genetically. Although RP patients share common clinical features, such as the progressive loss of rod photoreceptors followed by cone photoreceptor death and black bone–spicule pigmentation due to the death of retinal pigment epithelium cells, the time of disease onset, and disease progression vary dramatically among patients. Some patients exhibit phenotypes in their first decade, whereas others may not notice the symptoms until much later in life.1 In many cases, the degeneration of photoreceptor cells is so severe that the patients suffer complete blindness.2,3 The diversity of clinical appearances of RP is partially due to the underlying genetic heterogeneity of the disease. To date, mutations in over 50 genes have been linked to RP.4 These genes function in strikingly diverse biological pathways, including phototransduction, the retinoid (vitamin A) cycle, gene transcription, RNA splicing, and photoreceptor structure.1–3 Depending on the mutated genes and the nature of the alleles, the phenotypes may vary. Therefore, accurate molecular diagnosis of RP is critical for the diagnosis, prognosis, management, and, eventually, treatment of patients.
The comprehensive diagnosis of RP is made challenging by the large number of disease genes and alleles associated with RP. Until recently, the main method for RP molecular diagnosis was Sanger sequencing. However, because of its high reagent and labor costs, typically only a small set of genes can be Sanger sequenced for each patient, resulting in a low diagnostic rate. An alternative method is the Arrayed Primer Extension (APEX) method.5 Using this single-base extension technology, hundreds of known mutations can be detected in parallel. However, since the current APEX design covers only a small subset of known mutations, the diagnostic rate is typically less than 15%.6,7 Next-generation sequencing (NGS) provides a promising alternative for the molecular diagnosis of RP. Coupled with DNA capture technology, NGS enables rapid and cost-effective parallel sequencing of a large panel of disease genes. In several recent studies, approximately 100 retinal disease genes were targeted and sequenced using the capture-NGS method. A significantly higher rate of molecular diagnosis of approximately 50% was achieved.8–10
As one of the most common forms of inherited retinal degenerative disease, RP affects 1 in 3000 people worldwide.2,3 The prevalence of RP in China had been reported at 1 in 3800, resulting in an estimated total of 340,000 Chinese RP patients.11 It is known that both causative genes and mutation spectra can vary dramatically among different ethnic groups. For example, mutations in RHO (MIM #180380) are much less frequent in Chinese autosomal dominant RP (adRP) patients (4%–8%) than in adRP patients of European descent (16%–28%)12–17; similarly, the mutation spectra of several genes such as USH2A (MIM #608400) and EYS (MIM #612424) among Asian RP patients are different from those among European Caucasians.18,19 The majority of genetic studies of RP to date were conducted on patients of European descent and only a small subset of known RP disease genes have been investigated in Chinese patients.13,18,20,21 In this study, we report the first comprehensive molecular diagnosis of a Chinese RP patient cohort using capture-NGS. Thirty-one unrelated well-characterized arRP families were screened for mutations in 163 retinal disease genes using capture sequencing. We successfully identified causative mutations for 12 patient families, achieving a diagnosis rate of approximately 40%. Interestingly, 63% (10/16) of the alleles identified in this study are novel. In addition, three of the 12 families carry mutations in genes with little or no evidence for their association with RP, including CYP4V2 (MIM #608614) and ADAM9 (MIM #602713). Careful clinical reevaluation led to the clinical rediagnoses of two RP families to other retinal diseases. Collectively, this study underscores that comprehensive molecular diagnosis of RP is crucial for an accurate clinical classification. Further, studying patients from different ethnic backgrounds may greatly enhance our knowledge of the mutation spectrum of RP.
Materials and Methods
Clinical Diagnosis of arRP and Sample Collection
All subjects were identified at Peking Union Medical College Hospital (PUMCH, Beijing, China). Full medical and family histories were taken, pedigrees were drawn, and an ophthalmologic examination was performed. Each patient underwent standard ophthalmic examination: best correct visual acuity (BCVA) according to projected Snellen charts, slit-lamp biomicroscopy, dilated indirect ophthalmoscopy, fundus photography if possible, and visual field tests (Octopus; Interzeag, Schlieren, Switzerland). Retinal structure was examined by optical coherence tomography (OCT) (Topcon, Tokyo, Japan). Electroretinograms (ERGs) were performed (RetiPort ERG System; Roland Consult, Wiesbaden, Germany) using corneal “ERGjet” contact lens electrodes. The ERG protocol complied with the standards published by the International Society for Clinical Electrophysiology of Vision. The diagnosis of arRP was based on the presence of night blindness, fundus findings (retinal pigmentation, vessel attenuation, and various degrees of retinal atrophy), severe loss of peripheral visual field, abnormal ERG findings (dramatic diminution in amplitudes or complete absence of response), and family history. Written informed consents were obtained from all participating individuals or their guardians. Genomic DNA was isolated from peripheral leukocytes using a commercial kit (QIAamp DNA Blood Midi Kit; Qiagen, Hilden, Germany) according to the manufacturer's protocol. This study was approved by the Institutional Review Board of PUMCH and adhered to the tenets of the Declaration of Helsinki and the Guidance on Sample Collection of Human Genetic Diseases by the Ministry of Public Health of China.
Design of Our Capture Panel
A capture panel of retinal disease genes was developed in our group (Wang et al., unpublished data, 2013). A design file was submitted to Roche NimbleGen (Roche Applied Science, Penzberg, Upper Bavaria, Germany) to synthesize the hybridization probes. The probes covered 2560 exons and corresponding splice junctions of 163 known retinal disease genes, with a total of 649,804 bp in design region. In total, 47 RP genes were targeted, including all 29 arRP genes that had been reported at the time of panel design (Supplementary Table S1).
Library Preparation and Targeted Sequencing
Precapture libraries (Illumina, Inc., San Diego, CA) were generated according to the manufacturer's sample preparation protocol for genomic DNA. Briefly, 1 μg of each patient's genomic DNA was sheared into 300- to 500-bp fragments. The DNA fragments were end-repaired using polynucleotide kinase and Klenow fragment (large protein fragment). The 5′ ends of the DNA fragments were phosphorylated and a single adenine base was added to the 3′ ends using Klenow exonuclease. Y-shaped index adaptors (Illumina, Inc.) were ligated to the repaired ends, then the DNA fragments were PCR amplified for eight cycles and fragments of 200 to 500 bp were isolated by purification of beads. The precapture libraries were quantified (PicoGreen fluorescence assay kit; Life Technologies, Carlsbad, CA) and their size distributions determined by a commercial bioanalytical system (Agilent 2100 BioAnalyzer; Agilent Technologies, Santa Clara, CA). Fifty precapture libraries (60 ng/library) were pooled together for one capture reaction. Hybridization and wash kits (NimbleGen SeqCap EZ; Roche NimbleGen, Roche Applied Science) were used for panel capture, following the standard manufacturer's protocol. Captured libraries were sequenced (Illumina HiSeq 2000; Illumina, Inc.) as 100-bp paired-end reads, following the manufacturer's protocols.
Bioinformatics Analysis of Sequencing Results
Sequence reads were aligned to human hg19 reference by an aligner (Burrows-Wheeler Aligner, BWA version 0.5.9).22 After recalibration and local realignment using the genome analysis toolkit (GATK version 1.0.5974),23 the refined sequencing results were subject to variant calling using a toolkit (Atlas2).24 Several common variant databases (such as the 1000 Genome [Build 20110521 and 20101123],25 dbSNP135,26 NHLBI Exome Sequencing database,27 NIEHS Exome Sequencing database,28 and an internal control database of 997 exomes) were used to filter out common polymorphisms with an allele frequency higher than 0.5% in any of the above databases. ANNOVAR29 was used to remove synonymous mutations using RefSeq genes as reference. SIFT was used to make functional prediction of missense variants.30 dbNSFP was also used to generate comprehensive functional prediction of missense variants in unsolved cases.31 University of California Santa Cruz (UCSC) Genome Browser Vertebrate Multiz Alignment and Conservation track was used to assess whether missense variants affect conserved amino acids.32 The Human Gene Mutation Database (HGMD) professional database was used to search for known pathogenic mutations.33
Determination of Pathogenic Mutations
A stepwise strategy was utilized to systematically identify the putative pathogenic mutations for each of the arRP families. As shown in Figure 1, we first checked for mutations in known RP genes. If no clear pathogenic mutations were found, mutations in other retinal disease genes were considered. Within each step, we prioritized the mutations based on the order of “Known pathogenic mutations → Novel severe mutations → Novel missense mutations.” Recessive, dominant, X-linked, and digenic mutations were all considered. A more stringent frequency cutoff (no more than once in any variant database) was applied to novel heterozygous missense variants in dominant genes to reduce the risk of false positives. At any step, if pathogenic mutations were found, further Sanger validation, segregation, and clinical reevaluation (if necessary) were carried out.
Figure 1.
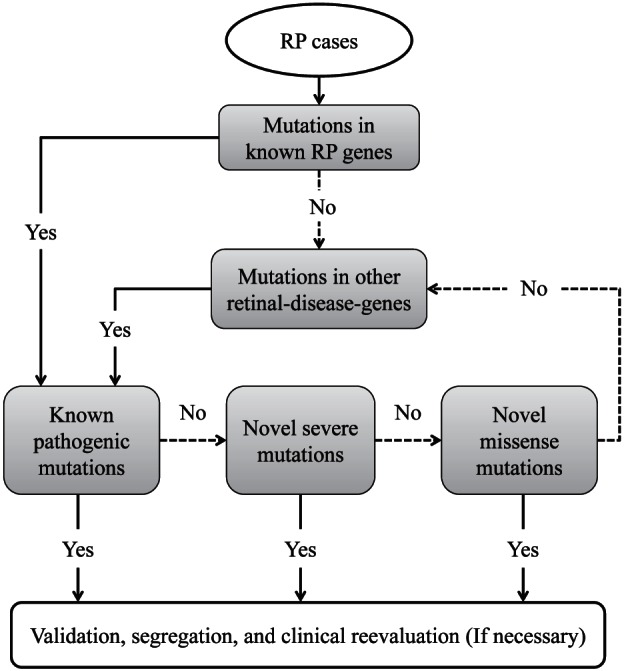
Schematic chart shows the stepwise strategy to identify pathogenic mutation.
Sanger Validation and Segregation Tests
All putative mutations found by NGS were validated by Sanger sequencing. DNA sequences were obtained from the UCSC Genome Browser. RepeatMasker was used to mask the repetitive regions.34 Primer3 was used to design the primers (Supplementary Table S2).35 The amplicons (300–500 bp) were sequenced on a commercial sequencing machine (ABI 3730xl capillary sequencing machine; Life Technologies). All the available family members were Sanger sequenced to perform segregation test.
Results
31 Chinese Families With arRP Were Recruited
A total of 59 patients (29 female, 30 male) from 31 unrelated Chinese arRP families were recruited for this study. Six families (19.4%) were consanguineous. More than 74% of the families (23/31) had two or more affected patients. The initial symptom was night blindness followed by poor vision. BCVA ranged from No Light Perception (NLP) to 0.9 (decimal Snellen); approximately 66% (29/59) patients have BCVA less than 0.3 (decimal Snellen) in their worse eyes. Patients show different degrees of retinal pigment deposits, retinal vascular attenuation, retinal pigment epithelium (RPE) atrophy, and choriocapillaris atrophy. Cystic macular edema, subcapsular cataract, and epiretinal membrane were observed complications. The summary of the observed clinical phenotypes is in Table 1 and the pedigrees are in Supplementary Figure S1.
Table 1. .
Clinical Summary of 31 arRP Families
|
Family ID |
Proband Age, y/Sex |
No. of Affected |
BCVA, OD/OS |
Additional Information of Proband |
| 4255 | 42/M | 2 | 0.4/0.1 | Photophobia for 2 years |
| 4257* | 29/M | 1 | 0.4/0.4 | −4.5DS; −5.0DS |
| 4258 | 44/F | 2 | 0.3/0.4 | Normal hearing; cystic macular edema |
| 4259 | 39/F | 1 | HM/HM | Photophobia for 4 years |
| 4263* | 9/M | 1 | 0.08/0.1 | −4.50DS–3.25DCX180; −4.50DS–3.25DCX180 |
| 4264 | 32/M | 1 | HM/HM | N/A |
| 4268 | 31/M | 3 | FC/0.03 | Nystagmus |
| 4270 | 39/M | 2 | FC/FC | N/A |
| 4271 | 28/M | 1 | HM/0.7 | OD is more severe than OS |
| 4273 | 43/M | 2 | 0.9/0.7 | N/A |
| 4274 | 52/M | 3 | LP/NLP | Dense cataracts; left eye trauma history |
| 4275 | 25/F | 2 | HM/HM | Nystagmus |
| 4277* | 48/M | 2 | LP/HM | N/A |
| 4278 | 41/M | 2 | HM/NLP | Dense cataracts |
| 4279 | 42/M | 1 | 0.1/0.2 | Dense confluent retinal pigments |
| 4280 | 27/M | 2 | 0.03/0.01 | Photophobia for 3 years |
| 4282* | 36/M | 2 | 0.06/0.1 | Epiretinal membrane |
| 4283 | 41/F | 2 | 0.2/0.2 | +0.75DS+1.75DCX90; +1.50DS+1.00DCX90; photophobia for 2 years |
| 4285 | 32/M | 2 | 1.0/1.0 | −2.00DS–1.75DCX80; −1.50DS; normal hearing |
| 4286 | 21/M | 3 | 0.9/0.2 | Emmetropia |
| 4287 | 55/F | 2 | 0.6/0.9 | Normal hearing |
| 4288 | 41/M | 2 | LP/LP | N/A |
| 4289 | 33/F | 2 | 1.0/0.9 | Emmetropia |
| 4290 | 28/M | 2 | 1.2/0.8 | Emmetropia |
| 4291 | 39/M | 2 | 0.08/0.05 | Affected brother has coats like retinal exudates |
| 4292 | 36/F | 2 | 1.0/0.7 | Cystic macular edema |
| 4293 | 40/M | 3 | 0.02/0.02 | Emmetropia |
| 4294* | 40/M | 1 | 0.1/LP | N/A |
| 4295* | 48/F | 2 | 0.6/0.6 | Emmetropia |
| 4296 | 53/F | 2 | 0.03/0.03 | Emmetropia |
| 4297 | 60/F | 2 | 0.03/0.03 | Dense cataracts |
BCVA (decimal Snellen); OD, right eye; OS, left eye; DS, diopter of spherical; DC, diopter of cylindrical; HM, hand motion; FC, finger counting; LP, light perception; NLP, no light perception.
Consanguineous families.
High-Quality Sequencing Results Were Obtained
Capture-NGS using our panel was performed on all 31 arRP families. For each arRP family, DNA from one affected member was selected, captured, and sequenced. High-quality sequencing results were obtained. On average, a mean coverage of 56× over the targeted region was achieved per sample and 90% of all the bases in the targeted region were covered by 10 or more reads (Supplementary Fig. S2). In addition, we assessed the evenness of read coverage across the entire target region by calculating the evenness score as in Mokry et al.36 An average evenness score of 0.78 (SD = 0.048) was obtained per sample, indicating uniform enrichment across the targeted region (Supplementary Fig. S2). Further, when only known arRP genes were assessed, a higher and more even coverage was observed. On average, 93% of all targeted bases in arRP genes were covered by 10 or more reads, with an average evenness score of 0.80 (SD = 0.052) was obtained per sample (Supplementary Fig. S2). Therefore, our capture-NGS provides nearly complete coverage of all coding bases of all 163 retinal disease genes included in the panel. Our approach enables highly sensitive and accurate mutation detection.
Automatic Variants Filtering and Annotation Was Performed
An automatic variant calling, filtering, and annotation pipeline (see Materials and Methods) was utilized to process the sequencing data from all 31 samples. An average of 455 variants, including 426 single nucleotide polymorphisms (SNPs) and 29 small insertions and deletions (INDELs), were initially identified for each sample in the targeted region. Polymorphisms that appear at a greater than 0.5% frequency in at least one of the following databases were considered too frequent to be pathogenic for RP and therefore excluded from further analysis: the 1000 Genome (Build 20110521 and 20101123),25 dbSNP135,26 NHLBI Exome Sequencing database,27 NIEHS Exome Sequencing database,28 and our internal control databases. On average, 27 rare variants were identified in each sample, among which six were predicted to affect protein coding using the ANNOVAR29 and were considered as candidate pathogenic mutations.
Putative Pathogenic Mutations in Known RP Genes Were Found in Nine arRP Families
One patient was found to carry known pathogenic mutations in a known RP gene. Specifically, patient 4279_1 from arRP family 4279 carries compound heterozygous missense mutations c.505C>T:p.(R169W) and c.226G>A:p.(G76R) in RDH12 (MIM #608830; NM_152443.2), both of which were previously found in patients with recessive Leber Congenital Amaurosis (LCA; MIM #204000), a more severe form of retinal dystrophy.37,38 A segregation test performed on the family showed that the two mutations cosegregated with the disease phenotypes (Fig. 2A). To rule out the possibility of a misdiagnosis, we clinically reevaluated patient 4279_1. During adolescence, the patient's visual acuity reached 0.8/0.8 (decimal Snellen), but his night vision was severely impaired. His daylight vision started to decrease at 18 years of age. At 39 years of age, his visual acuity was 0.3/0.3 (decimal Snellen). Very dense and confluent dark pigments and macular atrophy were observed (Supplementary Fig. S3). As a result, we confirmed that the patient indeed has early onset RP, not LCA.
Figure 2.

Three families (4279, 4283, and 4285) carry known pathogenic mutations in known RP genes. (A–C) The segregation results of the three families. The genotype of each evaluated individual is shown below the individual's symbol. Wild type is listed as a “+.” (D) Multispecies alignment shows the conservation of the amino acid affected by the novel missense mutation. Red box shows the affected amino acid.
In addition, as shown in Table 2, two patients, 4283_1 and 4285_1 from arRP families 4283 and 4285, respectively, were identified to carry a known mutation along with a novel rare variant. Patient 4283_1 presented night vision problem and poor visual acuity since childhood. Her BCVA never reached 0.5 (decimal Snellen) and she started experiencing photophobia when she was 41 years of age. Fundus examination reveals widespread pigment deposits, even in the macula. The retinal vessels were attenuated and there was waxy pallor of the optic disc. Patient 4283_1 was identified to carry compound heterozygous variants in RPE65 (MIM #180069; NM_000329.2). The first allele, c.200T>G:p.(L67R), is a known missense mutation previously found in patients with LCA.39 The second allele, c.434C>A:p.(A145D), is a novel missense variant that is likely to be pathogenic based on the following reasons. First, it is rare because it is not observed in any of the databases used in our filtering steps. Second, the amino acid affected by this variant is highly conserved (Fig. 2D). Third, the amino acid change was predicted to be detrimental with a SIFT score of 0.98. To further evaluate whether these two alleles are causative, a segregation test was performed. As shown in Figure 2B, both parents and the unaffected sibling of patient 4283_1 are carriers, whereas the other affected sibling who shows similar but more severe clinical manifestation carries both mutations, supporting that the disease phenotypes observed in this family are due to mutations in RPE65. Similarly, patient 4285_1 was found to carry compound heterozygous variants in USH2A (NM_206933.2). The first allele, c.99_100insT:p.(R34fs), is a frameshift mutation previously reported in Usher syndrome type II (MIM #276901) patients.40 The second allele, c.4627+2T>C, is a novel splicing variant that is likely to be pathogenic, in that it was predicted to impair splicing between exon 21 and exon 22, resulting in the truncation of Laminin G-like 1 domain. Missense mutation, c.4732C>T:p.(R1578C), in exon 22 has been reported in patients with Usher syndrome type II, suggesting the functional importance of this exon.41 Segregation testing indicated that the identified alleles cosegregated with the disease phenotypes in this family, further supporting the possibility that the variants are pathogenic (Fig. 2C). It is interesting to note that the known alleles, c.99_100insT:p.(R34fs), were previously reported in patients with Usher Syndrome but not nonsyndromic RP. Therefore, audiometry was performed to exclude the possibility of misdiagnosis. The results showed that both of the patient's ears had normal hearing sensitivity at 10 dBHL for most sound frequencies tested.
Table 2. .
Nine arRP Families Were Found to Have Putative Pathogenic Mutations in Known RP Genes
|
Family ID |
Patient ID |
Gene |
Allele1 |
Allele2 |
Reference |
| Solved with high confidence | |||||
| 4279 | 4279_1 | RDH12 (NM_152443.2) | c.505C> T:p.R(169W)* | c.226G> A:p.(G76R)* | (Mackay et al.,38 2011) (allele1) |
| (Aldahmesh et al.,37 2009) (allele2) | |||||
| 4283 | 4283_1 | RPE65 (NM_000329.2) | c.200T> G:p.(L67R)* | c.434C> A:p.(A145D)* | (Xu et al.,39 2012) (allele 1) |
| Novel (allele2) | |||||
| 4285 | 4285_1 | USH2A (NM_206933.2) | c.99_100insT:p.(R34fs)† | c.4627+2T>C‡ | (Dai et al.,40 2008) (allele1) |
| Novel (allele2) | |||||
| 4264 | 4264_1 | CERKL (NM_201548.4) | c.1404delA:p.(E468fs)† | c.1404delA:p.(E468fs)† | Novel |
| 4277 | 4277_1 | C2ORF71 (NM_001029883.1) | c.769A> T:p.(K257*)§|| | c.769A> T:p.(K257*)§|| | Novel |
| 4282 | 4282_1 | PRCD (NM_001077620.2) | c.2T>C:p.(M1?)¶ | c.2T>C:p.(M1?)¶ | Novel |
| 4295 | 4295_1 | PDE6B (NM_000283.3) | c.1467+1G>C‡ | c.1467+1G>C‡ | Novel |
| Likely solved | |||||
| 4268 | 4268_1 | RDH12 (NM_152443.2) | c.437T> A:p.(V146D)* | c.437T> A:p.(V146D)* | Novel |
| 4294 | 4294_1 | CNGB1 (NM_001297.4) | c.1589C> G:p.(P530R)* | c.1589C> G:p.(P530R)* | Novel |
Missense mutation; † frameshift mutation; ‡ splicing mutation; § nonsense mutation; || mutations; ¶ fail-to-start mutation identified in off-targeted regions.
Besides the three families described above, novel variants that are likely to result in loss-of-function in known RP disease genes were identified in four additional families (4264, 4277, 4282, and 4295) (Table 2). Specifically, patient 4264_1 from arRP family 4264 carries a homozygous frameshift variant, c.1404delA:p.(E468fs) in CERKL (MIM #608381; NM_201548.4), which causes a coding frameshift in exon 12. Patient 4277_1 from arRP family 4277 carries a homozygous nonsense variant, c.769A>T:p.(K257*), in C2ORF71 (MIM #613425; NM_001029883.1), which introduces a premature stop codon in the first exon of the gene, resulting in a truncated protein and/or nonsense-mediated decay. Patient 4282_1 from arRP family 4282 carries a homozygous fail-to-start variant, c.2T>C:p.(M1?), in PRCD (MIM #610598; NM_001077620.2), which disrupts the start codon of the gene. Patient 4295_1 from arRP family 4295 carries a homozygous splicing variant, c.1467+1G>C, in PDE6B (MIM #180072; NM_000283.3), which affects the donor splicing site between exon 11 and exon 12. Known missense mutation c.1580T>C:p.(L527P) in exon12 has been reported in RP patients, suggesting the functional importance of this exon.42 Segregation of these variants with the disease phenotypes was verified in all the families, supporting their pathogenicity (Figs. 3A–D).
Figure 3.

Four families (4264, 4277, 4282, and 4295) carry novel severe loss-of-function mutations in known RP genes. (A–D) The segregation results of the four families. The genotype of each evaluated individual is shown below the individual's symbol. Wild type is listed as a “+.”
In addition to families that carry known pathogenic or severe loss-of-function variants in known RP genes, families that carry solely novel missense variants were identified. Since the pathogenicity of single amino acid substitution is often harder to interpret compared with known or severe loss-of-function variants, conservation score and functional prediction were utilized to facilitate the interpretation. As shown in Table 2, presumably damaging missense variants in known RP disease genes were identified in two families (4268 and 4294). Patient 4268_1 from arRP family 4268 was identified to carry a novel homozygous missense variant c.437T>A:p.(V146D) in RDH12, affecting a conserved valine (Fig. 4B) and predicted to be detrimental, with a SIFT score of 1.0. Patient 4294_1 from arRP family 4294 carries a homozygous mutation, c.1589C>G:p.(P530R), in CNGB1 (MIM #600724; NM_001297.4). This missense mutation affects a conserved proline (Fig. 4D) and was predicted to be detrimental, with a SIFT score of 0.99. The identified missense variants in both families cosegregated with the disease phenotypes and were therefore considered as putative pathogenic mutations (Figs. 4A, 4C).
Figure 4.

Two families (4268 and 4294) carry presumably damaging missense mutations in known RP genes. (A, C) The segregation results of the two families. The genotype of each evaluated individual is shown below the individual's symbol. Wild type is listed as a “+.” (B, D) Multispecies alignments show the conservation of the amino acids affected by the novel missense mutations. Red boxes show the affected amino acids.
Three Families Were Identified to Carry Putative Pathogenic Mutations in Other Retinal Disease Genes
Because of the genetic heterogeneity of RP, it is entirely possible that mutations in other retinal disease genes can also be associated with RP. In our study, among the remaining unsolved families, a search for mutations in 116 other retinal disease genes yielded three families that carry putative pathogenic mutations in two different genes, CYP4V2 and ADAM9 (Table 3).
Table 3. .
Three Families Were Identified to Carry Putative Pathogenic Mutations in Other Retinal Disease Genes
|
Family ID |
Patient ID |
Gene |
Allele1 |
Allele2 |
Reference |
| 4274 | 4274_1 | CYP4V2 (NM_207352.3) | c.802-8_810del17insGC* | c.1091-2A>G† | (Wada et al.,44 2005) (allele1) |
| (Li et al.,43 2004) (allele2) | |||||
| 4293 | 4293_1 | CYP4V2 (NM_207352.3) | c.1091-2A>G† | c.1396A>G:p.(N466D)‡ | (Li et al.,43 2004) (allele1) |
| Novel (allele2) | |||||
| 4263 | 4263_1 | ADAM9 (NM_003816.2) | c.1786C> T:p.(R596*)§ | c.1786C>T:p.(R596*)§ | Novel |
Small insertion-and-deletion mutation.
Splicing mutation.
Missense mutation.
Nonsense mutation.
Mutations in CYP4V2 Found in Two Families.
CYP4V2 was first characterized as a causative gene for Bietti crystalline corneoretinal dystrophy (BCD; MIM #210370), an autosomal recessive retinal disease featured with crystals in the cornea, yellow and shiny deposits in the retina, and progressive atrophy of the RPE and choriocapillaris. A recent study showed that two pathogenic mutations in CYP4V2 (NM_207352.3), c.802-8_810del17insGC and c.1091-2A>G, previously known to cause BCD,43,44 were also found in a Chinese family with a potential clinical diagnosis of RP.21 In our study, putative disease-causing variants in CYP4V2 were identified in two families. Specifically, patient 4274_1 from family 4274 carries known compound heterozygous mutations in CYP4V2, c.802-8_810del17insGC and c.1091-2A>G, that were also found in the previously reported arRP family,21 suggesting that these alleles might be common pathogenic alleles in Chinese patients. Patient 4293_1 from family 4293 carries compound heterozygous variants in CYP4V2. The known splicing mutation c.1091-2A>G was observed again in this patient, further suggesting that it might be a common pathogenic allele in the Chinese population. In addition, a novel missense variant c.1396A>G:p.(N466D) affecting a conserved asparagine (Fig. 5D) and predicted to be detrimental, with a SIFT score of 1.0, was identified in patient 4293_1. Segregation tests indicated that the variants cosegregated with the disease phenotypes (Figs. 5A, 5B). To clarify the ambiguity between the molecular and clinical diagnosis, we reevaluated the clinical information for both patients. Patient 4274_1 started experiencing a night vision problem with fairly good visual acuity when he was in his 20s. At 52 years of age, his visual acuity ranged from light perception to no light perception. Due to dense cataracts at both eyes, no clear fundus observations could be made, preventing further clinical reevaluation for this patient. Patient 4293_1 experienced progressive night blindness and reduced visual acuity (0.02/0.02, decimal Snellen). Atrophy of the retinal pigment epithelium and choriocapillaris with scattered pigment clamps were observed, but no yellow crystals on the retina were found (Supplementary Fig. S3). However, we found yellowish shiny crystals in his affected younger sister. From our experience, sometimes the crystals vanish as the patients age and as the disease progresses. As a result, family 4293 was clinically rediagnosed with BCD.
Figure 5.

Three families (4274, 4293, and 4263) carry putative pathogenic mutations in other retinal disease genes. (A–C) The segregation results of the three families. (D) Multispecies alignment shows the conservation of the amino acid affected by the novel missense mutation. Red box shows the affected amino acid.
Mutation in ADAM9 Found in One Family.
ADAM9 was first identified as a causative gene for recessive cone–rod dystrophy (CRD; MIM #120970), which is a progressive retinal dystrophy featured with predominant or equal loss of cone compared with rod photoreceptors. In our study, a patient from arRP family 4263 was identified to carry a novel homozygous nonsense variant, 1786C>T:p.(R596*), in ADAM9 (NM_003816.2), which introduces a premature stop in exon16, resulting in a truncated protein or nonsense-mediated decay. A segregation test indicated that the nonsense variant cosegregated with the disease phenotype (Fig. 5C). To clarify the ambiguity between the molecular and clinical diagnosis, we reevaluated the clinical information for this patient. Patient 4263_1 was a 9-year-old boy from a consanguineous family. Initially, he experienced severe night blindness but no significant loss in daylight vision. His vision acuity was 0.3/0.3 (decimal Snellen) at first clinical visit and decreased rapidly to 0.08/0.1 (decimal Snellen). Full-field ERG showed both rod and cone functions were impaired. However, the amplitude of b-wave in dark-adapted rod response was more severely decreased compared with cone responses. Fundus image and OCT are shown in Supplementary Figure S3. Based on these clinical phenotypes and the ADAM9 mutation identified in the family, the patients were rediagnosed to have CRD with early-onset retinal dystrophy.
Nineteen arRP Families Remained Unsolved
Surprisingly, no clear pathogenic mutation was found in 19 arRP families. Each rare variant in every proband was excluded for one or more reasons. For example, novel heterozygous missense variant c.T616G:p.S206A in CRX was identified in proband 4257_1. However, this variant was predicted to be benign, with a SIFT score of 0.38, and does not affect conserved amino acid. Therefore, it was excluded from further analysis. Detailed variant information and our reasons for exclusion are listed in Supplementary Table S3.
Discussion
NGS allows for the comprehensive molecular diagnosis of heterogeneous genetic diseases. Using NGS, we have performed targeted sequencing of 31 arRP patient families, the first such study of a Chinese patient cohort. A total of 12 families were solved, achieving a diagnostic rate of approximately 40%.
One important point conveyed by this study is that the molecular diagnosis of patients from different ethnic backgrounds can expand our knowledge of the spectrum of human disease causing mutations. In this study, approximately 63% of the identified pathogenic mutations are novel. This is higher than that observed in similar studies on European descent where approximately 45% of pathogenic alleles are novel.9 Since the majority of relevant studies on RP were conducted with patients of European descent and only a small subset of known RP disease genes have been investigated in patients from other populations, ethnic bias in our current knowledge of the mutation spectrum underlying RP likely exists. By screening patients from different ethnic backgrounds, we can minimize this potential bias and learn more about the global spectrum of human disease-causing mutations.
Our study also suggests that NGS-based comprehensive molecular diagnosis, by screening both known RP genes and other retinal disease genes, can help clinicians to provide a more accurate clinical diagnosis. Different retinal diseases have many overlapping clinical features, making accurate diagnosis very challenging, especially in late-stage retinal dystrophy. A molecular diagnosis allows clinicians to perform a more accurate clinical evaluation. As illustrated in family 4293, the proband showed poor visual acuity, flat ERG, and no shiny crystals. This phenotype is consistent with late-stage RP. However, once the molecular diagnosis identified pathogenic mutations in CYP4V2, we searched for BCD symptoms in the family and found yellowish shiny crystals in the proband's affected younger sister, allowing the patients to be rediagnosed as BCD patients.
It is worth noting that 19 families remain unsolved. For these families, PLINK has been performed to check any large deletion.45 No additional pathogenic mutations were identified. Nevertheless, our study has technical and methodologic limitations, leaving the possibility that some of the unsolved families may harbor pathogenic mutations in known genes. For example, our approach cannot detect large copy-number variants or structural variations. Moreover, deep intronic regions are not covered; as a result, mutations in these regions were not identified. In addition, 60 of 2560 exons are underrepresented in our capture sequencing (Supplementary Table S4), making variant detection in these regions error prone. Also, RPGR-ORF15 might not be handled well by our capture-sequencing pipeline because of its highly repetitive sequences. Future Sanger sequencing of RPGR-ORF15 and those underrepresented exons may be useful to uncover pathogenic mutations in these regions.
Our comprehensive molecular diagnostic approach is more cost-effective than recently published NGS-based methods.8,9,46 First, we have developed and optimized a molecular barcoding protocol that allows us to pool as many as 96 samples together prior to capture, greatly reducing the cost and labor during target enrichment as well as reducing the amount of patient DNA required. Second, instead of array-based capture, our enrichment step is performed in the liquid phase, greatly simplifying the experimental procedure and shortening the hybridization time. Third, instead of 454, SOLiD or GAII sequencers used in other studies, we used the more cost-efficient system that batch-processes multiple samples in a single run (Illumina HiSeq System; Illumina, Inc.). Finally, we have optimized of our probe design leading to more efficient and uniform target enrichment.
In summary, powered by NGS technology, molecular diagnosis of heterogeneous genetic diseases is becoming more feasible and affordable. Comprehensive molecular testing can help to provide a more accurate clinical diagnosis. Molecularly diagnosing patients from different ethnic groups may help us to better understand the global spectrum of disease-causing mutations.
Supplementary Material
Acknowledgments
The authors thank all participating patients and their family members.
Supported by grants from the Retina Research Foundation and the National Eye Institute (NEI)/National Institutes of Health (NIH) Grants R01EY022356 and R01EY018571 (RC); NEI/NIH Training Grant T32 EY007102 (JEZ); the Ministry of Human Resource and Social Security of the People's Republic of China (2009) and the Foundation Fighting Blindness USA Grant CD-CL-0808-0470-PUMCH (RS); and Foundation Fighting Blindness Canada, Canadian Institutes for Health Research, Reseau de Vision, Fonds de Recherche Sante du Quebec (FRSQ), NIH, and the Foundation for Retinal Research (RKK). The authors alone are responsible for the content and writing of the paper.
Disclosure: Q. Fu, None; F. Wang, None; H. Wang, None; F. Xu, None; J.E. Zaneveld, None; H. Ren, None; V. Keser, None; I. Lopez, None; H.-F. Tuan, None; J.S. Salvo, None; X. Wang, None; L. Zhao, None; K. Wang, None; Y. Li, None; R.K. Koenekoop, None; R. Chen, None; R. Sui, None
References
- 1. Pagon RA, Daiger SP. Retinitis pigmentosa overview. In: Pagon RA, TD Bird, Dolan CR, Stephens K, Adam MP. eds GeneReviews. Seattle, WA: University of Washington; 1993. [Google Scholar]
- 2. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006; 1: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368: 1795–1809 [DOI] [PubMed] [Google Scholar]
- 4. Retnet. Available at: http://www.sph.uth.tmc.edu/Retnet. Accessed November 11, 2012. [Google Scholar]
- 5. Kurg A, Tonisson N, Georgiou I, Shumaker J, Tollett J, Metspalu A. Arrayed primer extension: solid-phase four-color DNA resequencing and mutation detection technology. Genetic Test. 2000; 4: 1–7 [DOI] [PubMed] [Google Scholar]
- 6. Avila-Fernandez A, Cantalapiedra D, Aller E, et al. Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol Vis. 2010; 16: 2550–2558 [PMC free article] [PubMed] [Google Scholar]
- 7. Blanco-Kelly F, Garcia-Hoyos M, Corton M, et al. Genotyping microarray: mutation screening in Spanish families with autosomal dominant retinitis pigmentosa. Mol Vis. 2012; 18: 1478–1483 [PMC free article] [PubMed] [Google Scholar]
- 8. Neveling K, Collin RW, Gilissen C, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat. 2012; 33: 963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. O'Sullivan J, Mullaney BG, Bhaskar SS, et al. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. J Med Genet. 2012; 49: 322–326 [DOI] [PubMed] [Google Scholar]
- 10. Shanks ME, Downes SM, Copley RR, et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur J Hum Genet. 2013; 21: 274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu DN. Prevalence and mode of inheritance of major genetic eye diseases in China. Journal of Medical Genetics. 1987; 24: 584–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chan WM, Yeung KY, Pang CP, et al. Rhodopsin mutations in Chinese patients with retinitis pigmentosa. Br J Ophthalmol. 2001; 85: 1046–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li S, Xiao X, Wang P, Guo X, Zhang Q. Mutation spectrum and frequency of the RHO gene in 248 Chinese families with retinitis pigmentosa. Biochem Biophys Res Commun. 2010; 401: 42–47 [DOI] [PubMed] [Google Scholar]
- 14. Dryja TP, Hahn LB, Cowley GS, McGee TL, Berson EL. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991; 88: 9370–9374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sung CH, Davenport CM, Hennessey JC, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991; 88: 6481–6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ziviello C, Simonelli F, Testa F, et al. Molecular genetics of autosomal dominant retinitis pigmentosa (ADRP): a comprehensive study of 43 Italian families. J Med Genet. 2005; 42: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sullivan LS, Bowne SJ, Birch DG, et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006; 47: 3052–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu W, Dai H, Lu T, Zhang X, Dong B, Li Y. Seven novel mutations in the long isoform of the USH2A gene in Chinese families with nonsyndromic retinitis pigmentosa and Usher syndrome Type II. Mol Vis. 2011; 17: 1537–1552 [PMC free article] [PubMed] [Google Scholar]
- 19. Hosono K, Ishigami C, Takahashi M, et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS One. 2012; 7: e31036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu T, Jin X, Zhang X, et al. A novel missense SNRNP200 mutation associated with autosomal dominant retinitis pigmentosa in a Chinese family. PLoS One. 2012; 7: e45464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Guo L, Cai SP, et al. Exome sequencing identifies compound heterozygous mutations in CYP4V2 in a pedigree with retinitis pigmentosa. PLoS One. 2012; 7: e33673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Challis D, Yu J, Evani US, et al. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012; 13: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Genomes Project C A map of human genome variation from population-scale sequencing. Nature. 2010; 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. National Center for Biotechnology Information NLoM Database of Single Nucleotide Polymorphisms (dbSNP). NCBI dbSNP Build 135. [Google Scholar]
- 27. NHLBI GO Exome Sequencing Project (ESP) S, WA. Available at: http://evs.gs.washington.edu/EVS/. Accessed November 23, 2012. [Google Scholar]
- 28. NIEHS Environmental Genome Project S, WA. Available at: http://evs.gs.washington.edu/niehsExome/. Accessed November 23, 2012. [Google Scholar]
- 29. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38: e164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003; 31: 3812–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu X, Jian X. Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011; 32: 894–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vertebrate Multiz Alignment & Conservation (46 Species), UCSC Genome Browser. Available at: https://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=332327213&g=cons46way&hgTracksConfigPage=configure. Accessed December 19, 2012 [Google Scholar]
- 33. Stenson PD, Ball EV, Mort M, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003; 21: 577–581 [DOI] [PubMed] [Google Scholar]
- 34. Smit A, Hubley R, Green P. RepeatMasker Open-3.0. Available at: http://www.repeatmasker.org 1996–2010. [Google Scholar]
- 35. Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000; 132: 365–386 [DOI] [PubMed] [Google Scholar]
- 36. Mokry M, Feitsma H, Nijman IJ, et al. Accurate SNP and mutation detection by targeted custom microarray-based genomic enrichment of short-fragment sequencing libraries. Nucleic Acids Res. 2010; 38: e116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aldahmesh MA, Safieh LA, Alkuraya H, et al. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol Vis. 2009; 15: 2464–2469 [PMC free article] [PubMed] [Google Scholar]
- 38. Mackay DS, Dev, Borman A, Moradi P, et al. RDH12 retinopathy: novel mutations and phenotypic description. Mol Vis. 2011; 17: 2706–2716 [PMC free article] [PubMed] [Google Scholar]
- 39. Xu F, Dong Q, Liu L, et al. Novel RPE65 mutations associated with Leber congenital amaurosis in Chinese patients. Mol Vis. 2012; 18: 744–750 [PMC free article] [PubMed] [Google Scholar]
- 40. Dai H, Zhang X, Zhao X, et al. Identification of five novel mutations in the long isoform of the USH2A gene in Chinese families with Usher syndrome type II. Mol Vis. 2008; 14: 2067–2075 [PMC free article] [PubMed] [Google Scholar]
- 41. Le Quesne Stabej P,, Saihan Z,, Rangesh N,, et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J Med Genet. 2012; 49: 27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci U S A. 1995; 92: 3249–3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li A, Jiao X, Munier FL, et al. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2. Am J Hum Genet. 2004; 74: 817–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wada Y, Itabashi T, Sato H, Kawamura M, Tada A, Tamai M. Screening for mutations in CYP4V2 gene in Japanese patients with Bietti's crystalline corneoretinal dystrophy. Am J Ophthalmol. 2005; 139: 894–899 [DOI] [PubMed] [Google Scholar]
- 45. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81: 559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Audo I, Bujakowska KM, Leveillard T, et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis. 2012; 7: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.